
A Theoretical Investigation on the Physical Properties of Zirconium Trichalcogenides, ZrS3, ZrSe3 and ZrTe3 Monolayers
 , , and
, , and
Abstract
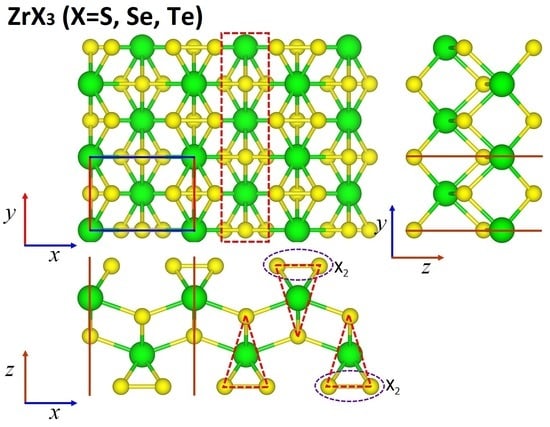
:
1. Introduction
2. Computational Methods
3. Results and Discussion
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Castro Neto, A.H.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K.; Guinea, F. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.-L.; Liu, Z.; Wang, L.; Zhou, T.; Ma, W.; Xu, C.; Feng, S.; Chen, L.; Chen, M.-L.; Sun, D.-M.; et al. Chemical vapor deposition of layered two-dimensional MoSi2N4 materials. Science 2020, 369, 670–674. [Google Scholar] [CrossRef]
- Novoselov, K.S. Discovery of 2D van der Waals layered MoSi2N4 family. Natl. Sci. Rev. 2020, 7, 1842–1844. [Google Scholar] [CrossRef]
- Bakharev, P.V.; Huang, M.; Saxena, M.; Lee, S.W.; Joo, S.H.; Park, S.O.; Dong, J.; Camacho-Mojica, D.C.; Jin, S.; Kwon, Y.; et al. Chemically induced transformation of chemical vapour deposition grown bilayer graphene into fluorinated single-layer diamond. Nat. Nanotechnol. 2020, 15, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Zhang, J.; Zhu, C.; Shen, W.; Hu, C.; Fu, W.; Yan, L.; Zhou, L.; Zheng, L.; Lei, H.; et al. Penta-PdPSe: A New 2D Pentagonal Material with Highly In-Plane Optical, Electronic, and Optoelectronic Anisotropy. Adv. Mater. 2021, 33, 2102541. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, F.; Wang, R.; Zhai, T.; Huang, F. 2D NbOI2: A Chiral Semiconductor with Highly In-Plane Anisotropic Electrical and Optical Properties. Adv. Mater. 2021, 33, 2101505. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xiong, T.; Zhao, K.; Zhou, Z.; Xin, K.; Deng, H.-X.; Kang, J.; Yang, J.; Liu, Y.-Y.; Wei, Z. Polarimetric Image Sensor and Fermi Level Shifting Induced Multi-Channel Transition Based on Two-dimensional PdPS. Adv. Mater. 2021, 33, 2107206. [Google Scholar] [CrossRef]
- Seo, T.H.; Lee, W.; Lee, K.S.; Hwang, J.Y.; Son, D.I.; Ahn, S.; Cho, H.; Kim, M.J. Dominant formation of h-BC2N in h-BxCyNz films: CVD synthesis and characterization. Carbon N. Y. 2021, 182, 791–798. [Google Scholar] [CrossRef]
- Bykov, M.; Bykova, E.; Ponomareva, A.V.; Tasnádi, F.; Chariton, S.; Prakapenka, V.B.; Glazyrin, K.; Smith, J.S.; Mahmood, M.F.; Abrikosov, I.A.; et al. Realization of an Ideal Cairo Tessellation in Nickel Diazenide NiN2: High-Pressure Route to Pentagonal 2D Materials. ACS Nano 2021, 15, 13539–13546. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sanchez-Santolino, G.; Puebla, S.; Frisenda, R.; Al-Enizi, A.M.; Nafady, A.; D’Agosta, R.; Castellanos-Gomez, A. Strongly Anisotropic Strain-Tunability of Excitons in Exfoliated ZrSe3. Adv. Mater. 2022, 34, 2103571. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zheng, C.; Gao, G. Bulk and Monolayer ZrS3 as Promising Anisotropic Thermoelectric Materials: A Comparative Study. J. Phys. Chem. C 2020, 124, 6536–6543. [Google Scholar] [CrossRef]
- Debnath, T.; Debnath, B.; Lake, R.K. Thermal conductivity of the quasi-one-dimensional materials TaSe3 and ZrTe3. Phys. Rev. Mater. 2021, 5, 34010. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Monkhorst, H.; Pack, J. Special points for Brillouin zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Shapeev, A.V. Moment tensor potentials: A class of systematically improvable interatomic potentials. Multiscale Model. Simul. 2016, 14, 1153–1173. [Google Scholar] [CrossRef]
- Mortazavi, B.; Shojaei, F.; Javvaji, B.; Rabczuk, T.; Zhuang, X. Outstandingly high thermal conductivity, elastic modulus, carrier mobility and piezoelectricity in two-dimensional semiconducting CrC2N4: A first-principles study. Mater. Today Energy 2021, 22, 100839. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, B.; Novikov, I.S.; Podryabinkin, E.V.; Roche, S.; Rabczuk, T.; Shapeev, A.V.; Zhuang, X. Exploring phononic properties of two-dimensional materials using machine learning interatomic potentials. Appl. Mater. Today 2020, 20, 100685. [Google Scholar] [CrossRef]
- Brattas, L.; Kjekshus, A. Others Properties of compounds with zrse 3-type structure. Acta Chem. Scand. 1972, 26, 3441–3449. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.; Guégan, F.; Yu, S.; Frapper, G. Prediction of allotropes of tellurium with molecular, one- and two-dimensional covalent nets for photofunctional applications. RSC Adv. 2021, 11, 29965–29975. [Google Scholar] [CrossRef]
- Tareen, A.K.; Khan, K.; Aslam, M.; Zhang, H.; Liu, X. Recent progress, challenges, and prospects in emerging group-VIA Xenes: Synthesis, properties and novel applications. Nanoscale 2021, 13, 510–552. [Google Scholar] [CrossRef]
- Zhu, Z.; Cai, X.; Yi, S.; Chen, J.; Dai, Y.; Niu, C.; Guo, Z.; Xie, M.; Liu, F.; Cho, J.-H.; et al. Multivalency-Driven Formation of Te-Based Monolayer Materials: A Combined First-Principles and Experimental study. Phys. Rev. Lett. 2017, 119, 106101. [Google Scholar] [CrossRef] [Green Version]
- Le, T.-H.; Oh, Y.; Kim, H.; Yoon, H. Exfoliation of 2D Materials for Energy and Environmental Applications. Chem. Eur. J. 2020, 26, 6360–6401. [Google Scholar] [CrossRef]
- Huo, C.; Yan, Z.; Song, X.; Zeng, H. 2D materials via liquid exfoliation: A review on fabrication and applications. Sci. Bull. 2015, 60, 1994–2008. [Google Scholar] [CrossRef]
- Biccai, S.; Barwich, S.; Boland, D.; Harvey, A.; Hanlon, D.; McEvoy, N.; Coleman, J.N. Exfoliation of 2D materials by high shear mixing. 2D Mater. 2018, 6, 15008. [Google Scholar] [CrossRef]
- Wang, W.; Dai, S.; Li, X.; Yang, J.; Srolovitz, D.J.; Zheng, Q. Measurement of the cleavage energy of graphite. Nat. Commun. 2015, 6, 7853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raeisi, M.; Mortazavi, B.; Podryabinkin, E.V.; Shojaei, F.; Zhuang, X.; Shapeev, A. V High Thermal Conductivity in Semiconducting Janus and Non-Janus Diamanes. Carbon N. Y. 2020, 167, 51–61. [Google Scholar] [CrossRef]
- Li, M.; Dai, J.; Zeng, X.C. Tuning the electronic properties of transition-metal trichalcogenides via tensile strain. Nanoscale 2015, 7, 15385–15391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarghami Dehaghani, M.; Molaei, F.; Spitas, C.; Hamed Mashhadzadeh, A. Thermal rectification in nozzle-like graphene/boron nitride nanoribbons: A molecular dynamics simulation. Comput. Mater. Sci. 2022, 207, 111320. [Google Scholar] [CrossRef]
- Dadrasi, A.; Albooyeh, A.; Fooladpanjeh, S.; Salmankhani, A.; Hamed Mashhadzadeh, A.; Saeb, M.R. Theoretical examination of the fracture behavior of BC3 polycrystalline nanosheets: Effect of crack size and temperature. Mech. Mater. 2022, 165, 104158. [Google Scholar] [CrossRef]
- Farzadian, O.; Dehaghani, M.Z.; Kostas, K.V.; Mashhadzadeh, A.H.; Spitas, C. A theoretical insight into phonon heat transport in graphene/biphenylene superlattice nanoribbons: A molecular dynamic study. Nanotechnology 2022, 33, 355705. [Google Scholar] [CrossRef]
- Mortazavi, B. Ultrahigh Thermal Conductivity and Strength in Direct-Gap Semiconducting Graphene-like BC6N: A First-Principles and Classical Investigation. Carbon N. Y. 2021, 182, 373–383. [Google Scholar] [CrossRef]
- Mortazavi, B.; Silani, M.; Podryabinkin, E.V.; Rabczuk, T.; Zhuang, X.; Shapeev, A. V First-Principles Multiscale Modeling of Mechanical Properties in Graphene/Borophene Heterostructures Empowered by Machine-Learning Interatomic Potentials. Adv. Mater. 2021, 33, 2102807. [Google Scholar] [CrossRef]
- Mortazavi, B.; Podryabinkin, E.V.; Roche, S.; Rabczuk, T.; Zhuang, X.; Shapeev, A. V Machine-Learning Interatomic Potentials Enable First-Principles Multiscale Modeling of Lattice Thermal Conductivity in Graphene/Borophene Heterostructures. Mater. Horizons 2020, 7, 2359–2367. [Google Scholar] [CrossRef]
- Arabha, S.; Rajabpour, A. Thermo-Mechanical Properties of Nitrogenated Holey Graphene (C2N): A Comparison of Machine-Learning-Based and Classical Interatomic Potentials. Int. J. Heat Mass Transf. 2021, 178, 121589. [Google Scholar] [CrossRef]
- Arabha, S.; Aghbolagh, Z.S.; Ghorbani, K.; Hatam-Lee, M.; Rajabpour, A. Recent Advances in Lattice Thermal Conductivity Calculation Using Machine-Learning Interatomic Potentials. J. Appl. Phys. 2021, 130, 210903. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
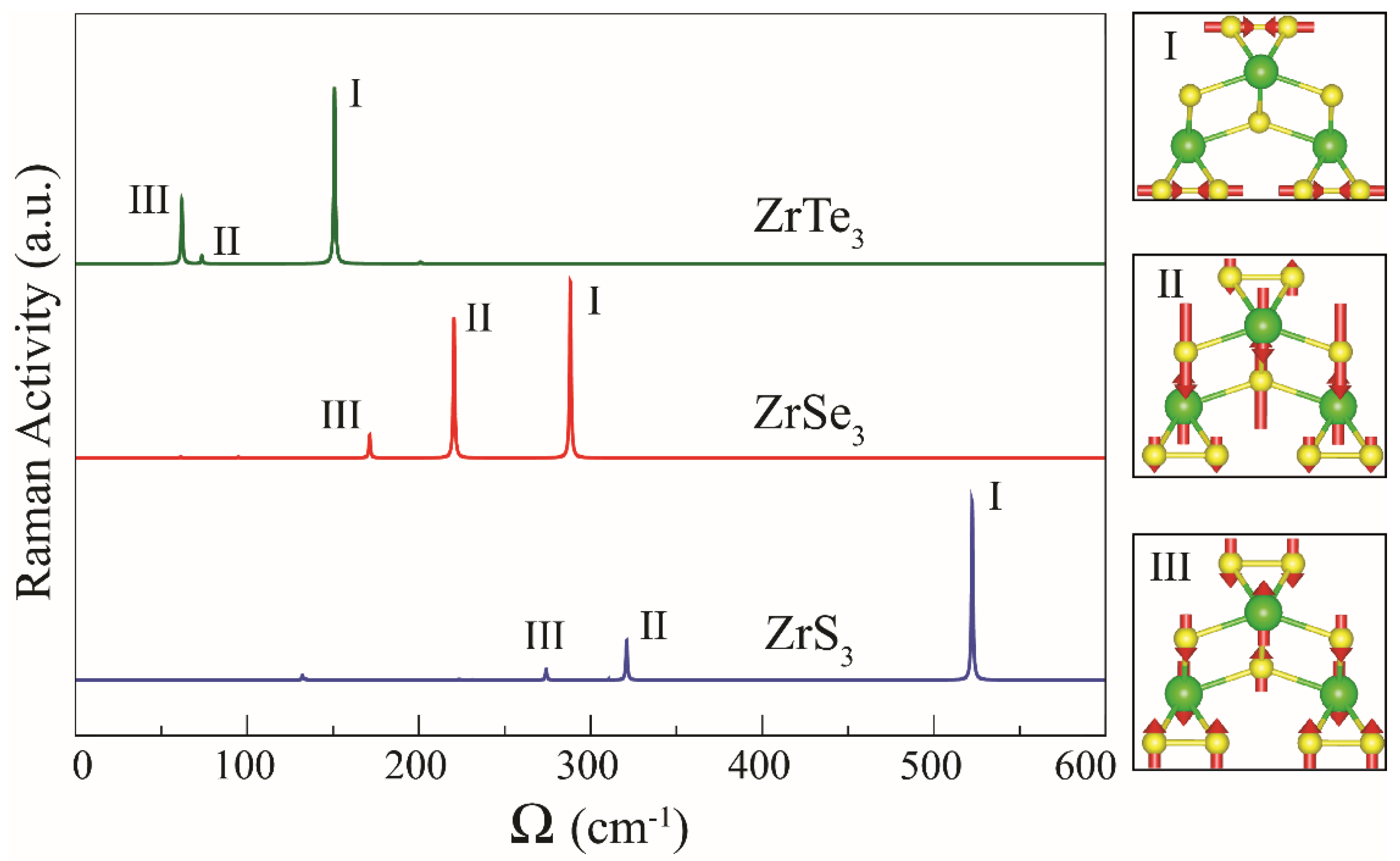{kind=link}
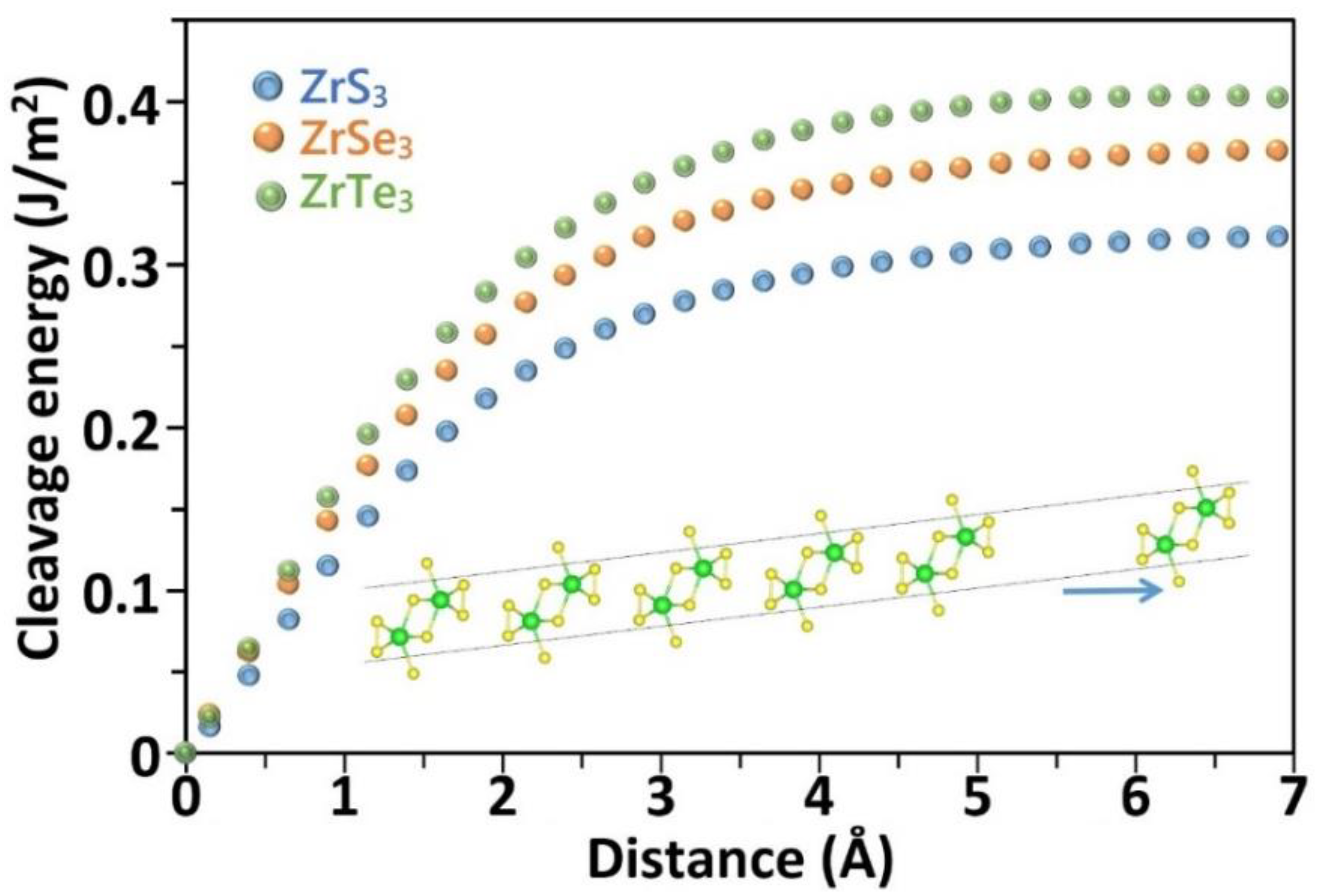{kind=link}
{kind=link}
{kind=link}
| Lattice | a,b (Å) 1 | Q(Zr,Xsurface,Xinternal) (e) 5 | ||||
|---|---|---|---|---|---|---|
| ZrS3 | 5.13,3.63 | 2.72, 2.61 | 2.07 | 3.06 | 1.77,−0.41,−0.95 | 1.06/1.06/1.93 (Γ-X)→Γ/1.93 |
| ZrSe3 | 5.41,3.76 | 2.87, 2.74 | 2.38 | 3.03 | 1.61,−0.38,−0.85 | 0.33/0.27/1.01 Γ→X/0.93 |
| ZrTe3 | 5.89,3.92 | 3.16, 2.98 | 2.83 | 3.05 | 1.33,−0.34,−0.64 | Metal/Metal/Metal/Metal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortazavi, B.; Shojaei, F.; Yagmurcukardes, M.; Makaremi, M.; Zhuang, X. A Theoretical Investigation on the Physical Properties of Zirconium Trichalcogenides, ZrS3, ZrSe3 and ZrTe3 Monolayers. Energies 2022, 15, 5479. https://doi.org/10.3390/en15155479
Mortazavi B, Shojaei F, Yagmurcukardes M, Makaremi M, Zhuang X. A Theoretical Investigation on the Physical Properties of Zirconium Trichalcogenides, ZrS3, ZrSe3 and ZrTe3 Monolayers. Energies. 2022; 15(15):5479. https://doi.org/10.3390/en15155479
Chicago/Turabian StyleMortazavi, Bohayra, Fazel Shojaei, Mehmet Yagmurcukardes, Meysam Makaremi, and Xiaoying Zhuang. 2022. "A Theoretical Investigation on the Physical Properties of Zirconium Trichalcogenides, ZrS3, ZrSe3 and ZrTe3 Monolayers" Energies 15, no. 15: 5479. https://doi.org/10.3390/en15155479