Polyamide 11/Poly(butylene succinate) Bio-Based Polymer Blends


Abstract
:
1. Introduction
2. Experimental Part
2.1. Materials
2.2. Blend Preparation
2.3. Preparation of Compression-Molded Sheets
2.4. Differential Scanning Calorimetry
2.5. Thermogravimetry
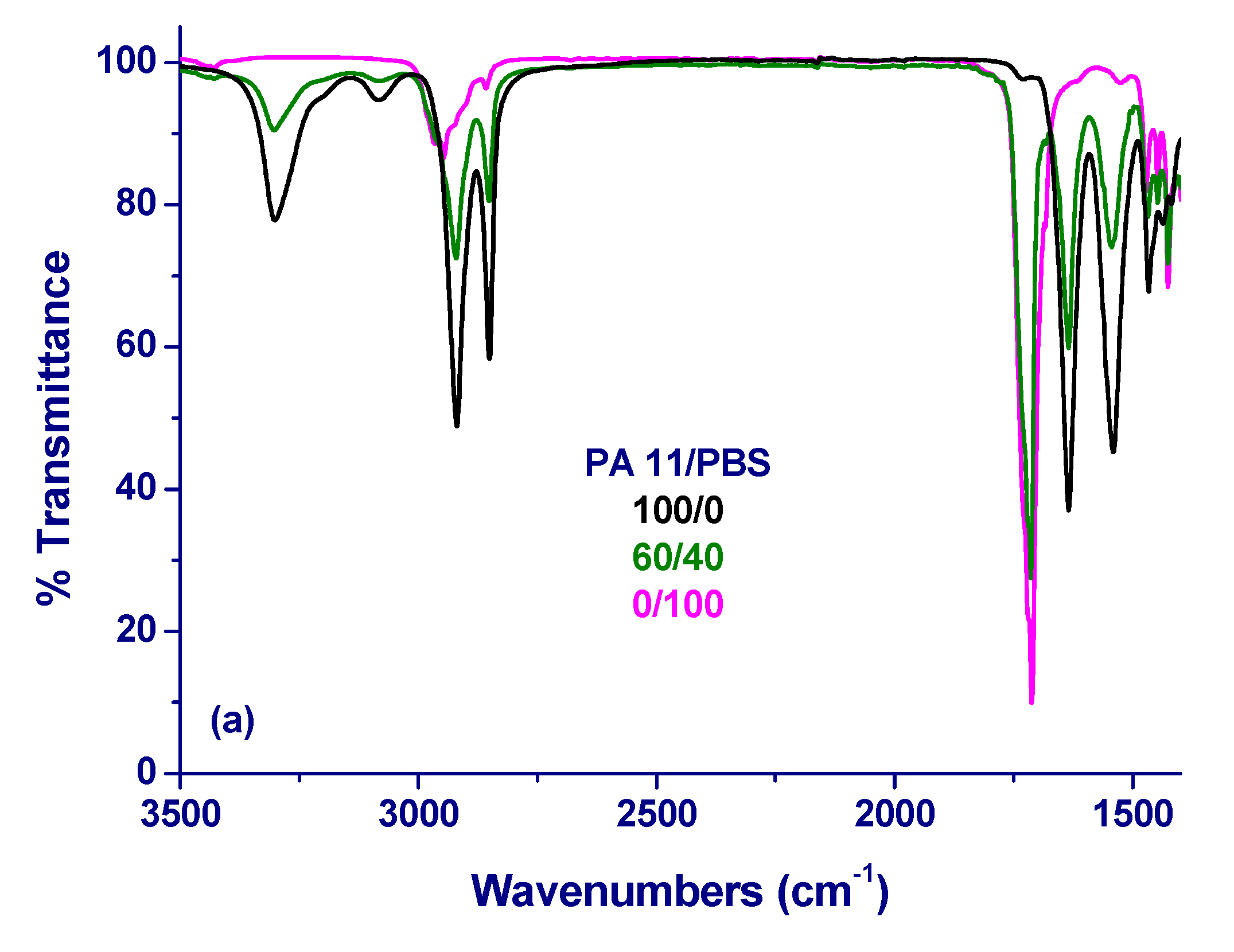
2.6. Fourier-Transform Infrared Spectroscopy
2.7. Scanning Electron Microscopy
2.8. Tensile Tests
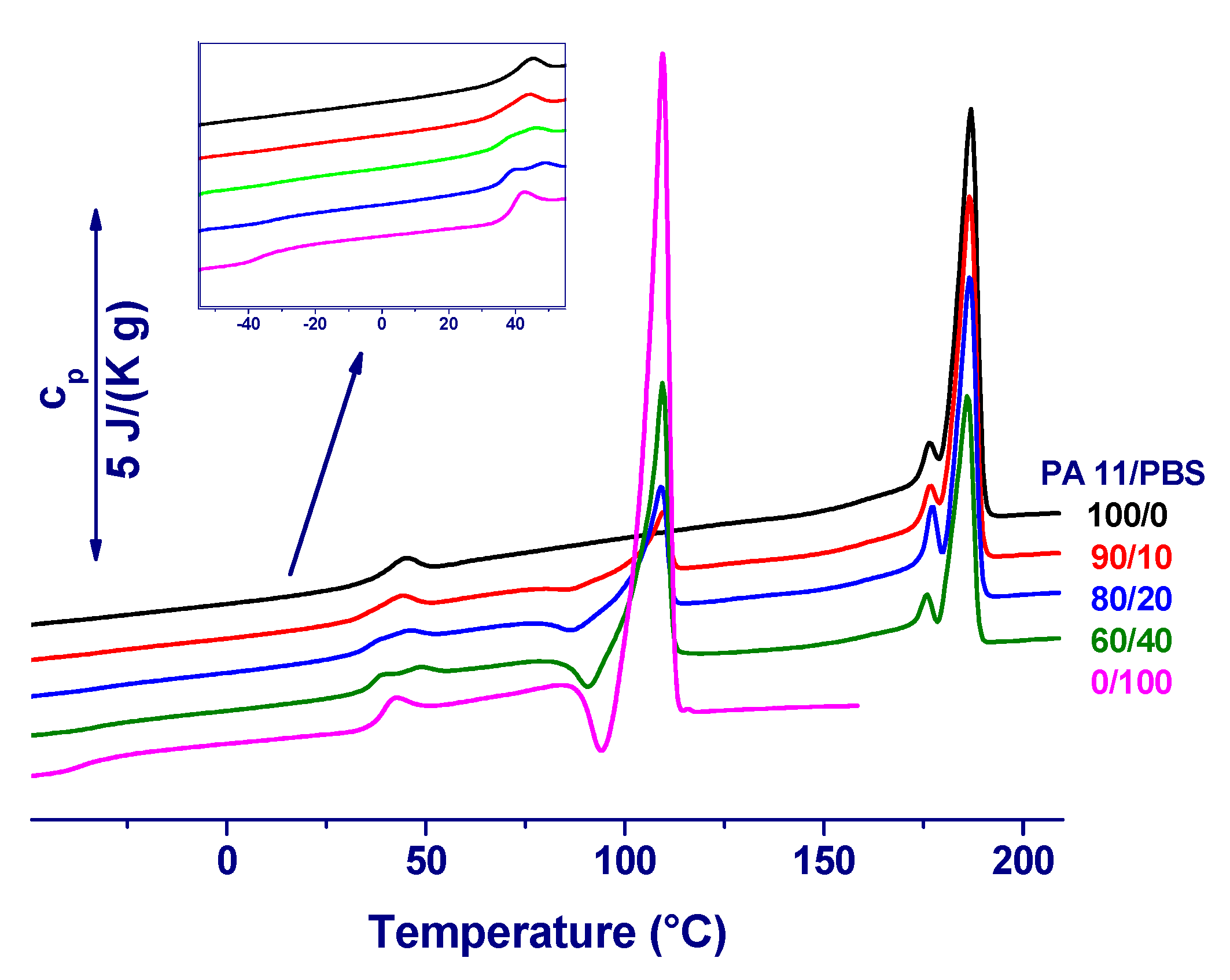
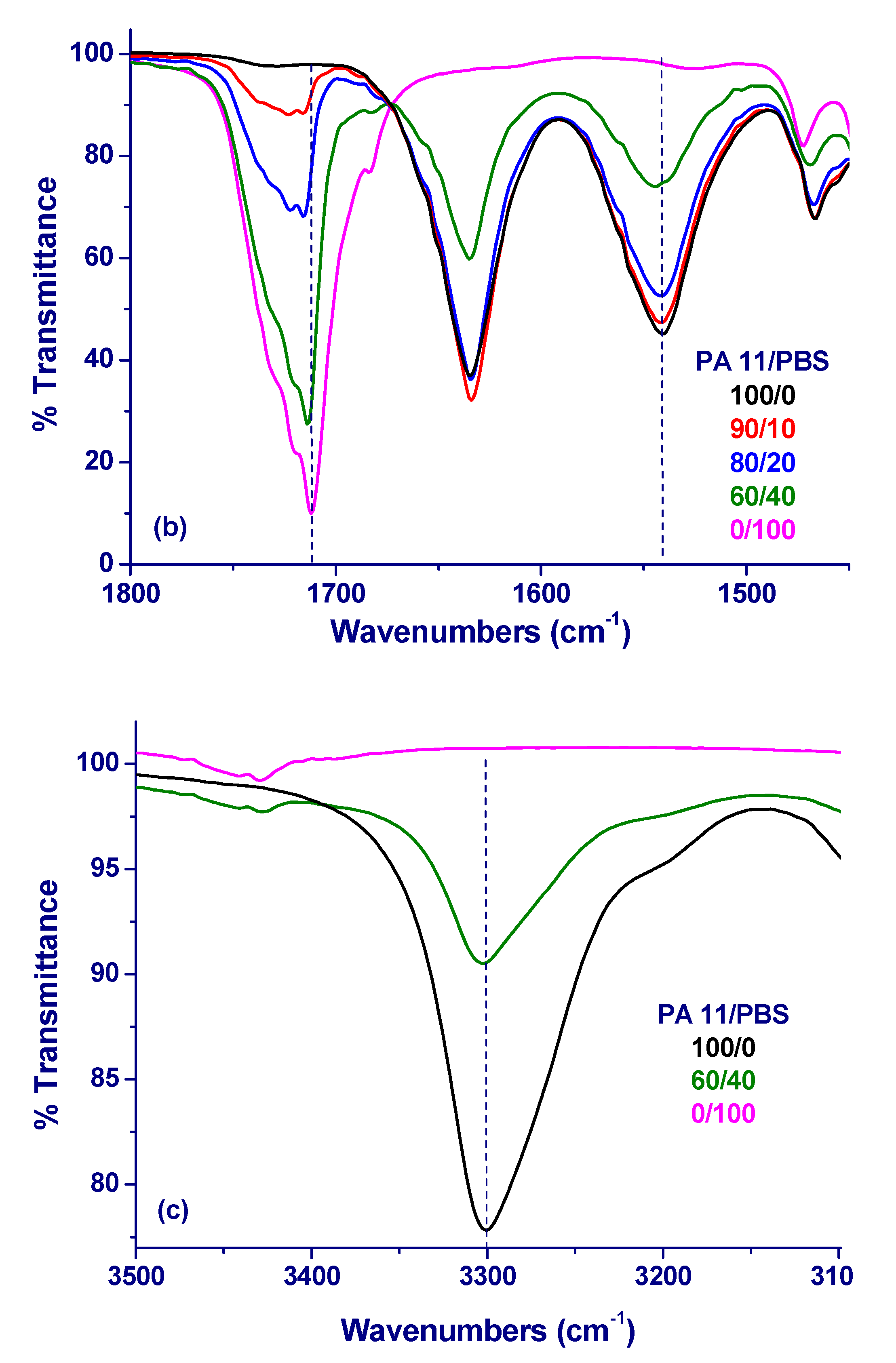
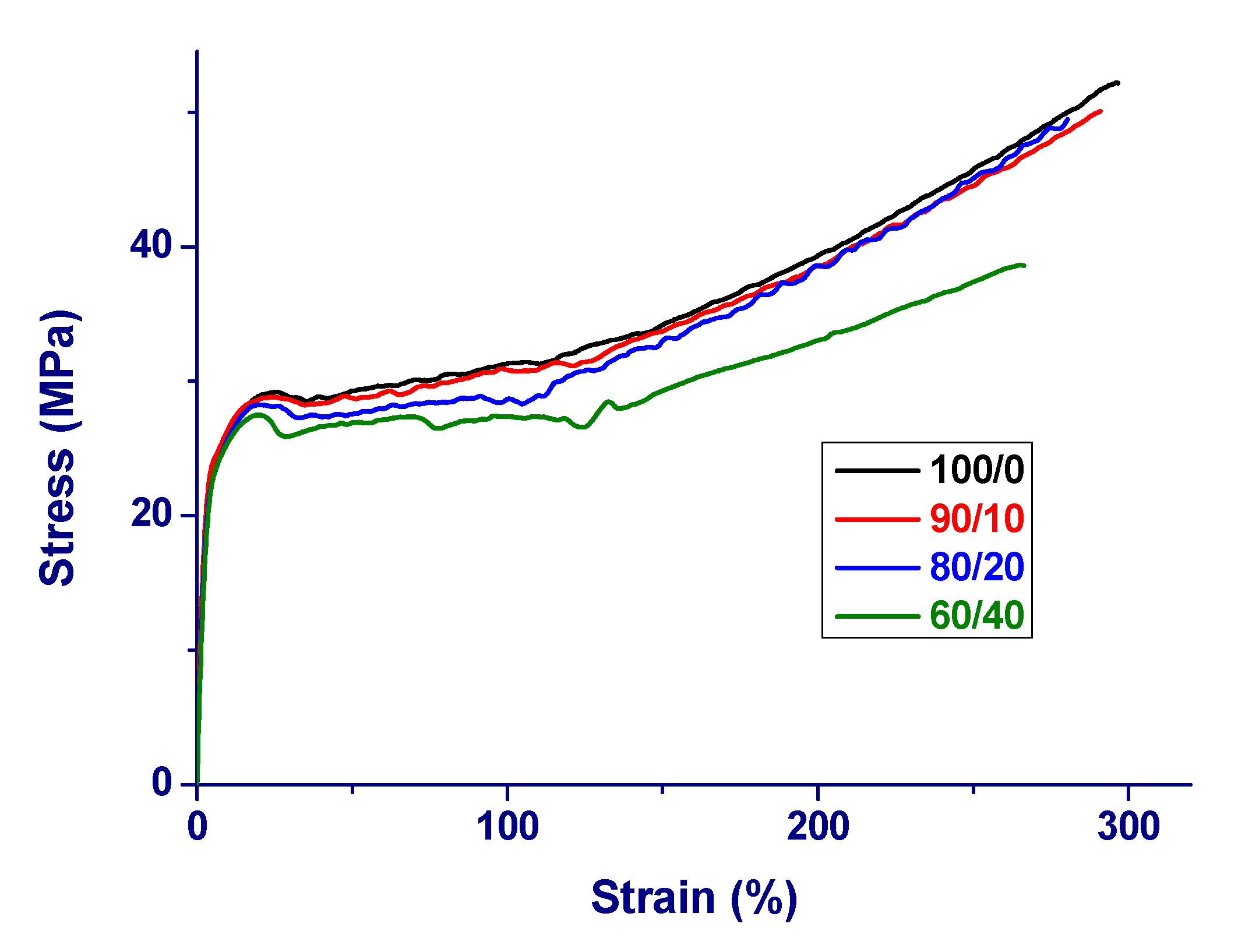
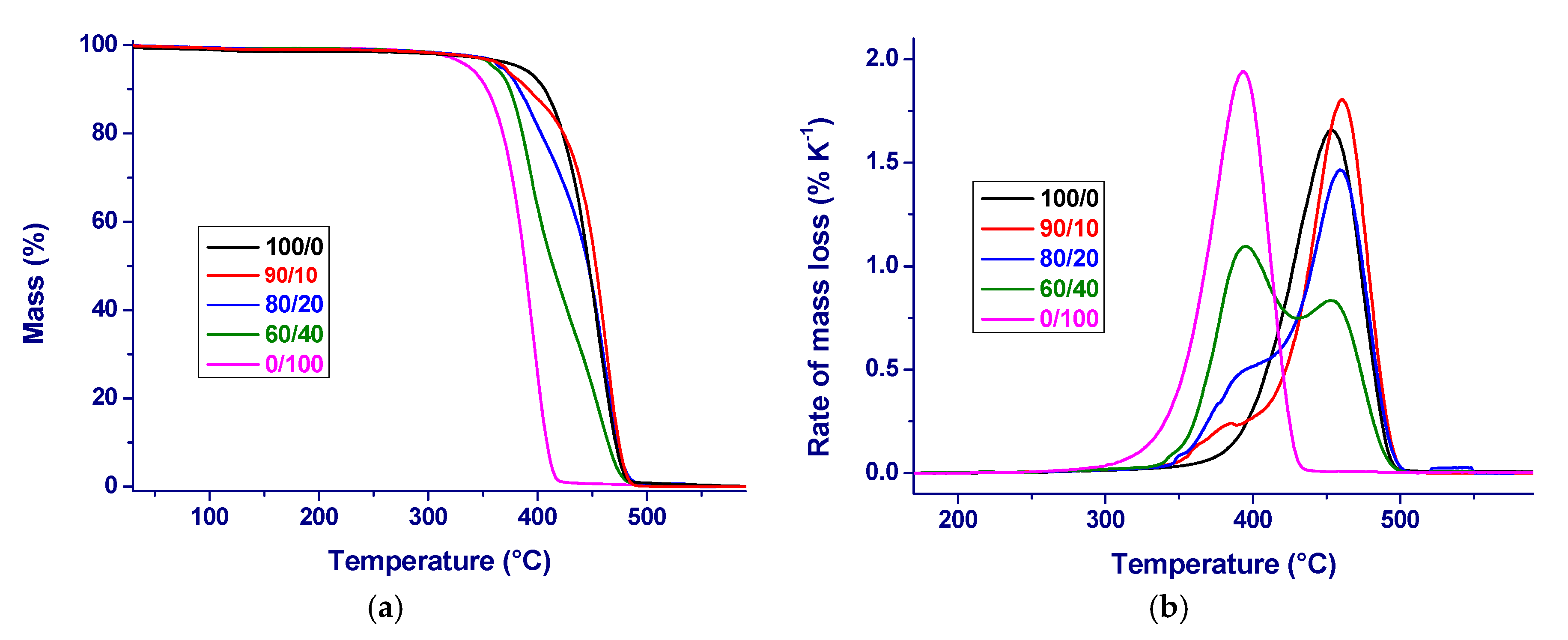
3. Results and Initial Discussion
4. Final Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bioplastic Market Data. Available online: www.european-bioplastics.org/market/ (accessed on 30 May 2019).
- Scott, J.L.; Buchard, A. Polymers from plants: Biomass fixed carbon dioxide as a resource. In Managing Global Warming—An Interface of Technology and Human Issues; Letcher, T.M., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 503–525. [Google Scholar]
- Imre, B.; Pukánszky, B. Compatibilization in bio-based and biodegradable polymer blends. Eur. Polym. J. 2013, 49, 1215–1233. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, M.L.; Raimo, M.; Cascone, E.; Martuscelli, E. Poly(3-hydroxybutyrate)-based copolymers and blends: Influence of a second component on crystallization and thermal behavior. J. Macromol. Sci.—Phys. 2001, 40, 639–667. [Google Scholar] [CrossRef]
- Nakajima, H.; Dijkstra, P.; Loos, K. The Recent Developments in Biobased Polymers toward General and Engineering Applications: Polymers That Are Upgraded from Biodegradable Polymers, Analogous to Petroleum-Derived Polymers, and Newly Developed. Polymers 2017, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, M.L.; Androsch, R. (Eds.) Industrial Applications of Poly(lactic acid); Springer International Publishing: Cham, Switzerland, 2018; p. 282. [Google Scholar]
- Paul, D.R.; Newman, S. Polymer Blends; Academic Press: New York, NY, USA, 1978. [Google Scholar]
- Sionkowska, A. Current research on the blends of natural and synthetic polymers as new biomaterials: Review. Progr. Polym. Sci. 2011, 36, 1254–1276. [Google Scholar] [CrossRef]
- Hamad, K.; Kaseem, M.; Ayyoob, M.; Joo, J.; Deri, F. Polylactic acid blends: The future of green, light and tough. Progr. Polym. Sci. 2018, 85, 83–127. [Google Scholar] [CrossRef]
- Martino, L.; Basilissi, L.; Farina, H.; Ortenzi, M.A.; Zini, E.; Di Silvestro, G.; Scandola, M. Bio-based polyamide 11: Synthesis, rheology and solid-state properties of star structures. Eur. Polym. J. 2014, 59, 69–77. [Google Scholar] [CrossRef]
- Patel, R.; Ruehle, D.A.; Dorgan, J.R.; Halley, P.; Martin, D. Biorenewable blends of polyamide-11 and polylactide. Polym. Eng. Sci. 2014, 54, 1523–1532. [Google Scholar] [CrossRef]
- He, X.; Yang, J.; Zhu, L.; Wang, B.; Sun, G.; Lv, P.; Phang, I.Y.; Liu, T. Morphology and melt rheology of nylon 11/clay nanocomposites. J. Appl. Polym. Sci. 2006, 102, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Lim, K.P.; Tjiu, W.C.; Pramoda, K.P.; Chen, Z.K. Preparation and characterization of nylon 11/organoclay nanocomposites. Polymer 2003, 44, 3529–3535. [Google Scholar] [CrossRef]
- Mancic, L.; Osman, R.F.M.; Costa, A.M.L.M.; d’Almeida, J.R.M.; Marinkovic, B.A.; Rizzo, F.C. Thermal and mechanical properties of polyamide 11 based composites reinforced with surface modified titanate nanotubes. Mater. Des. 2015, 83, 459–467. [Google Scholar] [CrossRef]
- Sahnoune, M.; Taguet, A.; Otazaghine, B.; Kaci, M.; Lopez-Cuesta, J.M. Effects of functionalized halloysite on morphology and properties of polyamide-11/SEBS-g-MA blends. Eur. Polym. J. 2017, 90, 418–430. [Google Scholar] [CrossRef]
- Yu, M.; Zhang, Q.; Fu, Q. Preparation and characterisation of polyamide 11/clay nanocomposites. Chin. J. Polym. Sci. 2004, 22, 43–47. [Google Scholar]
- Halim, K.A.A.; Farrell, J.B.; Kennedy, J.E. Preparation and characterisation of polyamide 11/montmorillonite (MMT) nanocomposites for use in angioplasty balloon applications. Mater. Chem. Phys. 2013, 143, 336–348. [Google Scholar] [CrossRef]
- Jariyavidyanont, K.; Focke, W.; Androsch, R. Crystallization kinetics of polyamide 11 in presence of sepiolite and montmorillonite nanofillers. Coll. Polym. Sci. 2016, 294, 1143–1151. [Google Scholar] [CrossRef]
- Tercjak, A.; Haponiuk, J.T.; Masiulanis, B. Study of thermal property changes of biopol/polyamide 11 blends during biodegradation in compost. J. Therm. Anal. Calor. 2003, 74, 605–608. [Google Scholar] [CrossRef]
- Walha, F.; Lamnawar, K.; Maazouz, A.; Jaziri, M. Rheological, Morphological and Mechanical Studies of Sustainably Sourced Polymer Blends Based on Poly(Lactic Acid) and Polyamide 11. Polymers 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Sumiya, N.; Saito, T. Impact strength improvement of polyamide 11 without flexural modulus reduction by dispersing poly(butylene succinate) particles. Polym. J. 2016, 48, 221–224. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Mizukoshi, T. Bionolle (Polybutylenesuccinate). Adv. Polym. Sci. 2012, 245, 285–313. [Google Scholar]
- Wu, Y.; Xiong, W.; Zhou, H.; Li, H.; Xu, G.; Zhao, J. Biodegradation of poly(butylene succinate) film by compost microorganisms and water soluble product impact on mung beans germination. Polym. Degrad. Stab. 2016, 126, 22–30. [Google Scholar] [CrossRef]
- Chanoine, P. Specialty Polyamides High Performance Materials Segment. Available online: https://www.arkema.com/export/sites/global/.content/medias/downloads/investorrelations/en/finance/arkema-investor-day-2012-specialty-polyamides-va.pdf (accessed on 1 July 2019).
- Schütte, A. Processing of Bioplastics: A Guideline. Available online: https://www.ifbb-hannover.de/files/IfBB/downloads/EV_Processing-of-Bioplastics-2016.pdf (accessed on 1 July 2019).
- Van den Oever, M.; Molenveld, K.; van der Zee, M.; Bos, H. Bio-Based and Biodegradable Plastics: Facts and Figures: Focus on Food Packaging in the Netherlands (Rapport nr. 1722); Wageningen Food Biobased Research: Wageningen, The Netherlands, 2017; ISBN 9789463431217. [Google Scholar]
- Jariyavidyanont, K.; Focke, W.; Androsch, R. Thermal Properties of Biobased Polyamide 11. In Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Xenopoulos, A.; Wunderlich, B. Thermodynamic properties of liquid and semicrystalline linear aliphatic polyamides. J. Polym. Sci. Part B Polym. Phys. 1990, 28, 2271–2290. [Google Scholar] [CrossRef]
- Mathias, L.J.; Powell, D.G.; Autran, J.P.; Porter, R.S. Nitrogen-15 nmr characterization of multiple crystal forms and phase transitions in polyundecanamide (nylon 11). Macromolecules 1990, 23, 963–967. [Google Scholar] [CrossRef]
- Zhang, G.; Li, Y.; Yan, D. Polymorphism in nylon-11/montmorillonite nanocomposite. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 253–259. [Google Scholar] [CrossRef]
- Nair, S.S.; Ramesh, C.; Tashiro, K. Crystalline phases in nylon-11: Studies using HTWAXS and HTFTIR. Macromolecules 2006, 39, 2841–2848. [Google Scholar] [CrossRef]
- Rhoades, A.M.; Wonderling, N.; Schick, C.; Androsch, R. Supercooling-controlled heterogeneous and homogenous crystal nucleation of polyamide 11 and its effect onto the crystal/mesophase polymorphism. Polymer 2016, 106, 29–34. [Google Scholar] [CrossRef]
- Jariyavidyanont, K.; Williams, J.L.; Rhoades, A.M.; Kühnert, I.; Focke, W.; Androsch, R. Crystallization of polyamide 11 during injection molding. Polym. Eng. Sci. 2018, 58, 1053–1061. [Google Scholar] [CrossRef]
- Mollova, A.; Androsch, R.; Mileva, D.; Schick, C.; Benhamida, A. Effect of supercooling on crystallization of polyamide 11. Macromolecules 2013, 46, 828–835. [Google Scholar] [CrossRef]
- Jariyavidyanont, K.; Janke, A.; Androsch, R. Crystal self-nucleation of polyamide 11. Thermochim. Acta 2019, 677, 139–143. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Androsch, R.; Righetti, M.C. Low-temperature crystallization of poly(butylene succinate). Eur. Polym. J. 2017, 94, 384–391. [Google Scholar] [CrossRef]
- Signori, F.; Pelagaggi, M.; Bronco, S.; Righetti, M.C. Amorphous/crystal and polymer/filler interphases in biocomposites from poly(butylene succinate). Thermochim. Acta 2012, 543, 74–81. [Google Scholar] [CrossRef]
- Gan, Z.; Abe, H.; Kurokawa, H.; Doi, Y. Solid-state microstructures, thermal properties, and crystallization of biodegradable poly(butylene succinate) (PBS) and its copolyesters. Biomacromolecules 2001, 2, 605–613. [Google Scholar] [CrossRef]
- Pyda, M. (Ed.) Nylon 11 (NYLON11) Heat Capacity, Enthalpy, Entropy, Gibbs Energy; Datasheet from “The Advanced THermal Analysis System (ATHAS) Databank—Polymer Thermodynamics”; Springer Materials, Springer-Verlag GmbH: Heidelberg, Germany, 2014. [Google Scholar]
- Zhang, Q.; Mo, Z.; Liu, S.; Zhang, H. Influence of annealing on structure of Nylon 11. Macromolecules 2000, 33, 5999–6005. [Google Scholar] [CrossRef]
- Verespej, M. NatureWorks Launches Biopolymer Alloys. Plast. Technol. 2012, 4. Available online: https://www.ptonline.com/articles/natureworks-launches-biopolymer-alloys (accessed on 2 July 2019).
- Rilsan BESNO TL Product Information. Available online: https://www.extremematerials-arkema.com/en/materials-database/ (accessed on 28 June 2019).
- Wunderlich, B. Thermal Analysis of Polymeric Materials; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Androsch, R.; Wunderlich, B. Scanning Calorimetry. In Macromolecular Engineering; Matyjaszewski, K., Gnanou, Y., Leibler, L., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; Volume 3, Chapter 12. [Google Scholar]
- Papageorgiou, D.G.; Zhuravlev, E.; Papageorgiou, G.Z.; Bikiaris, D.; Chrissafis, K.; Schick, C. Kinetics of nucleation and crystallization in poly(butylene succinate) nanocomposites. Polymer 2014, 55, 6725–6734. [Google Scholar] [CrossRef]
- Righetti, M.C.; Tombari, E.; Di Lorenzo, M.L. Crystalline, mobile amorphous and rigid amorphous fractions in isotactic polystyrene. Eur. Polym. J. 2008, 44, 2659–2667. [Google Scholar] [CrossRef]
- Righetti, M.C.; Laus, M.; Di Lorenzo, M.L. Temperature dependence of the rigid amorphous fraction in poly(ethylene terephthalate). Eur. Polym. J. 2014, 58, 60–68. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Righetti, M.C. Crystallization-induced formation of rigid amorphous fraction. Polym. Cryst. 2018, 1, e10023. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Bikiaris, D.N. Crystallization and melting behavior of three biodegradable poly(alkylene succinates). A comparative study. Polymer 2005, 46, 12081–12092. [Google Scholar] [CrossRef]
- Wu, S. Chain structure, phase morphology, and toughness relationships in polymers and blends. Polym. Eng. Sci. 1990, 30, 753–761. [Google Scholar] [CrossRef]
- Yao, S.F.; Chen, X.T.; Ye, H.M. Investigation of Structure and Crystallization Behavior of Poly(butylene succinate) by Fourier Transform Infrared Spectroscopy. J. Phys. Chem. B 2017, 121, 9476–9485. [Google Scholar] [CrossRef]
- Hong, K.; Rastogi, A.; Strobl, G. A Model Treating Tensile Deformation of Semicrystalline Polymers: Quasi-Static Stress−Strain Relationship and Viscous Stress Determined for a Sample of Polyethylene. Macromolecules 2004, 37, 10165–10173. [Google Scholar] [CrossRef]
- Patlazhan, S.; Remond, Y. Structural mechanics of semicrystalline polymers prior to the yield point: A review. J. Mater. Sci. 2012, 47, 6749–6767. [Google Scholar] [CrossRef]
- Pawlak, A.; Galeski, A.; Rozanski, A. Cavitation during deformation of semicrystalline polymers. Progr. Polym. Sci. 2014, 39, 921–958. [Google Scholar] [CrossRef]
- Stoclet, G.; Sclavons, M.; Devaux, J. Relations between structure and property of polyamide 11 nanocomposites based on raw clays elaborated by water-assisted extrusion. J. Appl. Polym. Sci. 2013, 127, 4809–4824. [Google Scholar] [CrossRef]
- Ruehle, D.A.; Perbix, C.; Castañeda, M.; Dorgan, J.R.; Mittal, V.; Halley, P.; Martin, D. Blends of biorenewable polyamide-11 and polyamide-6,10. Polymer 2013, 54, 6961–6970. [Google Scholar] [CrossRef]
- Li, H.; Chang, J.; Cao, A.; Wang, J. In vitro evaluation of biodegradable poly(butylene succinate) as a novel biomaterial. Macromol. Biosci. 2005, 5, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Yu, J.; Chang, P.R.; Li, J.; Huang, J. Poly(butylene succinate)-based biocomposites filled with polysaccharide nanocrystals: Structure and properties. Polym. Compos. 2011, 32, 472–482. [Google Scholar] [CrossRef]
- Yun, I.S.; Hwang, S.W.; Shim, J.K.; Seo, K.H. A Study on the Thermal and Mechanical Properties of Poly (Butylene Succinate)/Thermoplastic Starch Binary Blends. Int. J. Prec. Eng. Manuf.—Green Technol. 2016, 3, 289–296. [Google Scholar] [CrossRef]
- Totaro, G.; Sisti, L.; Celli, A.; Askanian, H.; Verney, V.; Leroux, F. Poly(butylene succinate) bionanocomposites: A novel bio-organo-modified layered double hydroxide for superior mechanical properties. RSC Adv. 2016, 6, 4780–4791. [Google Scholar] [CrossRef]
- Xu, J.; Guo, B.H. Microbial Succinic Acid, Its Polymer Poly(butylene succinate), and Applications. In Plastics from Bacteria; Microbiology Monographs Volume 14; Chen, G.Q., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 14. [Google Scholar]
- Puchalski, M.; Szparaga, G.; Biela, T.; Gutowska, A.; Sztajnowski, S.; Krucínska, I. Molecular and Supramolecular Changes in Polybutylene Succinate (PBS) and Polybutylene Succinate Adipate (PBSA) Copolymer during Degradation in Various Environmental Conditions. Polymers 2018, 10, 251. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Righetti, M.C. The three-phase structure of isotactic poly(1-butene). Polymer 2008, 49, 1323–1331. [Google Scholar] [CrossRef]
- Mileva, D.; Zia, Q.; Androsch, R. Tensile properties of random copolymers of polypropylene with ethylene and 1-butene: Effect of crystallinity and crystal habit. Polym. Bull. 2010, 65, 623–634. [Google Scholar] [CrossRef]
- Glüge, R.; Altenbach, H.; Kolesov, I.; Mahmood, N.; Beiner, M.; Androsch, R. On the effective elastic properties of isotactic polypropylene. Polymer 2019, 160, 291–302. [Google Scholar] [CrossRef]
- Cimmino, S.; Di Lorenzo, M.L.; Di Pace, E.; Silvestre, C. Isotactic Poly(1-butene)/Hydrogenated Oligo(cyclopentadiene) Blends: Miscibility, Morphology, and Thermal and Mechanical Properties. J. Appl. Polym. Sci. 1998, 67, 1369–1381. [Google Scholar] [CrossRef]
- Chrissafis, K.; Paraskevopoulos, K.M.; Bikiaris, D.N. Thermal degradation mechanism of poly(ethylene succinate) and poly(butylene succinate): Comparative study. Thermochim. Acta 2005, 435, 142–150. [Google Scholar] [CrossRef]
- Mallardo, S.; De Vito, V.; Malinconico, M.; Volpe, M.G.; Santagata, G.; Di Lorenzo, M.L. Poly(butylene succinate)-based composites containing β-cyclodextrin/d-limonene inclusion complex. Eur. Polym. J. 2016, 79, 82–96. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PA 11/PBS | E (MPa) | σy (MPa) | εy (%) | σr (MPa) | εr (%) |
|---|---|---|---|---|---|
| 100/0 | 930 ± 40 | 29 ± 1 | 25 ± 2 | 52 ± 3 | 300 ± 20 |
| 90/10 | 910 ± 30 | 29 ± 1 | 25 ± 3 | 50 ± 3 | 290 ± 20 |
| 80/20 | 890 ± 40 | 28 ± 2 | 23 ± 5 | 49 ± 5 | 280 ± 20 |
| 60/40 | 870 ± 30 | 27 ± 2 | 20 ± 3 | 39 ± 5 | 270 ± 30 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Lorenzo, M.L.; Longo, A.; Androsch, R. Polyamide 11/Poly(butylene succinate) Bio-Based Polymer Blends. Materials 2019, 12, 2833. https://doi.org/10.3390/ma12172833
Di Lorenzo ML, Longo A, Androsch R. Polyamide 11/Poly(butylene succinate) Bio-Based Polymer Blends. Materials. 2019; 12(17):2833. https://doi.org/10.3390/ma12172833
Chicago/Turabian StyleDi Lorenzo, Maria Laura, Alessandra Longo, and René Androsch. 2019. "Polyamide 11/Poly(butylene succinate) Bio-Based Polymer Blends" Materials 12, no. 17: 2833. https://doi.org/10.3390/ma12172833