Diferrocenyl Thioketone: Reactions with (Bisphosphane)Pt(0) Complexes—Electrochemical and Computational Studies

 ,
,
Abstract
:
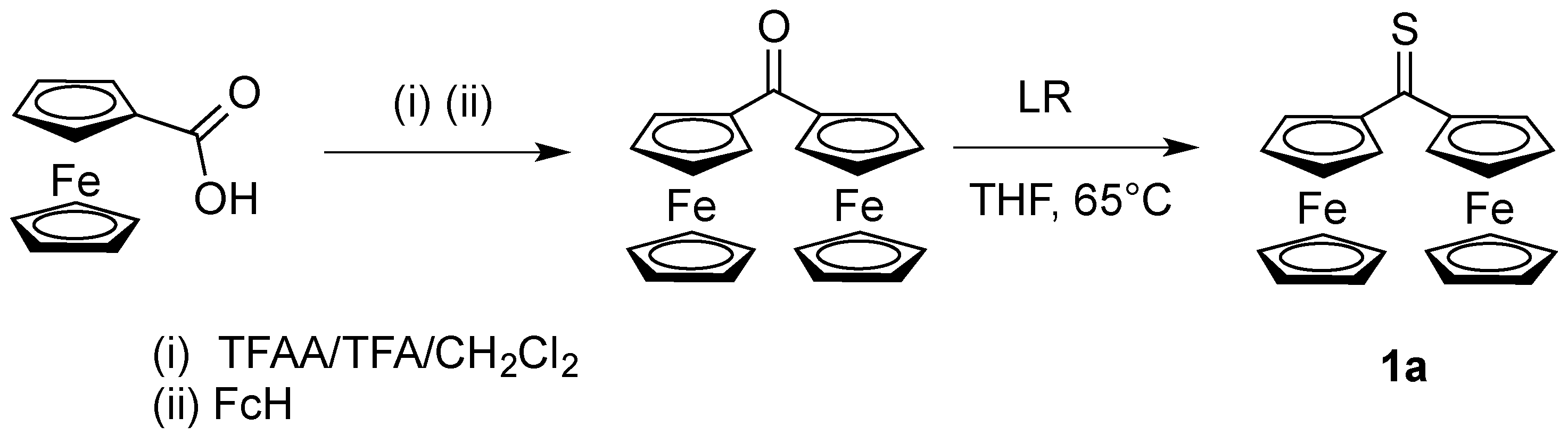
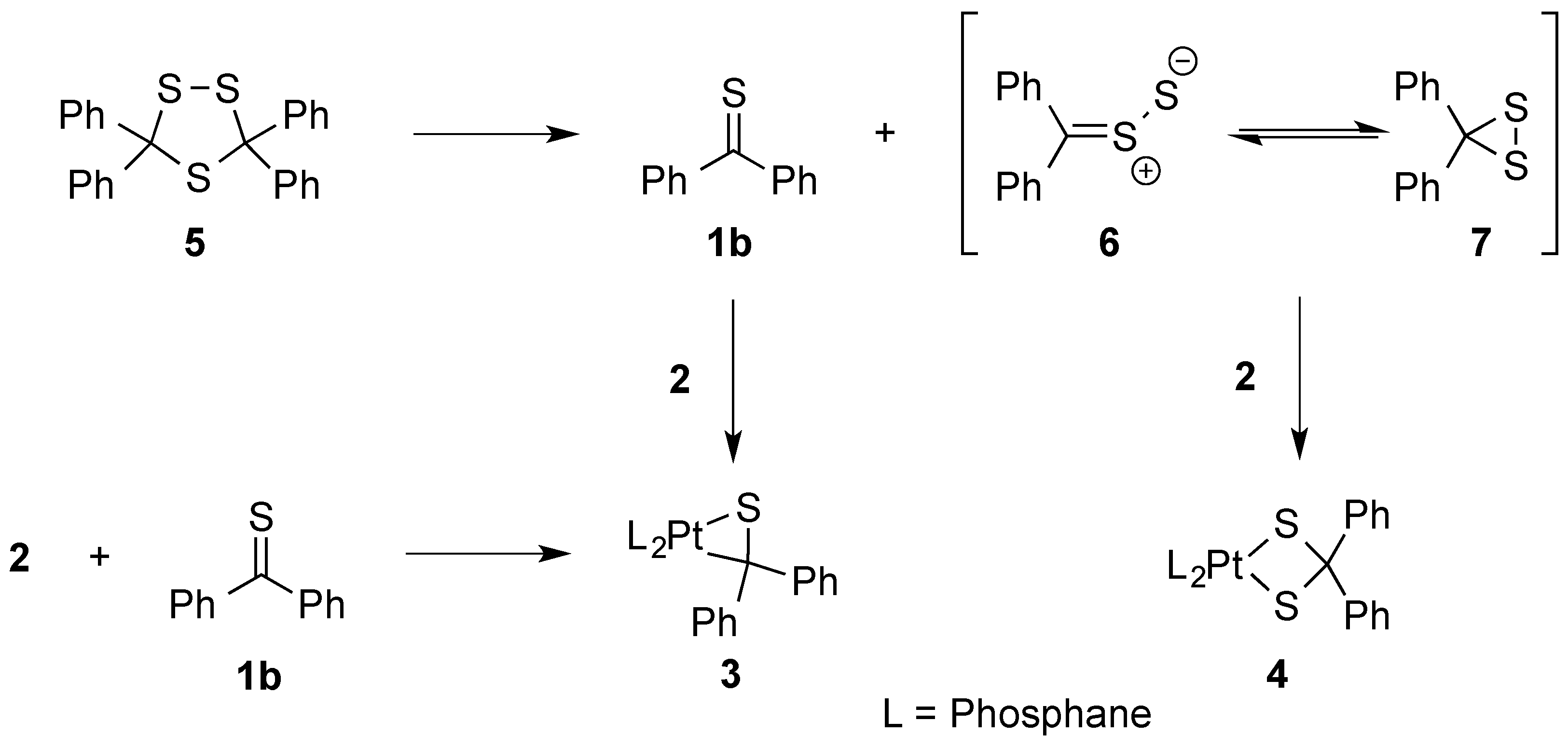
1. Introduction
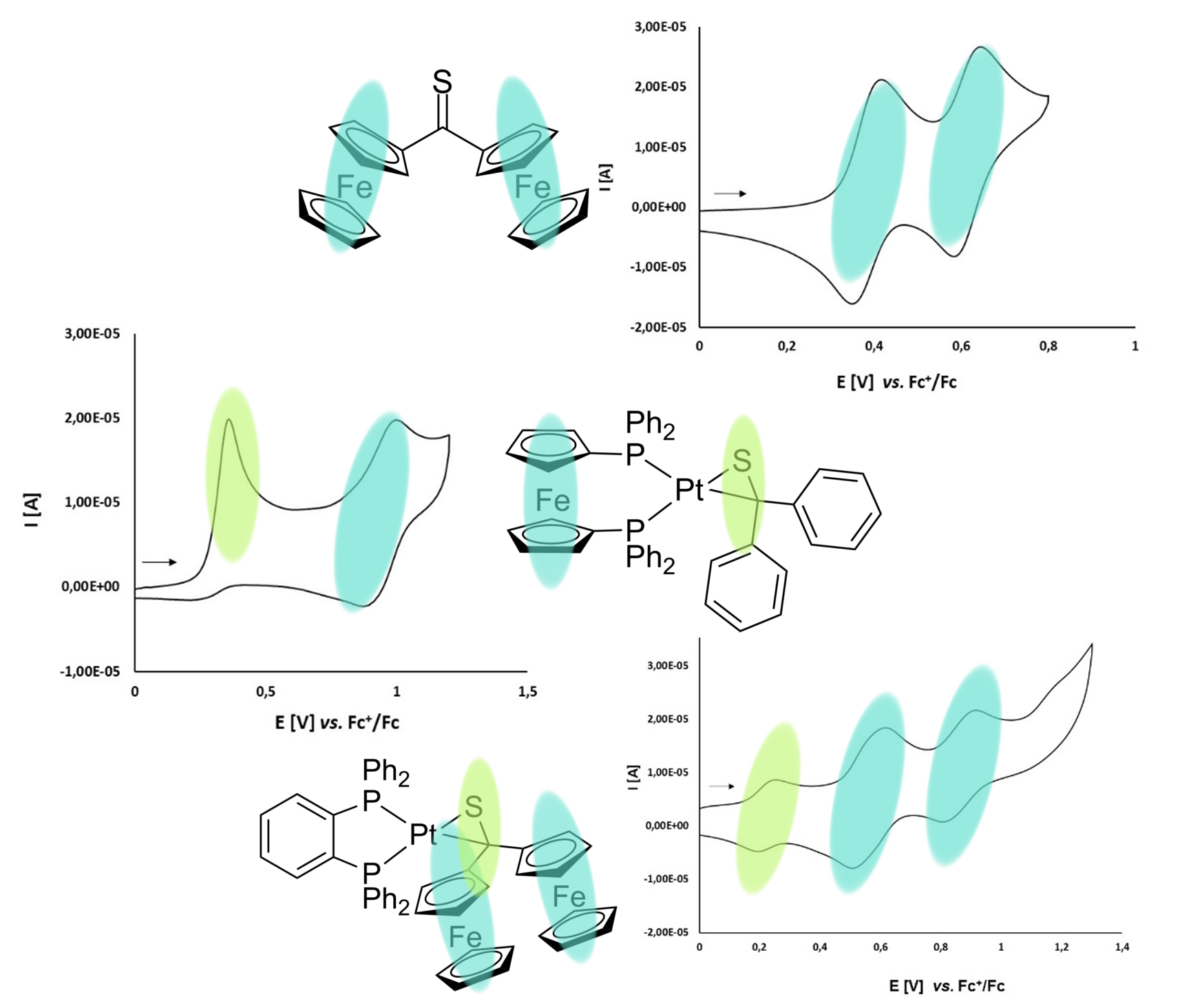
2. Materials and Methods
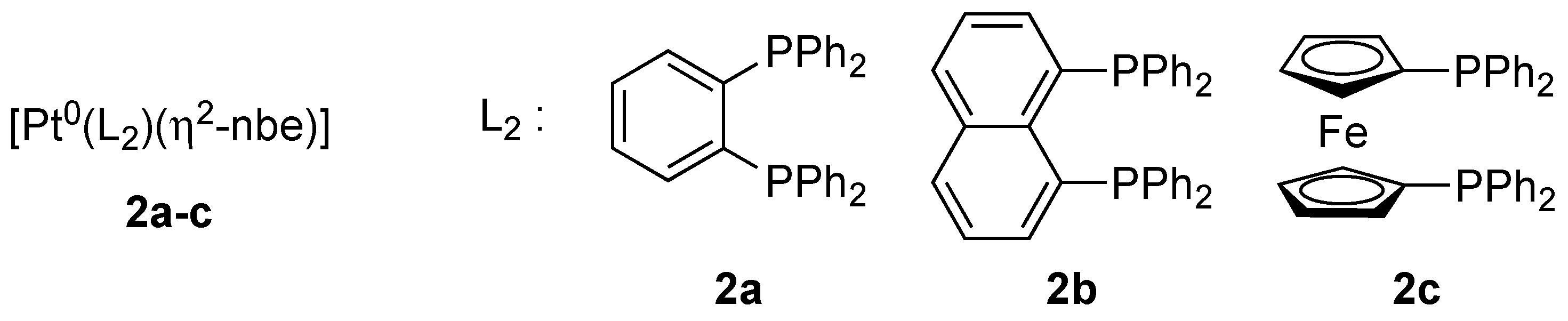
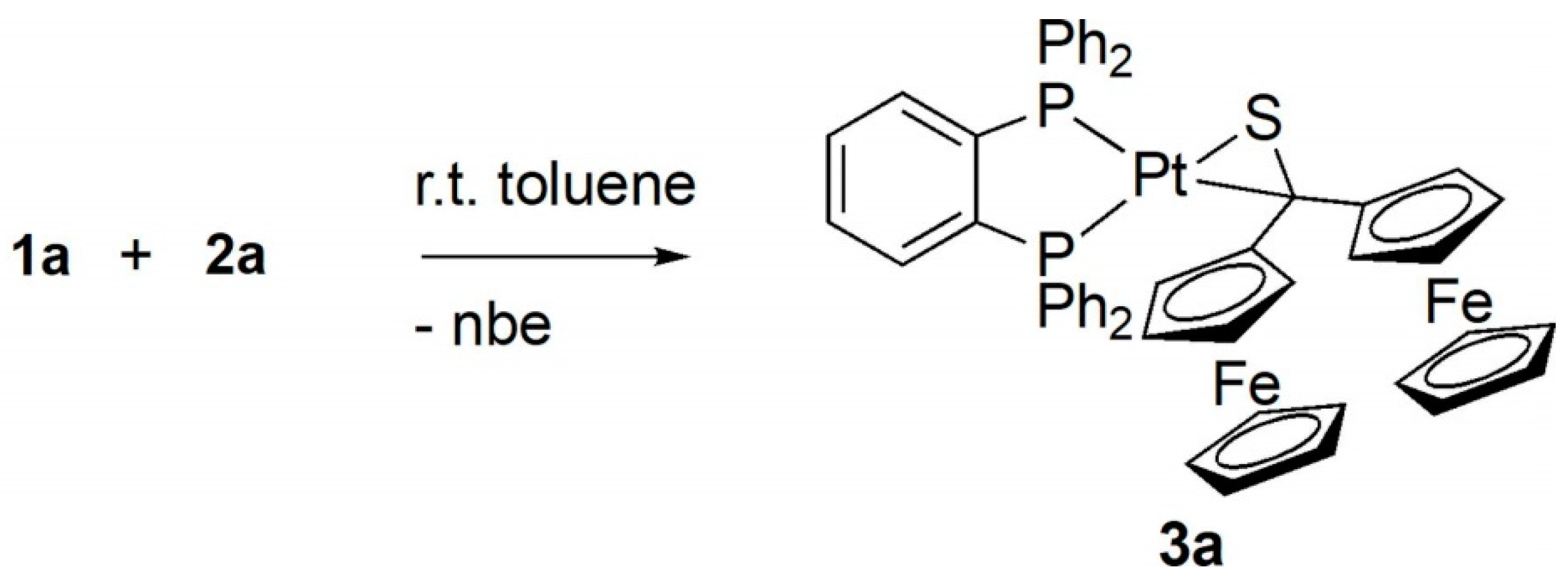
2.1. Preparation and Identification of Platinathiiranes 3a–d
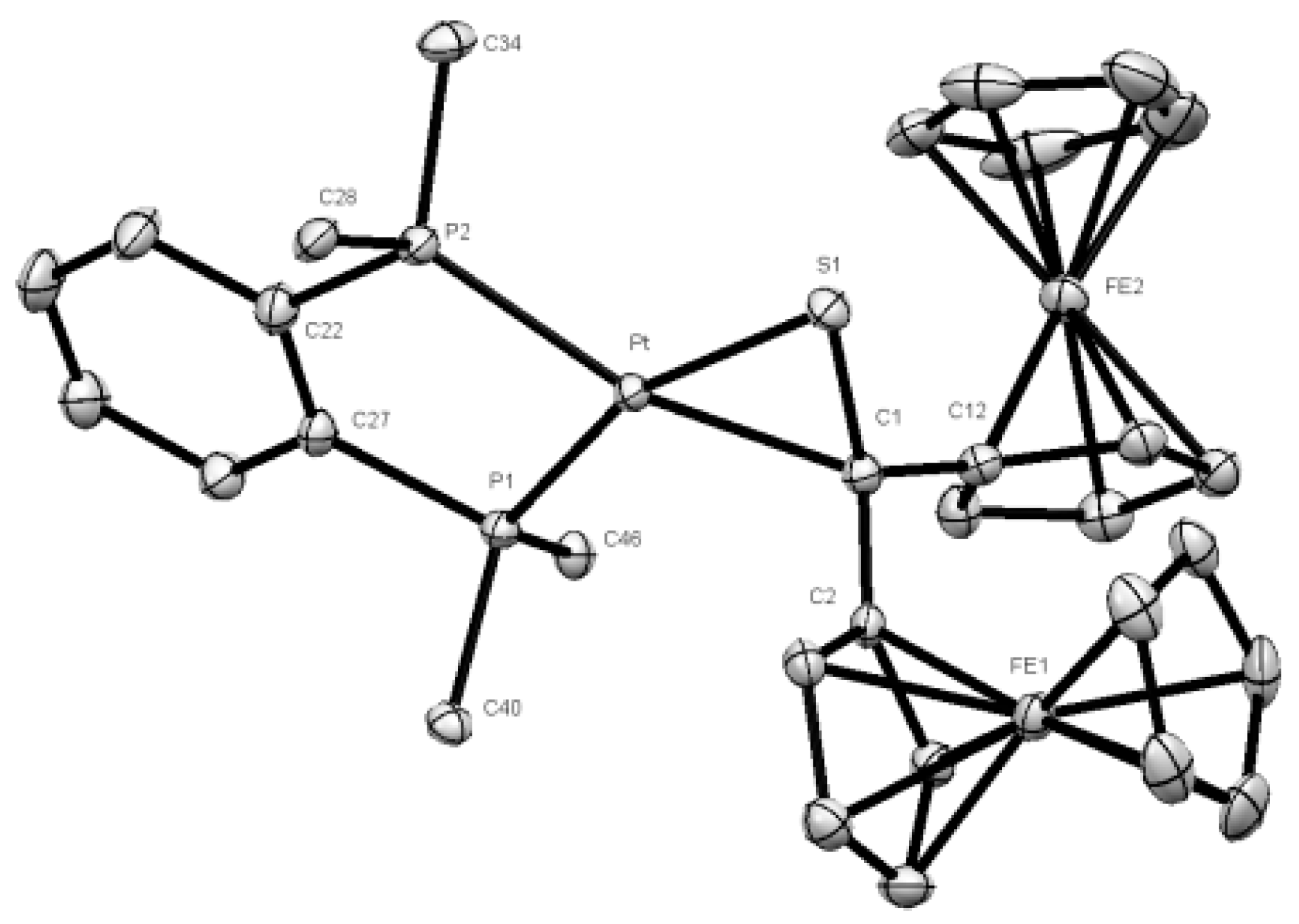
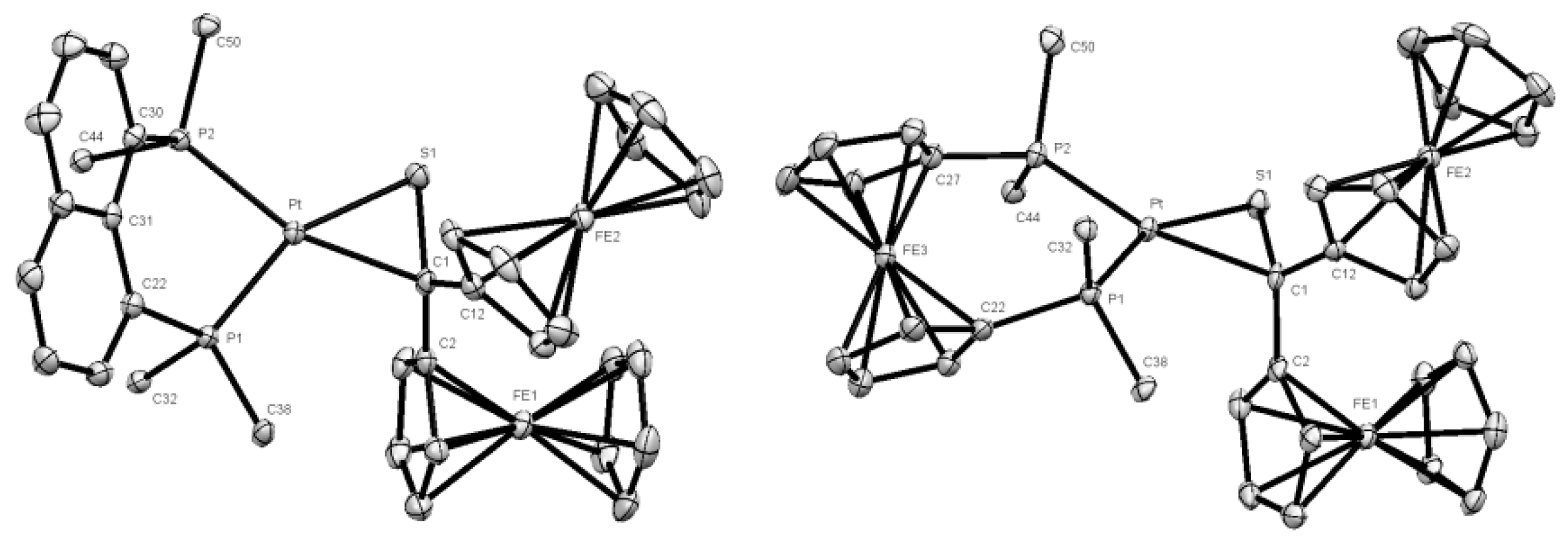
2.2. Crystal Structure Determination
2.3. Electrochemical Measurements
2.4. Theoretical Calculations
3. Results and Discussion
3.1. Synthesis and Characterization of Platinathiiranes 3a–d
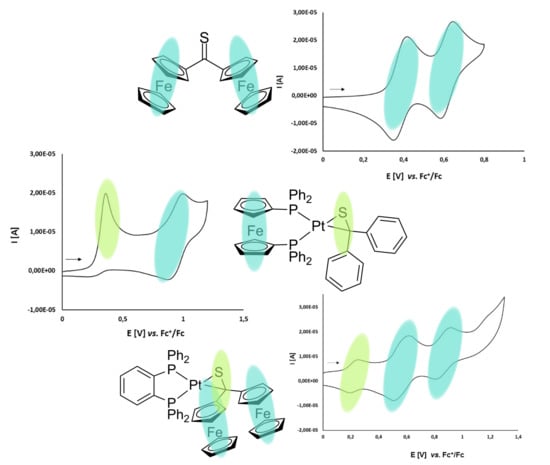
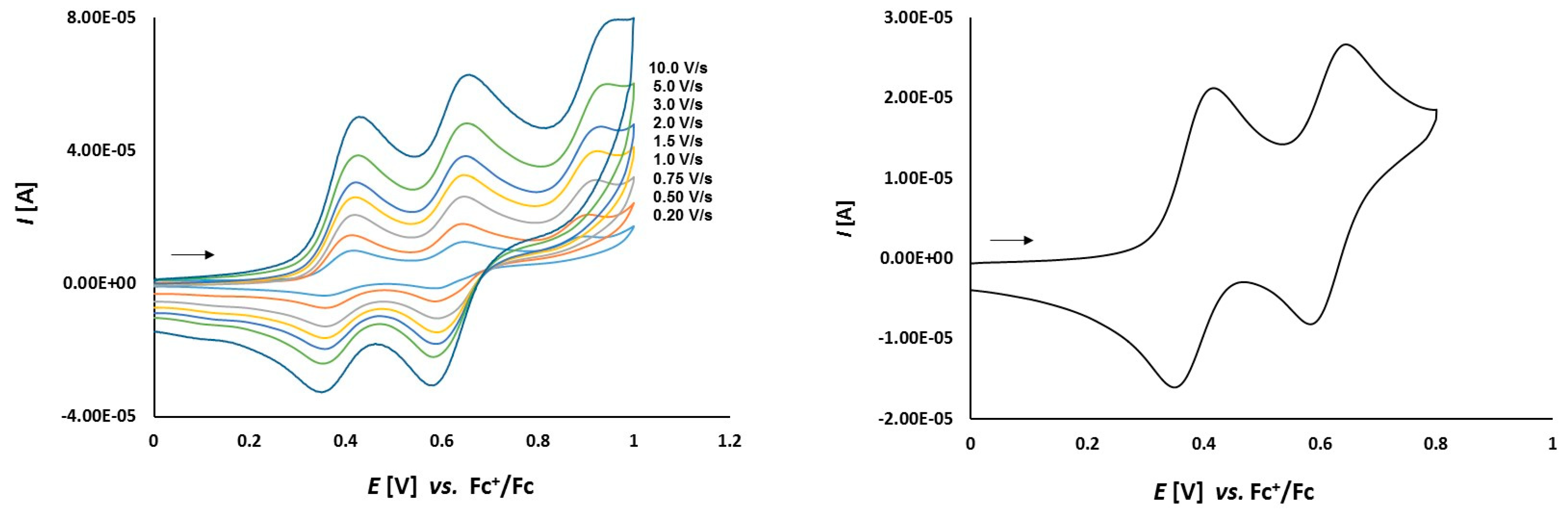
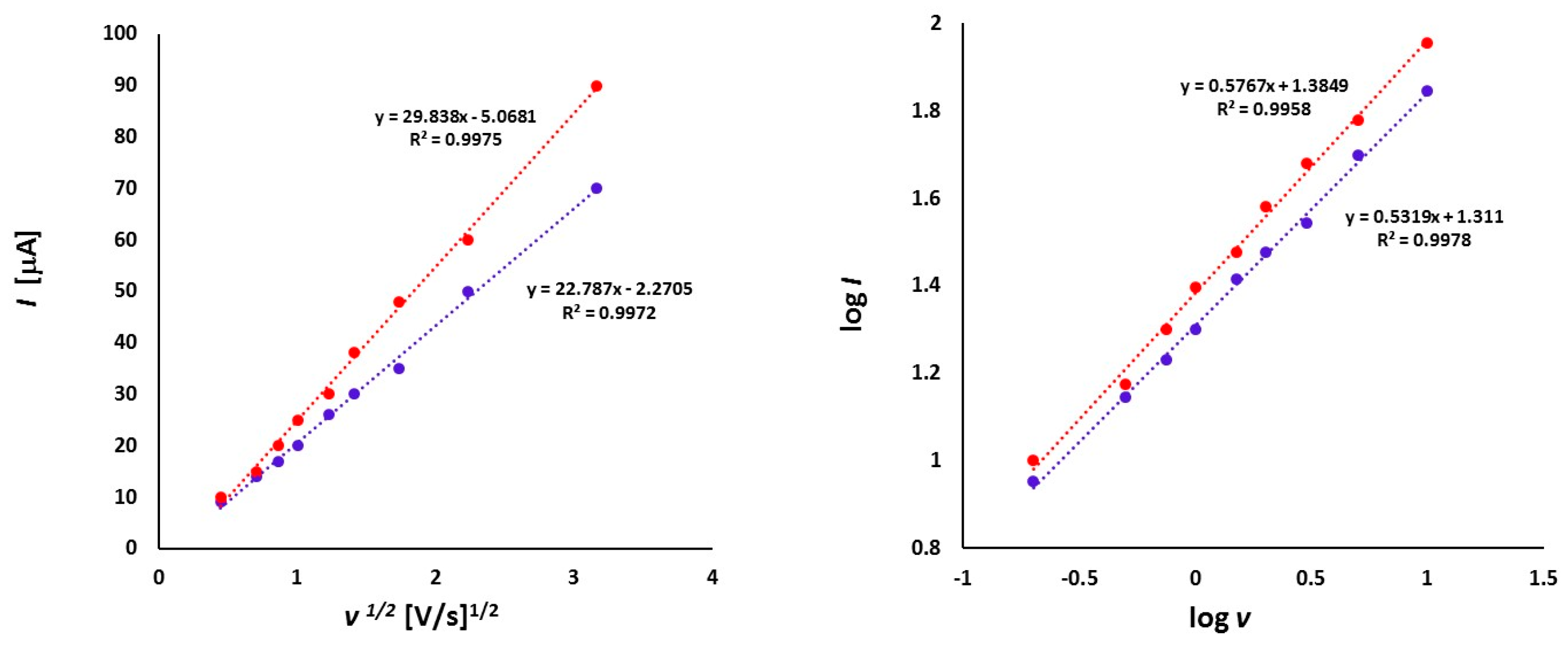
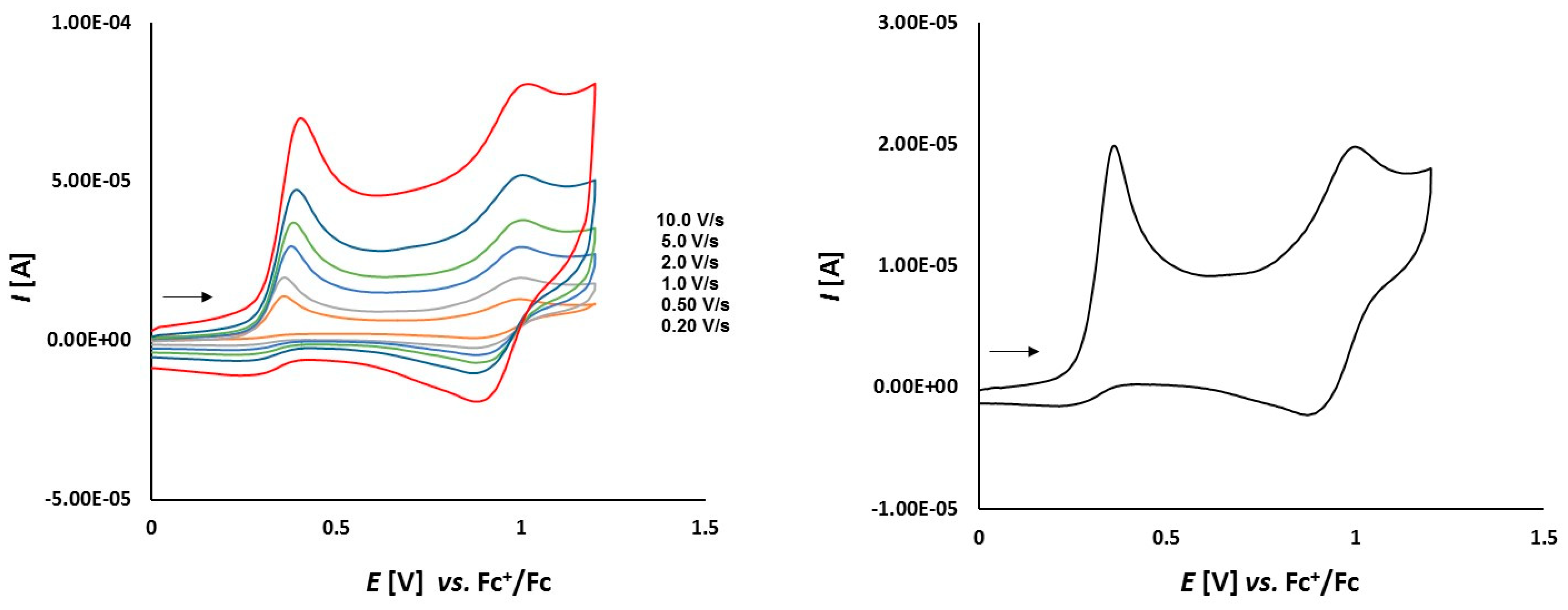
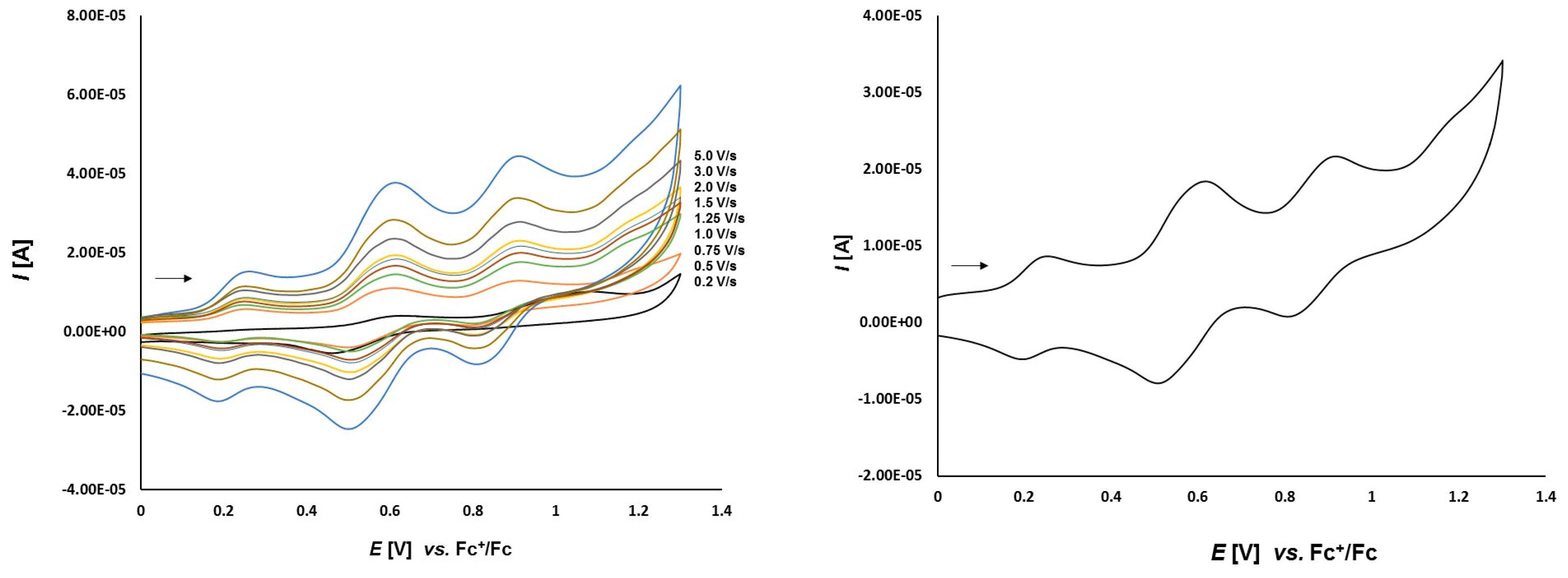
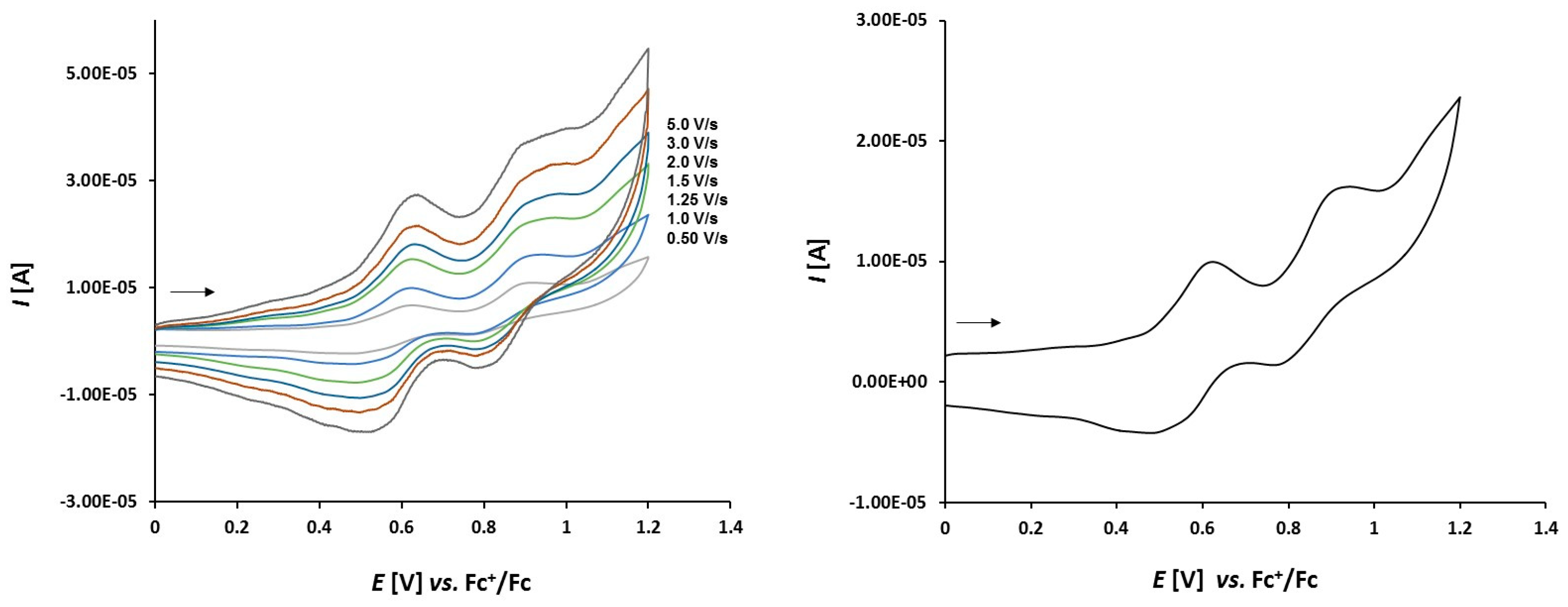
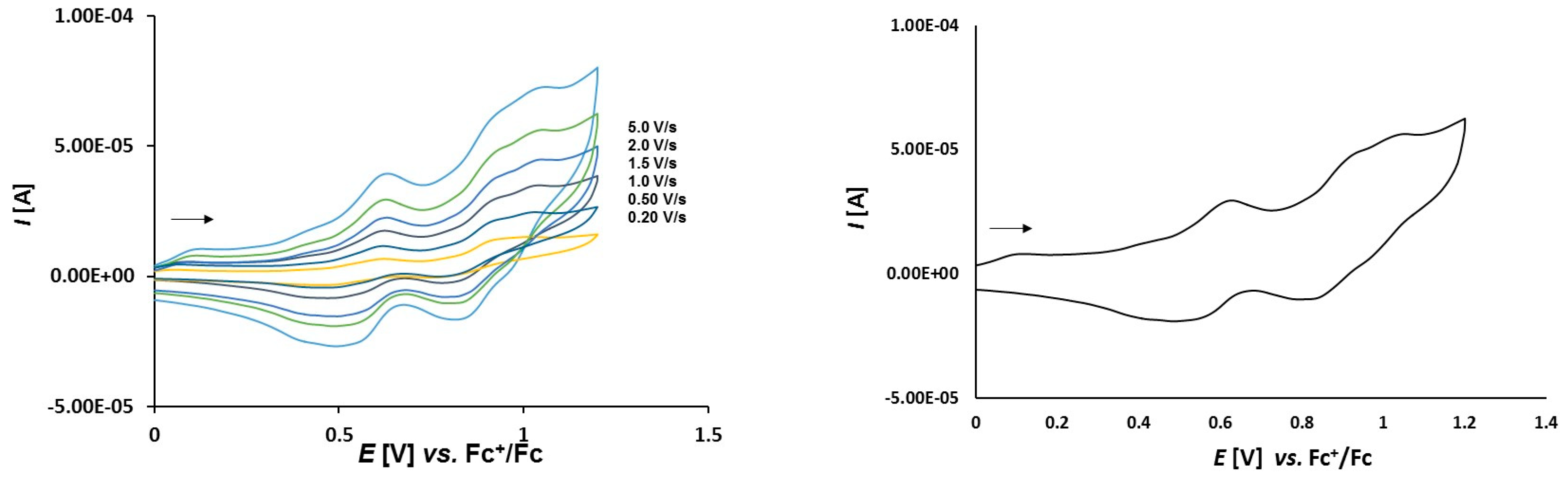
3.2. Electrochemical Studies
3.2.1. Thioketone 1a
3.2.2. Platinathiiranes 3a–d
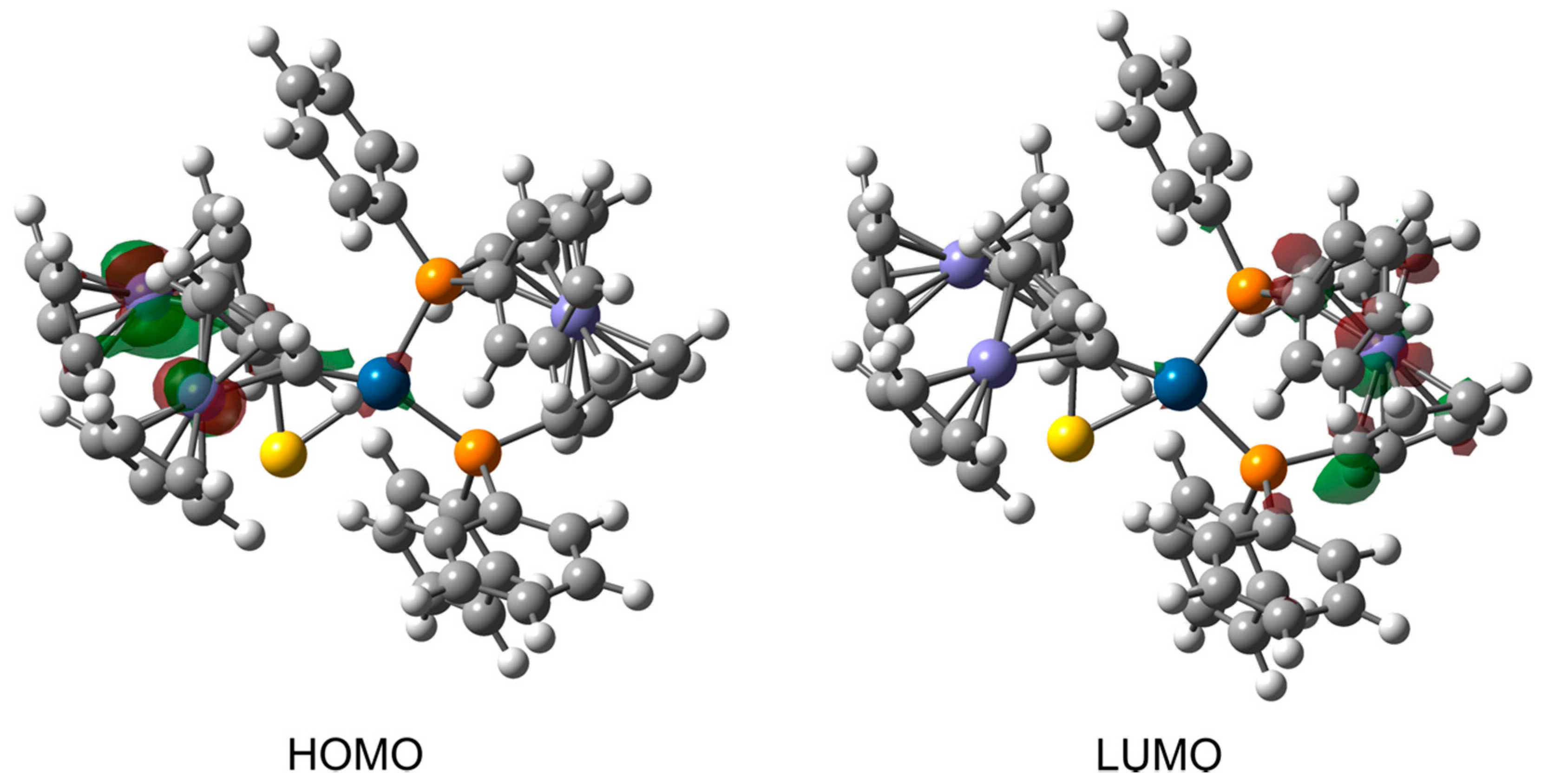
3.3. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Voss, J. Thioaldehyde bzw. Thioketone. In Houben-WeylMethoden der Organischen Chemie: Organische Schwefel-Verbindungen; Thieme Verlag: Stuttgart, Germany, 1985; Chapter VII; Volume E11, pp. 188–231. [Google Scholar]
- Paquer, D. Aliphatic Thioketones. Int. J. Sulfur Chem. B 1972, 7, 269–293. [Google Scholar]
- Ohno, A. Thiones. In Organic Chemistry of Sulfur; Oae, S., Ed.; Plenum Press: New York, NY, USA, 1977; Chapter 5; pp. 189–229. [Google Scholar]
- Mlostoń, G.; Grzelak, P.; Hamera-Fałdyga, R.; Jasiński, M.; Urbaniak, K.; Pipiak, P.; Albrecht, Ł.; Hejmanowska, J.; Heimgartner, H. Aryl, hetaryl, and ferrocenylthioketones as versatile building blocks for exploration in the organic chemistry of sulfur. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 204–211. [Google Scholar] [CrossRef]
- Huisgen, R.; Langhals, E. 1,3-Dipolar cycloadditions of diphenyldiazomethane to thioketones: Rate measurements disclose thiones to be superdipolarophiles. Heteroat. Chem. 2006, 17, 433–442. [Google Scholar] [CrossRef]
- Rohr, U.; Schatz, J.; Sauer, J. Thio-and selenocarbonyl compounds as “Superdienophiles” in cycloadditions. Eur. J. Org. Chem. 1998, 1998, 2875–2883. [Google Scholar] [CrossRef]
- Sugiyama, N.; Yoshioka, Y.; Aoyama, H.; Nishio, T. Photochemical reaction of thioacetophenone trimer (2,4,6-trimethyl-2,4,6-triphenyl-1,3,5-trithiolan). J. Chem. Soc. D 1971, 1971, 1063. [Google Scholar] [CrossRef]
- Mlostoń, G.; Hamera-Fałdyga, R.; Linden, A.; Heimgartner, H. Synthesis of ferrocenyl-substituted 1,3-dithiolanes via [3 + 2]-cycloadditions of ferrocenyl hetaryl thioketones with thiocarbonyl S-methanides. Beilstein J. Org. Chem. 2016, 12, 1421–1427. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Hamera, R.; Heimgartner, H. Synthesis of ferrocenyl thioketones and their reactions with diphenyldiazomethane. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 2125–2132. [Google Scholar] [CrossRef]
- Sato, M.; Asai, M. Synthesis and some properties of diferrocenyl thioketones and dynamic behavior of some [1.1] ferrocenophane derivatives. J. Organomet. Chem. 1992, 430, 105–110. [Google Scholar] [CrossRef]
- Rausch, M.D.; Fischer, E.O.; Grubert, H. The aromatic reactivity of ferrocene, ruthenocene and osmocene. J. Am. Chem. Soc. 1960, 82, 76–82. [Google Scholar] [CrossRef]
- Astruc, D. Why is ferrocene so exceptional? Eur. J. Inorg. Chem. 2017, 2017, 6–29. [Google Scholar] [CrossRef]
- Kowalski, K.; Karpowicz, R.; Mlostoń, G.; Miesel, D.; Hildebrandt, A.; Lang, H.; Czerwoniec, R.; Therrien, B. Synthesis and (spectro)electrochemistry of mixed-valent diferrocenyl-dihydrothiopyran derivatives. Dalton Trans. 2015, 44, 6268–6276. [Google Scholar] [CrossRef] [PubMed]
- Kursanov, D.N.; Setkina, V.N.; Dolgova, S.P.; Nefedova, M.N. Preparation of heterometallic binuclear complexes of transition metals from metallocenyl thioketones. Izv. Akad. Nauk. SSSR Ser. Khim. 1980, 29, 1360–1362. [Google Scholar] [CrossRef]
- Barnes, J.C.; Bell, W.; Glidewell, C.h.; Howie, R.A. Metal complexation of thioacylferocenes: Crystal structure of pentacarbonyl (thiobenzoylferrocene-S) chromium and benzoylferocene. J. Orgnomet. Chem. 1990, 385, 369–378. [Google Scholar] [CrossRef]
- Weigand, W.; Wünsch, R.; Robl, C.; Mlostoń, G.; Nöth, H.; Schmidt, M. Metallkomplexe mit funktionalisierten Schwefelliganden, XV [1]. Reaktionen von Platin(0)-Komplexen mit 1,2,4-Trithiolanen, 1,2,4,5-Tetrathianen, 1,2,3,5,6-Pentathiepanen sowie Thioketonen. Kristallstrukturanalyse von (Ph3P)2Pt(η2-Ph2C = S). Z. Naturforsch. B 2000, 55, 453–458. [Google Scholar] [CrossRef]
- Weisheit, T.; Kritz, A.; Görls, H.; Mlostoń, G.; Imhof, W.; Weigand, W. Reaction of ‘non-symmetrical’ 1,2,4-trithiolanes with phosphanePt0(η2-nbe) complexes. Chem. Asian J. 2012, 7, 1383–1393. [Google Scholar] [CrossRef]
- Weisheit, T.; Petzold, H.; Görls, H.; Mlostoń, G.; Weigand, W. Reaction of 3,3,5,5-tetraphenyl-1,2,4-trithiolane with Pt0(bisphosphine)(η2-nbe) complexes bearing bridged bisphosphine ligands with various bite angles. Eur. J. Inorg. Chem. 2009, 2009, 3545–3551. [Google Scholar] [CrossRef]
- Mlostoń, G.; Romański, J.; Weigand, W.; Heimgartner, H. Organic and coordination chemistry of 1,2,4-trithiolanes. Eur. J. Org. Chem. 2019, 2019, 1867–1875. [Google Scholar] [CrossRef]
- Petzold, H.; Görls, H.; Weigand, W. A simple and efficient synthesis of bisphosphine platinum(0) complexes with various P–Pt–P angles. J. Organomet. Chem. 2007, 692, 2736–2742. [Google Scholar] [CrossRef]
- COLLECT. Data Collection Software; Nonius B.V.: Delft, The Netherlands, 1998. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-Ray Diffraction Data Collected in Oscillation Mod. In Methods in Enzymology Macromolecular Crystallography, Part A; Carter, C.W., Sweet, R.M., Eds.; Academic Press: New York, NY, USA, 1997; Volume 276, pp. 307–326. [Google Scholar]
- Sheldrick, G.M. SADABS 2.10; Bruker-AXS Inc.: Madison, WI, USA, 2002. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. 2015, 71, 9–18. [Google Scholar]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1998, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 1995, 240, 283–290. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Armbruster, M.K.; Bachorz, R.A.; Bahmann, H.; Bär, M.; Baron, H.-P.; Bauernschmitt, R.; Bischoff, F.A.; Böcker, S.; Burow, A.M.; et al. TURBOMOLE 7.2, University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, 2017. Available online: http://www.turbomole.com (accessed on 29 August 2019).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon: Oxford, UK, 1990. [Google Scholar]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO 3.1; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 1993. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Calculation of molecular orbital composition. Acta Chim. Sin. 2011, 69, 2393–2406. [Google Scholar]
- Keith, T.A. AIMAll 14.11.23; TK Gristmill Software: Overland Park, KS, USA, 2014. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisheit, T.; Petzold, H.; Görls, H.; Mlostoń, G.; Weigand, W. Reactions of 1,2,4-trithiolanes and their 4-S-oxides with bisphosphane platinum(0) complexes. Eur. J. Inorg. Chem. 2010, 23, 3636–3641. [Google Scholar]
- Gorton, J.E.; Lentzner, H.L.; Watts, W.E. Bridged ferrocenes—VIII: Polarographic half-wave potentials of ferrocenophanes and related compounds. Tetrahedron 1971, 27, 4353–4360. [Google Scholar] [CrossRef]
- Brown, G.M.; Meyer, T.J.; Cowan, D.O.; LeVanda, C.; Kaufman, F.; Roling, P.V.; Rausch, M.D. Oxidation-state and electron-transfer properties of mixed-valence 1,1′-polyferrocene ions. Inorg. Chem. 1975, 14, 506–511. [Google Scholar] [CrossRef]
- Camire, N.; Mueller-Westerhoff, U.T.; Geiger, W.E. Improved electrochemistry of multi-ferrocenyl compounds: Investigation of biferrocene, terferrocene, bis(fulvalene)diiron and diferrocenylethane in dichloromethane using [NBu4][B(C6F5)4] as supporting electrolyte. J. Organomet. Chem. 2001, 637–639, 823–826. [Google Scholar] [CrossRef]
- Ferguson, G.; Glidewell, C.; Opromolla, G.; Zakaria, C.M.; Zanello, P. The redox behaviour of some bis-ferrocenyl compounds: Crystal and molecular structures of diferrocenylmethane and diferrocenylmethanol. J. Organomet. Chem. 1996, 517, 183–190. [Google Scholar] [CrossRef]
- Hedberg, F.L.; Rosenberg, H. Synthesis of 1,1′-biferrocenylene. J. Am. Chem. Soc. 1969, 91, 1258–1259. [Google Scholar] [CrossRef]
- Zanello, P.; Opromolla, G.; Herberhold, M.; Brendel, H.D. Redox behaviour of ferrocene derivatives VII. Chalcogen-bridged tri- and tetra-nuclear ferrocenes. J. Organomet. Chem. 1994, 484, 67–70. [Google Scholar] [CrossRef]
- Robin, M.B.; Day, P. Mixed valence chemistry—A survey and classification. Adv. Inorg. Chem. Radiochem. 1967, 10, 247–422. [Google Scholar]
- Xie, R.J.; Han, L.M.; Zhu, N.; Hong, H.L.; Suo, Q.L.; Ke, C.L. Synthesis, crystal structure and electrochemistry of carbon-bridged diferrocenyl compounds. Asian J. Chem. 2013, 25, 197–201. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the Pople-Santry-Segal CNDO method to the cyclopropylcarbinyl and cyclobutylcation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Angyan, J.G.; Loos, M.; Mayer, I. Covalent bond orders and atomic valence indices in the topological theory of atoms in molecules. J. Phys. Chem. 1994, 98, 5244–5248. [Google Scholar] [CrossRef]
- Macchi, P.; Sironi, A. Chemical bonding in transition metal carbonyl clusters: Complementary analysis of theoretical and experimental electron densities. Coord. Chem. Rev. 2003, 238–239, 383–412. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Minaev, B.F.; Minaeva, V.A.; Podgornaya, A.T.; Ågren, H. Application of Bader’s atoms in molecules theory to the description of coordination bonds in the complex compounds of Ca2+ and Mg2+ with methylidene rhodanine and its anion. Russ. J. Gen. Chem. 2012, 82, 1254–1262. [Google Scholar] [CrossRef]
- Alexiou, A.D.P.; Decandio, C.C.; da NAlmeida, S.; Ferreira, M.J.P.; Romoff, P.; Rocha, R.C. Metal-ligand coordination and antiradical activity of a trichromium(III) complex with the flavonoid naringenin. J. Coord. Chem. 2017, 70, 2148–2160. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essén, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical bonds without bonding electron density—Does the difference electron-density analysis suffice for a description of the chemical bond? Angew. Chem. Int. Ed. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H···F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Shahi, A.; Arunan, E. Hydrogen bonding, halogen bonding and lithium bonding: An atoms in molecules and natural bond orbital perspective towards conservation of total bond order, inter- and intra-molecular bonding. Phys. Chem. Chem. Phys. 2014, 16, 22935–22952. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, L.M.; Hill, A.F.; Stranger, R.; Terrett, R.N.L.; von Nessi, K.M.; Ward, J.S.; Willis, A.C. Thioxoethenylidene (CCS) as a Bridging Ligand. Organometallics 2015, 34, 328–334. [Google Scholar] [CrossRef]
- Heinze, K.; Lang, H. Ferrocene—Beauty and Functions. Organometallics 2013, 32, 5623–6164. [Google Scholar] [CrossRef]
- Dumas, P.; Girault, J.-P.; Guerin, P. Polymerization of methylthiirane initiated by platinum(II)- and palladium(II) complexes. Polym. Bull. 1980, 2, 257–261. [Google Scholar] [CrossRef]
- Schenk, W.A. The coordination chemistry of small sulfur-containing molecules: A personal perspective. J. Chem. Soc. Dalton Trans. 2011, 40, 1209–1219. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rauchfuss, T.B. Synthesis of Diiron(I) Dithiolato Carbonyl Complexes. Chem. Rev. 2016, 116, 7043–7077. [Google Scholar] [CrossRef] [PubMed]
- Daraosheh, A.Q.; Görls, H.; El-Khateeb, M.; Mloston, G.; Weigand, W. Reactions of selected aromatic thioketones with dodecarbonyltriiron. Eur. J. Inorg. Chem. 2011, 2011, 349–355. [Google Scholar] [CrossRef]
- Daraosheh, A.Q.; Apfel, U.-P.; Görls, H.; Friebe, C.; Schubert, U.S.; El-Khateeb, M.; Mloston, G.; Weigand, W. New approach to [FeFe]-hydrogenase models using aromatic thioketones. Eur. J. Inorg. Chem. 2012, 2012, 318–326. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoms | Bond Length (Å) | Atoms | Bond Angle (°) |
|---|---|---|---|
| Pt‒P(1) Pt‒P(2) Pt‒S(1) Pt‒C(1) C(1)‒S(1) | 2.246 (2.213) 2.282 (2.213) 2.326 2.155 1.788 (1.665) | P(1)‒Pt‒P(2) P(1)‒Pt‒C(1) P(2)‒Pt‒S(1) C(1)‒Pt‒S(1) | 99.8 (129.1) 105.7 107.7 46.8 |
| Atoms | WBI | DI |
|---|---|---|
| Pt‒P(1) Pt‒P(2) Pt‒S(1) Pt‒C(1) C(1)‒S(1) | 0.654 (0.835) 0.636 (0.835) 0.689 0.417 1.148 (1.656) | 1.057 (1.1865) 0.999 (1.1869) 0.926 0.701 1.157 (1.696) |
| Atoms | ρ (au) | ∇2ρ (au) | H (au) | ‒V/G | ‒λ1/λ3 |
|---|---|---|---|---|---|
| Pt‒P(1) | 0.1232 (0.1278) | 0.0755 (0.0584) | −0.0642 (−0.0712) | 1.7733 (1.8305) | 0.3711 (0.4011) |
| Pt‒P(2) | 0.1151 (0.1279) | 0.0911 (0.0584) | −0.0557 (−0.0713) | 1.7104 (1.8306) | 0.3413 (0.4011) |
| Pt‒S(1) Pt‒C(1) C(1)‒S(1) | 0.1010 0.1100 0.1839 | 0.1457 0.1239 −0.2656 | −0.0401 −0.0454 −0.1349 | 1.5242 1.5951 2.9701 | 0.2912 0.3625 1.1881 |
| (0.2269) | (−0.3901) | (−0.2612) | (2.5958) | (2.1543) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gröber, S.; Matczak, P.; Domagała, S.; Weisheit, T.; Görls, H.; Düver, A.; Mlostoń, G.; Weigand, W. Diferrocenyl Thioketone: Reactions with (Bisphosphane)Pt(0) Complexes—Electrochemical and Computational Studies. Materials 2019, 12, 2832. https://doi.org/10.3390/ma12172832
Gröber S, Matczak P, Domagała S, Weisheit T, Görls H, Düver A, Mlostoń G, Weigand W. Diferrocenyl Thioketone: Reactions with (Bisphosphane)Pt(0) Complexes—Electrochemical and Computational Studies. Materials. 2019; 12(17):2832. https://doi.org/10.3390/ma12172832
Chicago/Turabian StyleGröber, Sebastian, Piotr Matczak, Sławomir Domagała, Thomas Weisheit, Helmar Görls, Annika Düver, Grzegorz Mlostoń, and Wolfgang Weigand. 2019. "Diferrocenyl Thioketone: Reactions with (Bisphosphane)Pt(0) Complexes—Electrochemical and Computational Studies" Materials 12, no. 17: 2832. https://doi.org/10.3390/ma12172832