
Application of Porous Materials for CO2 Reutilization: A Review

 , , ,
, , ,
Abstract
:1. Introduction
2. Solar Thermochemical Conversion
2.1. Reduction/Oxidation Materials
2.1.1. Classification of RedOx Pairs
ZnO/Zn
SnO2/SnO
2.1.2. Non-Volatile RedOx Pairs
Iron-Oxide Based Cycle
Hercynite
CeO2/Ce2O3
Perovskites
2.2. Application of Porous Materials in Solar Thermochemical Conversion
2.3. Potentials and Critical Research Challenges
- RedOx material improvements are necessary to reduce both the required temperature of the RedOx reactions preferably to less than 1000 °C, and the degradation of the materials over a very large number of cycles. This is especially important for the reduction of the oxygen carriers used in the RedOx cycles (Equation (1)), because:
- a.
- The heat loss from the solar systems increases significantly with temperature [82,125,126,157]. The re-radiation heat losses from the cavity solar receiver and reactors increases with the fourth power of the reactor absolute temperature [7,63,75,162], while the challenges associated with the start-up and shut-down of the solar receiver and reactors and the associated parasitic losses also increase with the temperature [63].
- b.
- Material compatibility is a substantial challenge of the state-of-the-art reactors, which can be greatly reduced through lowering the operating temperature of the system. Commensurate with this, operating temperatures of less than ~1000 °C would enable the use of commercially available stainless steels with a lower cost of insolation and, hence, bring down the capital costs [68].
- c.
- To achieve temperatures of more than 1000 °C within the solar receiver and reactors there is need for high concentration ratios from the heliostat field, which, in turn, increase the spillage losses and capital costs of the solar concentrators.
- 2.
- Along with the progress in material synthesis and design, improvement in the performance of the reactors is needed to efficiently utilize the solar heat to drive the endothermic reduction reactions.
- i.
- The majority of the proposed and assessed RedOx cycles are based on the directly irradiated solar reactors [83,163,164,165], employing a quartz glass window [68]. Windows are often used in laboratory-based reactors [68]. In commercial application there are potential issues with the high cost of large quartz windows and the need to maintain/clean them in the field [68]. That is because the windows are vulnerable to particle deposition, thermal shock, and high/low pressures, while also they need effective sealing [166,167,168]. To avoid directly irradiated, windowed solar reactors, an approach is to use highly concentrated solar radiation to heat a heat transfer fluid (HTF), which is transported into an indirectly heated reduction reactor and used to provide the required heat of the endothermic reactions [158,159]. Nevertheless, the concept has recently begun to be explored such that the full extent of its potentials and challenges are yet to be identified. It is worth noting that potentially a relatively higher thermodynamic efficiency can be achieved in directly irradiated solar receiver/reactors relative to indirectly heated ones, due to the elimination of the exergy losses associated with the heating of the intermediate HTF and the temperature difference needed within the required heat exchangers to efficiently transfer heat from HTF to reactors and other components of the process [64]. It is also worth mentioning that a high-temperature pump has been recently developed and demonstrated to circulate molten tin at 1200–1400 °C in such a system [169]. Another potential approach is to use a windowless reactor [170]. This avoids the issues of using a window [64], although this would be achieved at the expense of a lower efficiency. The concept of windowless reactors has been recently explored at bench scale by Long et al. [170]. They characterized the isothermal flow-field within a vortex-based solar cavity receiver with an open aperture. However, further demonstration and assessments are needed to better understand the performance of these receiver/reactors under more realistic conditions, e.g., on solar towers, where they are well above the ground wind boundary layers and exposed to substantial turbulence from relatively permanent wind in different directions [82].
3. Photoreduction of CO2
3.1. TiO2 Photoreduction Catalyst
3.1.1. Metals’ and Non-Metals’ Doping and Cocatalysts
- (a)
- SB photoreactor: photoreactor tests were carried out in water media and gas mixture of 92% CO2 and 8% N2 at room temperature. Then, 1.5 g of photocatalyst and 600 mL of K2CO3 solution (0.1 M) were loaded.
- (b)
- PB photoreactor: 90% gas mixture (i.e., 92% CO2/N2) and 10% of water vapor at the saturated pressure of 46 °C were applied. The reaction was performed at room temperature and pipelines were heated at 50 °C to prevent water condensation.
- (c)
- MT photoreactor: This type of photoreactor was utilized to perform CO2 photoreduction tests in the industrial environment. For this purpose, a gas treatment unit was utilized to enrich CO2 content up to 60–80%. Thereafter, the MT photoreactor with 20 Pyrex reactor tubes connected in series was applied. The photoreaction was performed continuously with a stream of 60% CO2/N2 at 60–70% of relative humidity (RH).
3.1.2. Surface Photosensitization
3.1.3. Semiconductor Coupling
3.2. CO2 Photoreduction by Metal-Organic Frameworks (MOFs)
3.2.1. NH2-Modified MOF
3.2.2. Semiconductor MOF Composite
3.2.3. Metal-MOF Composite
3.2.4. Other Forms of MOF-Based Photocatalyst
3.3. Potentials and Critical Research Challenges
4. Electrochemical Reduction of CO2
Potentials and Critical Research Challenges
5. Conclusions
- The use of porous materials, made either entirely from or coated by the active reduction/oxidation materials, is a promising way to increase the conversion efficiency of solar to fuel in the solar thermochemical CDR cycle. The maximum reported solar-to-fuel efficiency is currently about 7.5%. Nevertheless, further research and development are needed to take the cyclic solar-to-fuel efficiency to about 20%, if the technology is to find a commercial use. This can be achieved through further optimization of both the porous materials’ intrinsic properties, such as pore density, size, shape, tortuosity, etc., and the geometrical configuration of the reactive porous structures inside a solar receiver/reactor. Moreover, there is a need to decrease the temperature of the RedOx reactions to mitigate the parasitic heat losses, i.e., re-radiation and convective heat losses from the solar receiver/reactors through the reduction reaction step. In doing so, the porous structures need to be precisely pore-engineered both to efficiently absorb the thermal energy and to achieve a high conversion during RedOx cycles. The porous structures need to be also properly configured inside the solar reactor to proficiently trap and absorb the solar radiation, in case a directly irradiated reactor is employed.
- Since CO2 solubility in water is very low and CO2 adsorption and activation/excitation are more difficult than H2O, finding a clean, non-toxic, and environmentally friendly solvent to increase CO2 solubility/selectivity for the photocatalytic reduction process is still challenging.
- Finding solar-active and stable photocatalysts, enabling a high selectivity and conversion efficiency to completely suppress the competition reaction of water reduction to hydrogen, is also still challenging and requires a deeper understanding of the mechanisms and reaction pathways of the reduction of CO2 on the heterogeneous photocatalysts within the porous structures.
- Development of a highly stable electrocatalyst for the long-term operation is challenging, while also the low solubility of CO2 in water (~0.034 M) hinders reactions when aqueous electrolytes are employed. Additionally, obtaining a high CO2 selectivity to favorable products is critical to reduce the costs and complexity of the down-stream process for the separation of products.
- There is a need to develop reactors facilitating the mass transfer from the gaseous CO2 phase into the electrolyte and from the electrolyte into the active sites within the porous cathode catalysts.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3DOM | Three-dimensionally ordered macroporous |
| AC | Activated carbon |
| AFC | Ammonium ferric citrate |
| ASU | Air separation unit |
| ATR | Auto-thermal reforming |
| bpy | Bipyridine |
| CB | Conduction band |
| CCD | Carbon dioxide dissociation |
| CDR | Carbon dioxide reutilization |
| CDS | Carbon dioxide splitting |
| CNNS | Carbon nitride nanosheets |
| Cp* | Pentamethylcyclopentadiene |
| CQD | Carbon quantum dots |
| CSP | Concentrated solar power |
| DMC | Dimethyl carbonate |
| DME | Di-methyl- ether |
| DMF | N,N-dimethylformamide |
| DMR | Dry-methane reforming |
| DMSO | Dimethyl sulfoxide |
| EGR | Enhanced gas recovery |
| EGS | Enhanced geothermal systems |
| EOR | Enhanced oil recovery |
| FE | Faradaic efficiencies |
| FT | Fischer–Tropsch |
| FTO | Fluorine-doped tin oxide |
| GC | Gas chromatography |
| GHG | Greenhouse gas |
| H2ATA | 2-aminoterephtalate acid |
| H2DTA | 2,5-diaminoterephthalic acid |
| HFSS | High flux solar simulator |
| IL | Ionic liquid |
| MeCN | Acetonitrile |
| MOF | Metal-organic framework |
| MPSZ | Magnesia partially stabilized zirconia |
| Mtonnes | Million tonnes |
| MWCNT | multi-walled carbon nanotubes |
| NHE | Normal hydrogen electrode |
| NOM | Nonordered macroporous |
| NPs | Nanoparticles |
| POX | Partial oxidation |
| RedOx | Reduction/oxidation |
| rGO | Reduced graphene oxide |
| RHE | Reversible hydrogen electrode |
| RPC | Reticulated porous ceramic |
| SDTR | Solar-driven thermochemical reactions |
| SMR | Steam methane reforming |
| SWCNT | Singe-walled carbon nanotubes |
| Syngas | Synthesis gas |
| TBAB | Tetrabutylammonium bromide |
| TBAP | Tetrabutylammonium perchlorate |
| TBATFB | Tetrabutylammonium tetrafluoroborate |
| TCPP | Tetrakis (4-carboxy phenyl) porphyrin |
| TEA | Triethylamine |
| TEOA | Triethanolamine |
| tpy | 2,4,6-tris(4-pyridyl)pyridine |
| VB | Valence band |
| WS | Water splitting |
| ZIF | Zeolitic imidazolate framework |
References
- Figueroa, J.D.; Fout, T.; Plasynski, S.; McIlvried, H.; Srivastava, R.D. Advances in CO2 capture technology—the us department of energy’s carbon sequestration program. Int. J. Greenh. Gas Control 2008, 2, 9–20. [Google Scholar] [CrossRef]
- Metz, B.; Davidson, O.; De Coninck, H. Carbon Dioxide Capture and Storage: Special Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- He, F.; Han, T.; Hong, H.; Jin, H. Solar thermochemical hybrid trigeneration system with CO2 capture using dimethyl ether-fueled chemical-looping combustion. In Proceedings of the ASME 2011 5th International Conference on Energy Sustainability, Washington, DC, USA, 7–10 August 2011; pp. 1651–1660. [Google Scholar]
- IEA. CO2 Emissions from Fuel Combustion; International Energy Agency: Paris, France, 2015. [Google Scholar]
- ProOxygen. Atmospheric CO2. Available online: https://www.CO2.earth/ (accessed on 25 September 2021).
- Rafiee, A.; Rajab Khalilpour, K.; Milani, D.; Panahi, M. Trends in CO2 conversion and utilization: A review from process systems perspective. J. Environ. Chem. Eng. 2018, 6, 5771–5794. [Google Scholar] [CrossRef]
- Jafarian, M.; Arjomandi, M.; Nathan, G.J. A hybrid solar chemical looping combustion system with a high solar share. Appl. Energy 2014, 126, 69–77. [Google Scholar] [CrossRef]
- Zhao, L.; Oleson, K.; Bou-Zeid, E.; Krayenhoff, E.S.; Bray, A.; Zhu, Q.; Zheng, Z.; Chen, C.; Oppenheimer, M. Global multi-model projections of local urban climates. Nat. Clim. Chang. 2021, 11, 152–157. [Google Scholar] [CrossRef]
- Huaman, R.N.E.; Jun, T.X. Energy related CO2 emissions and the progress on ccs projects: A review. Renew. Sustain. Energy Rev. 2014, 31, 368–385. [Google Scholar] [CrossRef]
- Adanez, J.; Abad, A.; Garcia-Labiano, F.; Gayan, P.; Luis, F. Progress in chemical-looping combustion and reforming technologies. Prog. Energy Combust. Sci. 2012, 38, 215–282. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Xu, Z.; Fan, M.; Gupta, R.; Slimane, R.B.; Bland, A.E.; Wright, I. Progress in carbon dioxide separation and capture: A review. J. Environ. Sci. 2008, 20, 14–27. [Google Scholar] [CrossRef]
- Rafiee, A.; Khalilpour, K.R.; Milani, D. Chapter 8—CO2 conversion and utilization pathways. In Polygeneration with Polystorage for Chemical and Energy Hubs; Khalilpour, K.R., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 213–245. [Google Scholar]
- Warsi, Y.; Kabanov, V.; Zhou, P.; Sinha, A. Novel carbon dioxide utilization technologies: A means to an end. Front. Energy Res. 2020, 8, 266. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Julbe, A.; Jouannaux, J.; Cartoixa, B. Two-step CO2 and H2O splitting using perovskite-coated ceria foam for enhanced green fuel production in a porous volumetric solar reactor. J. CO2 Util. 2020, 41, 101257. [Google Scholar] [CrossRef]
- Suter, S.; Steinfeld, A.; Haussener, S. Pore-level engineering of macroporous media for increased performance of solar-driven thermochemical fuel processing. Int. J. Heat Mass Transf. 2014, 78, 688–698. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, T.; Yan, Z.; Zhen, L.; Liu, J.; Wu, J.; Feng, Y. Mof-derived Cu2O/Cu nanospheres anchored in nitrogen-doped hollow porous carbon framework for increasing the selectivity and activity of electrochemical CO2-to-formate conversion. ACS Appl. Mater. Interfaces 2020, 12, 7030–7037. [Google Scholar] [CrossRef]
- Arifin, D.; Aston, V.J.; Liang, X.; McDaniel, A.H.; Weimer, A.W. CoFe2O4 on a porous AL2O3 nanostructure for solar thermochemical CO2 splitting. Energy Environ. Sci. 2012, 5, 9438–9443. [Google Scholar] [CrossRef]
- Parvanian, A.M.; Salimijazi, H.; Shabaninejad, M.; Kreider, P.; Saadatfar, M. Ca/Al doped lanthanum manganite perovskite coated porous sic for CO2 conversion. Mater. Chem. Phys. 2020, 253, 123306. [Google Scholar] [CrossRef]
- Scheffe, J.R.; Steinfeld, A. Oxygen exchange materials for solar thermochemical splitting of H2O and CO2: A review. Mater. Today 2014, 17, 341–348. [Google Scholar] [CrossRef]
- Guo, S.-H.; Qi, X.-J.; Zhou, H.-M.; Zhou, J.; Wang, X.-H.; Dong, M.; Zhao, X.; Sun, C.-Y.; Wang, X.-L.; Su, Z.-M. A bimetallic-mof catalyst for efficient CO2 photoreduction from simulated flue gas to value-added formate. J. Mater. Chem. A 2020, 8, 11712–11718. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Sun, Q.; Wang, Z.; Huang, B.; Dai, Y.; Qin, X.; Zhang, X. Chemical adsorption enhanced CO2 capture and photoreduction over a copper porphyrin based metal organic framework. ACS Appl. Mater. Interfaces 2013, 5, 7654–7658. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, J.; Tan, X.; Zheng, L.; Tan, D.; Liu, L.; Chen, G.; Wan, Q.; Zhang, B.; Zhang, F.; et al. Improved photocatalytic performance of metal–organic frameworks for CO2 conversion by ligand modification. Chem. Commun. 2020, 56, 7637–7640. [Google Scholar] [CrossRef]
- Loutzenhiser, P.G.; Meier, A.; Steinfeld, A. Review of the two-step H2O/CO2-splitting solar thermochemical cycle based on zn/zno redox reactions. Materials 2010, 3, 4922. [Google Scholar] [CrossRef] [Green Version]
- Kovačič, Ž.; Likozar, B.; Huš, M. Photocatalytic CO2 reduction: A review of ab initio mechanism, kinetics, and multiscale modeling simulations. ACS Catal. 2020, 10, 14984–15007. [Google Scholar] [CrossRef]
- Shehzad, N.; Tahir, M.; Johari, K.; Murugesan, T.; Hussain, M. A critical review on TiO2 based photocatalytic CO2 reduction system: Strategies to improve efficiency. J. CO2 Util. 2018, 26, 98–122. [Google Scholar] [CrossRef]
- Ola, O.; Maroto-Valer, M.M. Review of material design and reactor engineering on TiO2 photocatalysis for CO2 reduction. J. Photochem. Photobiol. C Photochem. Rev. 2015, 24, 16–42. [Google Scholar] [CrossRef] [Green Version]
- Low, J.; Cheng, B.; Yu, J. Surface modification and enhanced photocatalytic CO2 reduction performance of TiO2: A review. Appl. Surf. Sci. 2017, 392, 658–686. [Google Scholar] [CrossRef]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Li, K.; An, X.; Park, K.H.; Khraisheh, M.; Tang, J. A critical review of CO2 photoconversion: Catalysts and reactors. Catal. Today 2014, 224, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Thompson, W.A.; Sanchez Fernandez, E.; Maroto-Valer, M.M. Review and analysis of CO2 photoreduction kinetics. ACS Sustain. Chem. Eng. 2020, 8, 4677–4692. [Google Scholar] [CrossRef] [Green Version]
- Luu, M.T.; Milani, D.; Bahadori, A.; Abbas, A. A comparative study of CO2 utilization in methanol synthesis with various syngas production technologies. J. CO2 Util. 2015, 12, 62–76. [Google Scholar] [CrossRef]
- Milani, D.; Khalilpour, R.; Zahedi, G.; Abbas, A. A model-based analysis of CO2 utilization in methanol synthesis plant. J. CO2 Util. 2015, 10, 12–22. [Google Scholar] [CrossRef]
- Luu, M.T.; Milani, D.; Sharma, M.; Zeaiter, J.; Abbas, A. Model-based analysis of CO2 revalorization for di-methyl ether synthesis driven by solar catalytic reforming. Appl. Energy 2016, 177, 863–878. [Google Scholar] [CrossRef]
- Rieks, M.; Bellinghausen, R.; Kockmann, N.; Mleczko, L. Experimental study of methane dry reforming in an electrically heated reactor. Int. J. Hydrogen Energy 2015, 40, 15940–15951. [Google Scholar] [CrossRef]
- Aasberg-Petersen, K.; Christensen, T.S.; Dybkjaer, I.; Sehested, J.; Østberg, M.; Coertzen, R.M.; Keyser, M.J.; Steynberg, A.P. Chapter 4—synthesis gas production for ft synthesis. In Studies in Surface Science and Catalysis; Steynberg, A., Dry, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 152, pp. 258–405. [Google Scholar]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Hillestad, M. Systematic staging in chemical reactor design. Chem. Eng. Sci. 2010, 65, 3301–3312. [Google Scholar] [CrossRef]
- Anwar, M.N.; Fayyaz, A.; Sohail, N.F.; Khokhar, M.F.; Baqar, M.; Yasar, A.; Rasool, K.; Nazir, A.; Raja, M.U.F.; Rehan, M.; et al. CO2 utilization: Turning greenhouse gas into fuels and valuable products. J. Environ. Manag. 2020, 260, 110059. [Google Scholar] [CrossRef]
- Ateka, A.; Pérez-Uriarte, P.; Gamero, M.; Ereña, J.; Aguayo, A.T.; Bilbao, J. A comparative thermodynamic study on the CO2 conversion in the synthesis of methanol and of dme. Energy 2017, 120, 796–804. [Google Scholar] [CrossRef]
- Dieterich, V.; Buttler, A.; Hanel, A.; Spliethoff, H.; Fendt, S. Power-to-liquid via synthesis of methanol, dme or fischer–tropsch-fuels: A review. Energy Environ. Sci. 2020, 13, 3207–3252. [Google Scholar] [CrossRef]
- Jadhav, S.G.; Vaidya, P.D.; Bhanage, B.M.; Joshi, J.B. Catalytic carbon dioxide hydrogenation to methanol: A review of recent studies. Chem. Eng. Res. Des. 2014, 92, 2557–2567. [Google Scholar] [CrossRef]
- Park, N.; Park, M.-J.; Ha, K.-S.; Lee, Y.-J.; Jun, K.-W. Modeling and analysis of a methanol synthesis process using a mixed reforming reactor: Perspective on methanol production and CO2 utilization. Fuel 2014, 129, 163–172. [Google Scholar] [CrossRef]
- Rafiee, A. Staging of di-methyl-ether (dme) synthesis reactor from synthesis gas (syngas): Direct versus indirect route. Chem. Eng. Res. Des. 2020, 163, 157–168. [Google Scholar] [CrossRef]
- Bonura, G.; Cannilla, C.; Frusteri, L.; Mezzapica, A.; Frusteri, F. Dme production by CO2 hydrogenation: Key factors affecting the behaviour of cuznzr/ferrierite catalysts. Catal. Today 2017, 281, 337–344. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Zhang, S.; Wang, K.; Wu, J. CO2 hydrogenation to dimethyl ether over cuo–zno–al2o3/hzsm-5 prepared by combustion route. RSC Adv. 2014, 4, 16391–16396. [Google Scholar] [CrossRef]
- Ziaei, M.; Panahi, M.; Fanaei, M.A.; Rafiee, A.; Khalilpour, K.R. Maximizing the profitability of integrated fischer-tropsch gtl process with ammonia and urea synthesis using response surface methodology. J. CO2 Util. 2020, 35, 14–27. [Google Scholar] [CrossRef]
- Otto, A.; Grube, T.; Schiebahn, S.; Stolten, D. Closing the loop: Captured CO2 as a feedstock in the chemical industry. Energy Environ. Sci. 2015, 8, 3283–3297. [Google Scholar] [CrossRef] [Green Version]
- Bose, A.; Jana, K.; Mitra, D.; De, S. Co-production of power and urea from coal with CO2 capture: Performance assessment. Clean Technol. Environ. Policy 2015, 17, 1271–1280. [Google Scholar] [CrossRef]
- Langanke, J.; Wolf, A.; Hofmann, J.; Bohm, K.; Subhani, M.A.; Muller, T.E.; Leitner, W.; Gurtler, C. Carbon dioxide (CO2) as sustainable feedstock for polyurethane production. Green Chem. 2014, 16, 1865–1870. [Google Scholar] [CrossRef]
- Zarandi, M.; Panahi, M.; Rafiee, A. Simulation of a natural gas-to-liquid process with a multitubular fischer–tropsch reactor and variable chain growth factor for product distribution. Ind. Eng. Chem. Res. 2020, 59, 19322–19333. [Google Scholar] [CrossRef]
- Panzone, C.; Philippe, R.; Chappaz, A.; Fongarland, P.; Bengaouer, A. Power-to-liquid catalytic CO2 valorization into fuels and chemicals: Focus on the fischer-tropsch route. J. CO2 Util. 2020, 38, 314–347. [Google Scholar] [CrossRef]
- Dimitriou, I.; Garcia-Gutierrez, P.; Elder, R.H.; Cuellar-Franca, R.M.; Azapagic, A.; Allen, R.W.K. Carbon dioxide utilisation for production of transport fuels: Process and economic analysis. Energy Environ. Sci. 2015, 8, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Agrafiotis, C.; Roeb, M.; Sattler, C. A review on solar thermal syngas production via redox pair-based water/carbon dioxide splitting thermochemical cycles. Renew. Sustain. Energy Rev. 2015, 42, 254–285. [Google Scholar] [CrossRef]
- Yadav, D.; Banerjee, R. A review of solar thermochemical processes. Renew. Sustain. Energy Rev. 2016, 54, 497–532. [Google Scholar] [CrossRef]
- Yuan, L.; Xu, Y.-J. Photocatalytic conversion of CO2 into value-added and renewable fuels. Appl. Surf. Sci. 2015, 342, 154–167. [Google Scholar] [CrossRef]
- Karamian, E.; Sharifnia, S. On the general mechanism of photocatalytic reduction of CO2. J. CO2 Util. 2016, 16, 194–203. [Google Scholar] [CrossRef]
- Xu, D.; Dong, L.; Ren, J. Chapter 2—introduction of hydrogen routines. In Hydrogen Economy; Scipioni, A., Manzardo, A., Ren, J., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 35–54. [Google Scholar]
- Laursen, S.; Poudyal, S. Chapter 8—photo- and electro-catalysis: CO2 mitigation technologies. In Novel Materials for Carbon Dioxide Mitigation Technology; Shi, F., Morreale, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 233–268. [Google Scholar]
- Al-Rowaili, F.N.; Jamal, A.; Ba Shammakh, M.S.; Rana, A. A review on recent advances for electrochemical reduction of carbon dioxide to methanol using metal–organic framework (mof) and non-mof catalysts: Challenges and future prospects. ACS Sustain. Chem. Eng. 2018, 6, 15895–15914. [Google Scholar] [CrossRef]
- Duan, X.; Xu, J.; Wei, Z.; Ma, J.; Guo, S.; Wang, S.; Liu, H.; Dou, S. Metal-free carbon materials for CO2 electrochemical reduction. Adv. Mater. 2017, 29, 1701784. [Google Scholar] [CrossRef]
- Rackley, S.A. 22—CO2 utilization and other sequestration options. In Carbon Capture and Storage, 2nd ed.; Rackley, S.A., Ed.; Butterworth-Heinemann: Boston, MA, USA, 2017; pp. 577–591. [Google Scholar]
- Steinfeld, A.; Weimer, A.W. Thermochemical production of fuels with concentrated solar energy. Opt. Express 2010, 18, A100–A111. [Google Scholar] [CrossRef]
- Nathan, G.J.; Jafarian, M.; Dally, B.B.; Saw, W.L.; Ashman, P.J.; Hu, E.; Steinfeld, A. Solar thermal hybrids for combustion power plant: A growing opportunity. Prog. Energy Combust. Sci. 2018, 64, 4–28. [Google Scholar] [CrossRef]
- Jafarian, M.; Arjomandi, M.; Nathan, G.J. Thermodynamic potential of molten copper oxide for high temperature solar energy storage and oxygen production. Appl. Energy 2017, 201, 69–83. [Google Scholar] [CrossRef]
- Schunk, L.O.; Steinfeld, A. Kinetics of the thermal dissociation of zno exposed to concentrated solar irradiation using a solar-driven thermogravimeter in the 1800–2100 k range. AIChE J. 2009, 55, 1497–1504. [Google Scholar] [CrossRef]
- Silakhori, M.; Jafarian, M.; Arjomandi, M.; Nathan, G.J. The energetic performance of a liquid chemical looping cycle with solar thermal energy storage. Energy 2019, 170, 93–101. [Google Scholar] [CrossRef]
- Takeshita, T.; Yamaji, K. Important roles of fischer–tropsch synfuels in the global energy future. Energy Policy 2008, 36, 2773–2784. [Google Scholar] [CrossRef]
- Nathan, G.; Dally, B.; Alwahabi, Z.; Van Eyk, P.; Jafarian, M.; Ashman, P. Research challenges in combustion and gasification arising from emerging technologies employing directly irradiated concentrating solar thermal radiation. Proc. Combust. Inst. 2017, 36, 2055–2074. [Google Scholar] [CrossRef]
- Steinfeld, A. Solar thermochemical production of hydrogen––A review. Sol. Energy 2005, 78, 603–615. [Google Scholar] [CrossRef]
- Chueh, W.C.; Falter, C.; Abbott, M.; Scipio, D.; Furler, P.; Haile, S.M.; Steinfeld, A. High-flux solar-driven thermochemical dissociation of CO2 and H2O using nonstoichiometric ceria. Science 2010, 330, 1797–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Henao, C.A.; Johnson, T.A.; Dedrick, D.E.; Miller, J.E.; Stechel, E.B.; Maravelias, C.T. Methanol production from CO2 using solar-thermal energy: Process development and techno-economic analysis. Energy Environ. Sci. 2011, 4, 3122–3132. [Google Scholar] [CrossRef]
- Kim, J.; Johnson, T.A.; Miller, J.E.; Stechel, E.B.; Maravelias, C.T. Fuel production from CO2 using solar-thermal energy: System level analysis. Energy Environ. Sci. 2012, 5, 8417–8429. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Julbe, A.; Jouannaux, J.; Cartoixa, B. Solar thermochemical fuel production from H2O and CO2 splitting via two-step redox cycling of reticulated porous ceria structures integrated in a monolithic cavity-type reactor. Energy 2020, 201, 117649. [Google Scholar] [CrossRef]
- Alxneit, I. Assessing the feasibility of separating a stoichiometric mixture of zinc vapor and oxygen by a fast quench—Model calculations. Sol. Energy 2008, 82, 959–964. [Google Scholar] [CrossRef]
- Steinfeld, A.; Larson, C.; Palumbo, R.; Foley, M., III. Thermodynamic analysis of the co-production of zinc and synthesis gas using solar process heat. Energy 1996, 21, 205–222. [Google Scholar] [CrossRef]
- Silakhori, M.; Jafarian, M.; Arjomandi, M.; Nathan, G.J. Comparing the thermodynamic potential of alternative liquid metal oxides for the storage of solar thermal energy. Sol. Energy 2017, 157, 251–258. [Google Scholar] [CrossRef]
- Weidenkaff, A.; Steinfeld, A.; Wokaun, A.; Auer, P.; Eichler, B.; Reller, A. Direct solar thermal dissociation of zinc oxide: Condensation and crystallisation of zinc in the presence of oxygen. Sol. Energy 1999, 65, 59–69. [Google Scholar] [CrossRef]
- Steinfeld, A. Solar hydrogen production via a two-step water-splitting thermochemical cycle based on zn/zno redox reactions. Int. J. Hydrogen Energy 2002, 27, 611–619. [Google Scholar] [CrossRef]
- Schunk, L.O.; Haeberling, P.; Wepf, S.; Wuillemin, D.; Meier, A.; Steinfeld, A. A receiver-reactor for the solar thermal dissociation of zinc oxide. J. Sol. Energy Eng. 2008, 130, 021009. [Google Scholar] [CrossRef]
- Fletcher, E.A. Solarthermal and solar quasi-electrolytic processing and separations: Zinc from zinc oxide as an example. Ind. Eng. Chem. Res. 1999, 38, 2275–2282. [Google Scholar] [CrossRef]
- Stamatiou, A.; Loutzenhiser, P.; Steinfeld, A. Solar syngas production via H2O/CO2-splitting thermochemical cycles with Zn/ZnO and FeO/Fe3O4 redox reactions. Chem. Mater. 2010, 22, 851–859. [Google Scholar] [CrossRef]
- Lee, K.L.; Jafarian, M.; Ghanadi, F.; Arjomandi, M.; Nathan, G.J. An investigation into the effect of aspect ratio on the heat loss from a solar cavity receiver. Sol. Energy 2017, 149, 20–31. [Google Scholar] [CrossRef]
- Davis, D.; Jafarian, M.; Chinnici, A.; Saw, W.L.; Nathan, G.J. Thermal performance of vortex-based solar particle receivers for sensible heating. Sol. Energy 2019, 177, 163–177. [Google Scholar] [CrossRef]
- Perkins, C.; Lichty, P.R.; Weimer, A.W. Thermal zno dissociation in a rapid aerosol reactor as part of a solar hydrogen production cycle. Int. J. Hydrogen Energy 2008, 33, 499–510. [Google Scholar] [CrossRef]
- Abanades, S.; Charvin, P.; Lemont, F.; Flamant, G. Novel two-step SnO2/sno water-splitting cycle for solar thermochemical production of hydrogen. Int. J. Hydrogen Energy 2008, 33, 6021–6030. [Google Scholar] [CrossRef]
- Charvin, P.; Abanades, S.; Lemont, F.; Flamant, G. Experimental study of SnO2/SnO/Sn thermochemical systems for solar production of hydrogen. AIChE J. 2008, 54, 2759–2767. [Google Scholar] [CrossRef]
- Le Gal, A.; Abanades, S. Dopant incorporation in ceria for enhanced water-splitting activity during solar thermochemical hydrogen generation. J. Phys. Chem. C 2012, 116, 13516–13523. [Google Scholar] [CrossRef]
- Levêque, G.; Abanades, S.; Jumas, J.-C.; Olivier-Fourcade, J. Characterization of two-step tin-based redox system for thermochemical fuel production from solar-driven CO2 and H2O splitting cycle. Ind. Eng. Chem. Res. 2014, 53, 5668–5677. [Google Scholar] [CrossRef]
- Ketteler, G.; Weiss, W.; Ranke, W.; Schlögl, R. Bulk and surface phases of iron oxides in an oxygen and water atmosphere at low pressure. Phys. Chem. Chem. Phys. 2001, 3, 1114–1122. [Google Scholar] [CrossRef]
- Jafarian, M.; Arjomandi, M.; Nathan, G.J. Thermodynamic potential of high temperature chemical looping combustion with molten iron oxide as the oxygen carrier. Chem. Eng. Res. Des. 2017, 120, 69–81. [Google Scholar] [CrossRef]
- Jafarian, M.; Arjomandi, M.; Nathan, G.J. Influence of the type of oxygen carriers on the performance of a hybrid solar chemical looping combustion system. Energy Fuels 2014, 28, 2914–2924. [Google Scholar] [CrossRef]
- Tofighi, A.; Sibieude, F. Note on the condensation of the vapor phase above a melt of iron oxide in a solar parabolic concentrator. Int. J. Hydrogen Energy 1980, 5, 375–381. [Google Scholar] [CrossRef]
- Sibieude, F.; Ducarroir, M.; Tofighi, A.; Ambriz, J. High temperature experiments with a solar furnace: The decomposition of Fe3O4, Mn3O4, CdO. Int. J. Hydrogen Energy 1982, 7, 79–88. [Google Scholar] [CrossRef]
- Tofighi, A.; Sibieude, F. Dissociation of magnetite in a solar furnace for hydrogen-production—Tentative production evaluation of a 1000 kw concentrator from small-scale (2 kw) experimental results. Int. J. Hydrogen Energy 1984, 9, 293–296. [Google Scholar] [CrossRef]
- Haseli, P.; Jafarian, M.; Nathan, G.J. High temperature solar thermochemical process for production of stored energy and oxygen based on Cuo/Cu2O redox reactions. Sol. Energy 2017, 153, 1–10. [Google Scholar] [CrossRef]
- Allen, K.M.; Klausner, J.F.; Coker, E.N.; AuYeung, N.; Mishra, R. Synthesis and analysis of cobalt ferrite in ysz for use as reactive material in solar thermochemical water and carbon dioxide splitting. In Proceedings of the ASME 2013 7th International Conference on Energy Sustainability collocated with the ASME 2013 Heat Transfer Summer Conference and the ASME 2013 11th International Conference on Fuel Cell Science, Engineering and Technology, Minneapolis, MN, USA, 14–19 July 2013. [Google Scholar]
- Gokon, N.; Murayama, H.; Nagasaki, A.; Kodama, T. Thermochemical two-step water splitting cycles by monoclinic zro2-supported nife2o4 and fe3o4 powders and ceramic foam devices. Sol. Energy 2009, 83, 527–537. [Google Scholar] [CrossRef]
- Coker, E.N.; Ambrosini, A.; Rodriguez, M.A.; Miller, J.E. Ferrite-ysz composites for solar thermochemical production of synthetic fuels: In operando characterization of CO2 reduction. J. Mater. Chem. 2011, 21, 10767–10776. [Google Scholar] [CrossRef]
- Coker, E.N.; Ambrosini, A.; Miller, J.E. Compositional and operational impacts on the thermochemical reduction of CO2 to co by iron oxide/yttria-stabilized zirconia. RSC Adv. 2021, 11, 1493–1502. [Google Scholar] [CrossRef]
- Scheffe, J.R.; Li, J.; Weimer, A.W. A spinel ferrite/hercynite water-splitting redox cycle. Int. J. Hydrogen Energy 2010, 35, 3333–3340. [Google Scholar] [CrossRef]
- Kaneko, H.; Yokoyama, T.; Fuse, A.; Ishihara, H.; Hasegawa, N.; Tamaura, Y. Synthesis of new ferrite, Al–Cu ferrite, and its oxygen deficiency for solar H2 generation from H2O. Int. J. Hydrogen Energy 2006, 31, 2256–2265. [Google Scholar] [CrossRef]
- Lougou, B.G.; Shuai, Y.; Zhang, H.; Ahouannou, C.; Zhao, J.; Kounouhewa, B.B.; Tan, H. Thermochemical CO2 reduction over nife2o4@ alumina filled reactor heated by high-flux solar simulator. Energy 2020, 197, 117267. [Google Scholar] [CrossRef]
- Millican, S.L.; Androshchuk, I.; Tran, J.T.; Trottier, R.M.; Bayon, A.; Al Salik, Y.; Idriss, H.; Musgrave, C.B.; Weimer, A.W. Oxidation kinetics of hercynite spinels for solar thermochemical fuel production. Chem. Eng. J. 2020, 401, 126015. [Google Scholar] [CrossRef]
- Otsuka, K.; Hatano, M.; Morikawa, A. Hydrogen from water by reduced cerium oxide. J. Catal. 1983, 79, 493–496. [Google Scholar] [CrossRef]
- Otsuka, K.; Hatano, M.; Morikawa, A. Decomposition of water by cerium oxide of δ-phase. Inorg. Chim. Acta 1985, 109, 193–197. [Google Scholar] [CrossRef]
- Abanades, S.; Flamant, G. Thermochemical hydrogen production from a two-step solar-driven water-splitting cycle based on cerium oxides. Sol. Energy 2006, 80, 1611–1623. [Google Scholar] [CrossRef]
- Chueh, W.C.; Haile, S.M. Ceria as a thermochemical reaction medium for selectively generating syngas or methane from H2O and CO2. ChemSusChem 2009, 2, 735–739. [Google Scholar] [CrossRef] [Green Version]
- Abanades, S.; Legal, A.; Cordier, A.; Peraudeau, G.; Flamant, G.; Julbe, A. Investigation of reactive cerium-based oxides for H2 production by thermochemical two-step water-splitting. J. Mater. Sci. 2010, 45, 4163–4173. [Google Scholar] [CrossRef]
- Chueh, W.C.; Haile, S.M. A thermochemical study of ceria: Exploiting an old material for new modes of energy conversion and CO2 mitigation. Philos. Trans. R. Soc. A 2010, 368, 3269–3294. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.L.; Lee, C.I.; Ishihara, T.; Kaneko, H.; Tamaura, Y. Reactivity of ceo(2)-based ceramics for solar hydrogen production via a two-step water-splitting cycle with concentrated solar energy. Int. J. Hydrogen Energy 2011, 36, 13435–13441. [Google Scholar] [CrossRef]
- Scheffe, J.R.; Steinfeld, A. Thermodynamic analysis of cerium-based oxides for solar thermochemical fuel production. Energy Fuels 2012, 26, 1928–1936. [Google Scholar] [CrossRef]
- Jang, J.T.; Yoon, K.J.; Han, G.Y. Methane reforming and water splitting using zirconia-supported cerium oxide in a volumetric receiver–reactor with different types of foam devices. Sol. Energy 2014, 101, 29–39. [Google Scholar] [CrossRef]
- Venstrom, L.J.; Petkovich, N.; Rudisill, S.; Stein, A.; Davidson, J.H. The effects of morphology on the oxidation of ceria by water and carbon dioxide. J. Sol. Energy Eng. 2011, 134, 011005. [Google Scholar] [CrossRef]
- Furler, P.; Scheffe, J.; Gorbar, M.; Moes, L.; Vogt, U.; Steinfeld, A. Solar thermochemical CO2 splitting utilizing a reticulated porous ceria redox system. Energy Fuels 2012, 26, 7051–7059. [Google Scholar] [CrossRef]
- Le Gal, A.; Abanades, S.; Bion, N.; Le Mercier, T.; Harlé, V. Reactivity of doped ceria-based mixed oxides for solar thermochemical hydrogen generation via two-step water-splitting cycles. Energy Fuels 2013, 27, 6068–6078. [Google Scholar] [CrossRef]
- Call, F.; Roeb, M.; Schmücker, M.; Bru, H.; Curulla-Ferre, D.; Sattler, C.; Pitz-Paal, R. Thermogravimetric analysis of zirconia-doped ceria for thermochemical production of solar fuel. Am. J. Anal. Chem. 2013, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.; Venstrom, L.J.; Davidson, J.H.; Lipiński, W. Thermodynamic analysis of isothermal redox cycling of ceria for solar fuel production. Energy Fuels 2013, 27, 5533–5544. [Google Scholar] [CrossRef]
- Furler, P.; Scheffe, J.; Marxer, D.; Gorbar, M.; Bonk, A.; Vogt, U.; Steinfeld, A. Thermochemical CO2 splitting via redox cycling of ceria reticulated foam structures with dual-scale porosities. Phys. Chem. Chem. Phys. 2014, 16, 10503–10511. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.T.; Yoon, K.J.; Bae, J.W.; Han, G.Y. Cyclic production of syngas and hydrogen through methane-reforming and water-splitting by using ceria-zirconia solid solutions in a solar volumetric receiver-reactor. Sol. Energy 2014, 109, 70–81. [Google Scholar] [CrossRef]
- Call, F.; Roeb, M.; Schmücker, M.; Sattler, C.; Pitz-Paal, R. Ceria doped with zirconium and lanthanide oxides to enhance solar thermochemical production of fuels. J. Phys. Chem. C 2015, 119, 6929–6938. [Google Scholar] [CrossRef]
- Muhich, C.L.; Evanko, B.W.; Weston, K.C.; Lichty, P.; Liang, X.; Martinek, J.; Musgrave, C.B.; Weimer, A.W. Efficient generation of h2 by splitting water with an isothermal redox cycle. Science 2013, 341, 540. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, R.R. Thermodynamic efficiency analysis of zinc oxide based solar driven thermochemical H2O splitting cycle: Effect of partial pressure of O2, thermal reduction and H2O splitting temperatures. Int. J. Hydrogen Energy 2018, 43, 14915–14924. [Google Scholar] [CrossRef]
- Bhosale, R.R.; Takalkar, G.; Sutar, P.; Kumar, A.; AlMomani, F.; Khraisheh, M. A decade of ceria based solar thermochemical H2O/CO2 splitting cycle. Int. J. Hydrogen Energy 2019, 44, 34–60. [Google Scholar] [CrossRef]
- Bhosale, R.R.; Kumar, A.; AlMomani, F.; Ghosh, U.; Al-Muhtaseb, S.; Gupta, R.; Alxneit, I. Assessment of CexZryHfzO2 based oxides as potential solar thermochemical CO2 splitting materials. Ceram. Int. 2016, 42, 9354–9362. [Google Scholar] [CrossRef]
- Lee, K.L.; Chinnici, A.; Jafarian, M.; Arjomandi, M.; Dally, B.; Nathan, G. The influence of wind speed, aperture ratio and tilt angle on the heat losses from a finely controlled heated cavity for a solar receiver. Renew. Energy 2019, 143, 1544–1553. [Google Scholar] [CrossRef]
- Lee, K.L.; Chinnici, A.; Jafarian, M.; Arjomandi, M.; Dally, B.; Nathan, G. The influence of wall temperature distribution on the mixed convective losses from a heated cavity. Appl. Therm. Eng. 2019, 155, 157–165. [Google Scholar] [CrossRef]
- Miller, J.E.; Allendorf, M.D.; Diver, R.B.; Evans, L.R.; Siegel, N.P.; Stuecker, J.N. Metal oxide composites and structures for ultra-high temperature solar thermochemical cycles. J. Mater. Sci. 2008, 43, 4714–4728. [Google Scholar] [CrossRef]
- Scheffe, J.R.; Weibel, D.; Steinfeld, A. Lanthanum–strontium–manganese perovskites as redox materials for solar thermochemical splitting of H2O and CO2. Energy Fuels 2013, 27, 4250–4257. [Google Scholar] [CrossRef]
- McDaniel, A.H.; Miller, E.C.; Arifin, D.; Ambrosini, A.; Coker, E.N.; O’Hayre, R.; Chueh, W.C.; Tong, J. Sr- and mn-doped LaAlO3−δ for solar thermochemical H2 and co production. Energy Environ. Sci. 2013, 6, 2424–2428. [Google Scholar] [CrossRef]
- Nalbandian, L.; Evdou, A.; Zaspalis, V. La(1−x)Sr(x)Mo(3) (m = mn, fe) perovskites as materials for thermochemical hydrogen production in conventional and membrane reactors. Int. J. Hydrogen Energy 2009, 34, 7162–7172. [Google Scholar] [CrossRef]
- McDaniel, A.H.; Ambrosini, A.; Coker, E.N.; Miller, J.E.; Chueh, W.C.; O’Hayre, R.; Tong, J. Nonstoichiometric perovskite oxides for solar thermochemical H2 and co production. Energy Procedia 2014, 49, 2009–2018. [Google Scholar] [CrossRef] [Green Version]
- Demont, A.; Abanades, S. Solar thermochemical conversion of CO2 into fuel via two-step redox cycling of non-stoichiometric mn-containing perovskite oxides. J. Mater. Chem. A 2015, 3, 3536–3546. [Google Scholar] [CrossRef]
- Bork, A.H.; Kubicek, M.; Struzik, M.; Rupp, J.L.M. Perovskite La0.6Sr0.4Cr1−xCoxO3−δ solid solutions for solar-thermochemical fuel production: Strategies to lower the operation temperature. J. Mater. Chem. A 2015, 3, 15546–15557. [Google Scholar] [CrossRef] [Green Version]
- Cooper, T.; Scheffe, J.R.; Galvez, M.E.; Jacot, R.; Patzke, G.; Steinfeld, A. Lanthanum manganite perovskites with ca/sr a-site and al b-site doping as effective oxygen exchange materials for solar thermochemical fuel production. Energy Technol. Ger 2015, 3, 1130–1142. [Google Scholar] [CrossRef]
- Takacs, M.; Hoes, M.; Caduff, M.; Cooper, T.; Scheffe, J.R.; Steinfeld, A. Oxygen nonstoichiometry, defect equilibria, and thermodynamic characterization of lamno3 perovskites with ca/sr a-site and al b-site doping. Acta Mater 2016, 103, 700–710. [Google Scholar] [CrossRef] [Green Version]
- Parvanian, A.M.; Salimijazi, H.; Shabaninejad, M.; Troitzsch, U.; Kreider, P.; Lipiński, W.; Saadatfar, M. Thermochemical CO2 splitting performance of perovskite coated porous ceramics. RSC Adv. 2020, 10, 23049–23057. [Google Scholar] [CrossRef]
- Gokon, N.; Hasegawa, T.; Takahashi, S.; Kodama, T. Thermochemical two-step water-splitting for hydrogen production using fe-ysz particles and a ceramic foam device. Energy 2008, 33, 1407–1416. [Google Scholar] [CrossRef]
- Kawakami, S.; Myojin, T.; Cho, H.S.; Hatamachi, T.; Gokon, N.; Kodama, T. Thermochemical two-step water splitting cycle using ni-ferrite and CeO2 coated ceramic foam devices by concentrated xe-light radiation. Energy Procedia 2014, 49, 1980–1989. [Google Scholar] [CrossRef] [Green Version]
- Marxer, D.; Furler, P.; Takacs, M.; Steinfeld, A. Solar thermochemical splitting of CO2 into separate streams of co and O2 with high selectivity, stability, conversion, and efficiency. Energy Environ. Sci. 2017, 10, 1142–1149. [Google Scholar] [CrossRef] [Green Version]
- Guene Lougou, B.; Shuai, Y.; Pan, R.; Chaffa, G.; Ahouannou, C.; Zhang, H.; Tan, H. Radiative heat transfer and thermal characteristics of fe-based oxides coated sic and alumina rpc structures as integrated solar thermochemical reactor. Sci. China Technol. Sci. 2018, 61, 1788–1801. [Google Scholar] [CrossRef]
- Agrafiotis, C.; Roeb, M.; Konstandopoulos, A.G.; Nalbandian, L.; Zaspalis, V.T.; Sattler, C.; Stobbe, P.; Steele, A.M. Solar water splitting for hydrogen production with monolithic reactors. Sol. Energy 2005, 79, 409–421. [Google Scholar] [CrossRef]
- Walker, L.S.; Miller, J.E.; Hilmas, G.E.; Evans, L.R.; Corral, E.L. Coextrusion of zirconia–iron oxide honeycomb substrates for solar-based thermochemical generation of carbon monoxide for renewable fuels. Energy Fuels 2012, 26, 712–721. [Google Scholar] [CrossRef]
- Gokon, N.; Kodama, T.; Imaizumi, N.; Umeda, J.; Seo, T. Ferrite/zirconia-coated foam device prepared by spin coating for solar demonstration of thermochemical water-splitting. Int. J. Hydrogen Energy 2011, 36, 2014–2028. [Google Scholar] [CrossRef]
- Rouquerol, J.; Avnir, D.; Fairbridge, C.W.; Everett, D.H.; Haynes, J.M.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Recommendations for the characterization of porous solids (technical report). Pure Appl. Chem 1994, 66, 1739–1758. [Google Scholar] [CrossRef]
- Levy, M.; Rubin, R.; Rosin, H.; Levitan, R. Methane reforming by direct solar irradiation of the catalyst. Energy 1992, 17, 749–756. [Google Scholar] [CrossRef]
- Levy, M.; Rosin, H.; Levitan, R. Chemical reactions in a solar furnace by direct solar irradiation of the catalyst. J. Sol. Energy Eng. 1989, 111, 96–97. [Google Scholar] [CrossRef]
- Roeb, M.; Sattler, C.; Klüser, R.; Monnerie, N.; de Oliveira, L.; Konstandopoulos, A.G.; Agrafiotis, C.; Zaspalis, V.T.; Nalbandian, L.; Steele, A.; et al. Solar hydrogen production by a two-step cycle based on mixed iron oxides. J. Sol. Energy Eng. 2005, 128, 125–133. [Google Scholar] [CrossRef]
- Diver, R.B.; Miller, J.E.; Allendorf, M.D.; Siegel, N.P.; Hogan, R.E. Solar thermochemical water-splitting ferrite-cycle heat engines. J. Sol. Energy Eng. 2008, 130, 041001. [Google Scholar] [CrossRef] [Green Version]
- Roeb, M.; Säck, J.P.; Rietbrock, P.; Prahl, C.; Schreiber, H.; Neises, M.; de Oliveira, L.; Graf, D.; Ebert, M.; Reinalter, W.; et al. Test operation of a 100 kw pilot plant for solar hydrogen production from water on a solar tower. Sol. Energy 2011, 85, 634–644. [Google Scholar] [CrossRef]
- Furler, P.; Scheffe, J.R.; Steinfeld, A. Syngas production by simultaneous splitting of H2O and CO2 via ceria redox reactions in a high-temperature solar reactor. Energy Environ. Sci. 2012, 5, 6098–6103. [Google Scholar] [CrossRef]
- Rudisill, S.G.; Venstrom, L.J.; Petkovich, N.D.; Quan, T.; Hein, N.; Boman, D.B.; Davidson, J.H.; Stein, A. Enhanced oxidation kinetics in thermochemical cycling of CeO2 through templated porosity. J. Phys. Chem. C 2013, 117, 1692–1700. [Google Scholar] [CrossRef]
- Rhodes, N.R.; Bobek, M.M.; Allen, K.M.; Hahn, D.W. Investigation of long term reactive stability of ceria for use in solar thermochemical cycles. Energy 2015, 89, 924–931. [Google Scholar] [CrossRef] [Green Version]
- Marxer, D.; Furler, P.; Scheffe, J.; Geerlings, H.; Falter, C.; Batteiger, V.; Sizmann, A.; Steinfeld, A. Demonstration of the entire production chain to renewable kerosene via solar thermochemical splitting of H2O and CO2. Energy Fuels 2015, 29, 3241–3250. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S.; Rodat, S. Syngas production via solar-driven chemical looping methane reforming from redox cycling of ceria porous foam in a volumetric solar reactor. Chem. Eng. J. 2019, 356, 756–770. [Google Scholar] [CrossRef]
- Luyten, J.; Cooymans, J.; Smolders, C.; Vercauteren, S.; Vansant, E.; Leysen, R. Shaping of multilayer ceramic membranes by dip coating. J. Eur. Ceram. Soc. 1997, 17, 273–279. [Google Scholar] [CrossRef]
- Diver, R. Receiver/Reactor Concepts for Thermochemical Transport of Solar Energy. ASME. J. Sol. Energy Eng. 1987, 109, 199–204. [Google Scholar] [CrossRef]
- Lee, K.L.; Chinnici, A.; Jafarian, M.; Arjomandi, M.; Dally, B.; Nathan, G. Experimental investigation of the effects of wind speed and yaw angle on heat losses from a heated cavity. Sol. Energy 2018, 165, 178–188. [Google Scholar] [CrossRef]
- Yuan, C.; Jarrett, C.; Chueh, W.; Kawajiri, Y.; Henry, A. A new solar fuels reactor concept based on a liquid metal heat transfer fluid: Reactor design and efficiency estimation. Sol. Energy 2015, 122, 547–561. [Google Scholar] [CrossRef] [Green Version]
- Jafarian, M.; Abdollahi, M.R.; Nathan, G.J. Preliminary evaluation of a novel solar bubble receiver for heating a gas. Sol. Energy 2019, 182, 264–277. [Google Scholar] [CrossRef]
- Silakhori, M.; Jafarian, M.; Arjomandi, M.; Nathan, G.J. Experimental assessment of copper oxide for liquid chemical looping for thermal energy storage. J. Energy Storage 2019, 21, 216–221. [Google Scholar] [CrossRef]
- Jafarian, M.; Chisti, Y.; Nathan, G.J. Gas-lift circulation of a liquid between two inter-connected bubble columns. Chem. Eng. Sci. 2020, 218, 115574. [Google Scholar] [CrossRef]
- Jafarian, M.; Arjomandi, M.; Nathan, G.J. A hybrid solar and chemical looping combustion system for solar thermal energy storage. Appl. Energy 2013, 103, 671–678. [Google Scholar] [CrossRef]
- Jafarian, M.; Arjomandi, M.; Nathan, G.J. The influence of high intensity solar radiation on the temperature and reduction of an oxygen carrier particle in hybrid chemical looping combustion. Chem. Eng. Sci. 2013, 95, 331–342. [Google Scholar] [CrossRef]
- Romero, M.; Steinfeld, A. Concentrating solar thermal power and thermochemical fuels. Energy Environ. Sci. 2012, 5, 9234–9245. [Google Scholar] [CrossRef]
- Lu, Z.; Jafarian, M.; Arjomandi, M.; Nathan, G.J. Analytical assessment of a novel rotating fluidized bed solar reactor for steam gasification of char particles. Sol. Energy 2016, 140, 113–123. [Google Scholar] [CrossRef]
- Maag, G.; Lipiński, W.; Steinfeld, A. Particle–gas reacting flow under concentrated solar irradiation. Int. J. Heat Mass Transf. 2009, 52, 4997–5004. [Google Scholar] [CrossRef]
- Z’Graggen, A.; Haueter, P.; Trommer, D.; Romero, M.; De Jesus, J.; Steinfeld, A. Hydrogen production by steam-gasification of petroleum coke using concentrated solar power—ii reactor design, testing, and modeling. Int. J. Hydrogen Energy 2006, 31, 797–811. [Google Scholar] [CrossRef]
- Piatkowski, N.; Wieckert, C.; Steinfeld, A. Experimental investigation of a packed-bed solar reactor for the steam-gasification of carbonaceous feedstocks. Fuel Process. Technol. 2009, 90, 360–366. [Google Scholar] [CrossRef]
- Amy, C.; Budenstein, D.; Bagepalli, M.; England, D.; DeAngelis, F.; Wilk, G.; Jarrett, C.; Kelsall, C.; Hirschey, J.; Wen, H.; et al. Pumping liquid metal at high temperatures up to 1,673 kelvin. Nature 2017, 550, 199–203. [Google Scholar] [CrossRef]
- Long, S.; Lau, T.C.; Chinnici, A.; Nathan, G.J. The flow-field within a vortex-based solar cavity receiver with an open aperture. Exp. Therm. Fluid Sci. 2021, 123, 110314. [Google Scholar] [CrossRef]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, D. Photocatalysis: Basic principles, diverse forms of implementations and emerging scientific opportunities. Adv. Energy Mater. 2017, 7, 1700841. [Google Scholar] [CrossRef] [Green Version]
- Shtyka, O.; Ciesielski, R.; Kedziora, A.; Maniukiewicz, W.; Dubkov, S.; Gromov, D.; Maniecki, T. Photocatalytic reduction of CO2 over me (Pt, Pd, Ni, Cu)/TiO2 catalysts. Top. Catal. 2020, 63, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, C.S. 17—photocatalytic reduction of carbon dioxide (CO2). In Developments and Innovation in Carbon Dioxide (CO2) Capture and Storage Technology; Maroto-Valer, M.M., Ed.; Woodhead Publishing: Cambridge, UK, 2010; Volume 2, pp. 463–501.(CO2). In Developments and Innovation in Carbon Dioxide (CO2) Capture and Storage Technology; Maroto-Valer, M.M., Ed.; Woodhead Publishing: Cambridge, UK, 2010; Volume 2, pp. 463–501. [Google Scholar]
- Abdullah, H.; Khan, M.M.R.; Ong, H.R.; Yaakob, Z. Modified TiO2 photocatalyst for CO2 photocatalytic reduction: An overview. J. CO2 Util. 2017, 22, 15–32. [Google Scholar] [CrossRef]
- Dong, H.; Zeng, G.; Tang, L.; Fan, C.; Zhang, C.; He, X.; He, Y. An overview on limitations of TiO2-based particles for photocatalytic degradation of organic pollutants and the corresponding countermeasures. Water Res. 2015, 79, 128–146. [Google Scholar] [CrossRef]
- Tasbihi, M.; Călin, I.; Šuligoj, A.; Fanetti, M.; Lavrenčič Štangar, U. Photocatalytic degradation of gaseous toluene by using TiO2 nanoparticles immobilized on fiberglass cloth. J. Photochem. Photobiol. A Chem. 2017, 336, 89–97. [Google Scholar] [CrossRef]
- Baloyi, J.; Seadira, T.; Raphulu, M.; Ochieng, A. Preparation, characterization and growth mechanism of dandelion-like TiO2 nanostructures and their application in photocatalysis towards reduction of cr(vi). Mater. Today Proc. 2015, 2, 3973–3987. [Google Scholar] [CrossRef]
- Tsang, C.H.A.; Li, K.; Zeng, Y.; Zhao, W.; Zhang, T.; Zhan, Y.; Xie, R.; Leung, D.Y.C.; Huang, H. Titanium oxide based photocatalytic materials development and their role of in the air pollutants degradation: Overview and forecast. Environ. Int. 2019, 125, 200–228. [Google Scholar] [CrossRef]
- Liu, X.; Xing, Z.; Zhang, Y.; Li, Z.; Wu, X.; Tan, S.; Yu, X.; Zhu, Q.; Zhou, W. Fabrication of 3d flower-like black n-TiO2-x@mos2 for unprecedented-high visible-light-driven photocatalytic performance. Appl. Catal. B Environ. 2017, 201, 119–127. [Google Scholar] [CrossRef]
- Carp, O.; Huisman, C.L.; Reller, A. Photoinduced reactivity of titanium dioxide. Prog. Solid State Chem. 2004, 32, 33–177. [Google Scholar] [CrossRef]
- Tahir, M.; Amin, N.S. Indium-doped TiO2 nanoparticles for photocatalytic CO2 reduction with H2O vapors to CH4. Appl. Catal. B Environ. 2015, 162, 98–109. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Navalon, S.; Corma, A.; Garcia, H. Photocatalytic CO2 reduction by TiO2 and related titanium containing solids. Energy Environ. 2012, 5, 9217–9233. [Google Scholar] [CrossRef]
- Liu, C.; Yu, T.; Tan, X.; Huang, X. Comparison n-cu–codoped nanotitania and n-doped nanotitania in photocatalytic reduction of CO2 under uv light. Inorg. Nano Met. Chem. 2017, 47, 9–14. [Google Scholar] [CrossRef]
- Zaleska, A. Doped-TiO2: A review. Recent Pat. Eng. 2008, 2, 157–164. [Google Scholar] [CrossRef]
- Nematollahi, R.; Ghotbi, C.; Khorasheh, F.; Larimi, A. Ni-bi co-doped TiO2 as highly visible light response nano-photocatalyst for CO2 photo-reduction in a batch photo-reactor. J.CO2 Util. 2020, 41, 101289. [Google Scholar] [CrossRef]
- Poznyak, S.K.; Talapin, D.V.; Kulak, A.I. Structural, optical, and photoelectrochemical properties of nanocrystalline TiO2−In2O3 composite solids and films prepared by sol−gel method. J. Phys. Chem. B 2001, 105, 4816–4823. [Google Scholar] [CrossRef]
- Kuo, Y.; Frye, C.D.; Ikenberry, M.; Klabunde, K.J. Titanium–indium oxy(nitride) with and without ruo2 loading as photocatalysts for hydrogen production under visible light from water. Catal. Today 2013, 199, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Ran, J.; Jaroniec, M.; Qiao, S.-Z. Cocatalysts in semiconductor-based photocatalytic CO2 reduction: Achievements, challenges, and opportunities. Adv. Mater. 2018, 30, 1704649. [Google Scholar] [CrossRef]
- Wang, W.-N.; An, W.-J.; Ramalingam, B.; Mukherjee, S.; Niedzwiedzki, D.M.; Gangopadhyay, S.; Biswas, P. Size and structure matter: Enhanced CO2 photoreduction efficiency by size-resolved ultrafine pt nanoparticles on TiO2 single crystals. J. Am. Chem. Soc. 2012, 134, 11276–11281. [Google Scholar] [CrossRef]
- Xie, S.; Wang, Y.; Zhang, Q.; Deng, W.; Wang, Y. Mgo- and pt-promoted TiO2 as an efficient photocatalyst for the preferential reduction of carbon dioxide in the presence of water. ACS Catal. 2014, 4, 3644–3653. [Google Scholar] [CrossRef]
- Feng, X.; Sloppy, J.D.; LaTempa, T.J.; Paulose, M.; Komarneni, S.; Bao, N.; Grimes, C.A. Synthesis and deposition of ultrafine pt nanoparticles within high aspect ratio TiO2 nanotube arrays: Application to the photocatalytic reduction of carbon dioxide. J. Mater. Chem. 2011, 21, 13429–13433. [Google Scholar] [CrossRef]
- Tasbihi, M.; Fresno, F.; Simon, U.; Villar-García, I.J.; Pérez-Dieste, V.; Escudero, C.; de la Peña O’Shea, V.A. On the selectivity of CO2 photoreduction towards CH4 using pt/TiO2 catalysts supported on mesoporous silica. Appl. Catal. B Environ. 2018, 239, 68–76. [Google Scholar] [CrossRef]
- Larimi, A.; Rahimi, M.; Khorasheh, F. Carbonaceous supports decorated with pt–TiO2 nanoparticles using electrostatic self-assembly method as a highly visible-light active photocatalyst for CO2 photoreduction. Renew. Energy 2020, 145, 1862–1869. [Google Scholar] [CrossRef]
- Elahifard, M.R.; Ahmadvand, S.; Mirzanejad, A. Effects of ni-doping on the photo-catalytic activity of TiO2 anatase and rutile: Simulation and experiment. Mater. Sci. Semicond. Process. 2018, 84, 10–16. [Google Scholar] [CrossRef]
- Ranjitha, A.; Muthukumarasamy, N.; Thambidurai, M.; Velauthapillai, D.; Balasundaraprabhu, R.; Agilan, S. Fabrication of ni-doped TiO2 thin film photoelectrode for solar cells. Sol. Energy 2014, 106, 159–165. [Google Scholar] [CrossRef]
- Yoshinaga, M.; Yamamoto, K.; Sato, N.; Aoki, K.; Morikawa, T.; Muramatsu, A. Remarkably enhanced photocatalytic activity by nickel nanoparticle deposition on sulfur-doped titanium dioxide thin film. Appl. Catal. B Environ. 2009, 87, 239–244. [Google Scholar] [CrossRef]
- Tseng, H.-H.; Wei, M.-C.; Hsiung, S.-F.; Chiou, C.-W. Degradation of xylene vapor over ni-doped TiO2 photocatalysts prepared by polyol-mediated synthesis. Chem. Eng. J. 2009, 150, 160–167. [Google Scholar] [CrossRef]
- Ren, C.; Qiu, W.; Zhang, H.; He, Z.; Chen, Y. Degradation of benzene on TiO2/SiO2/Bi2O3 photocatalysts under uv and visible light. J. Mol. Catal. A Chem. 2015, 398, 215–222. [Google Scholar] [CrossRef]
- He, R.A.; Cao, S.; Zhou, P.; Yu, J. Recent advances in visible light bi-based photocatalysts. Chin. J. Catal. 2014, 35, 989–1007. [Google Scholar] [CrossRef]
- Murcia-López, S.; Hidalgo, M.C.; Navío, J.A. Synthesis, characterization and photocatalytic activity of bi-doped TiO2 photocatalysts under simulated solar irradiation. Appl. Catal. A Gen. 2011, 404, 59–67. [Google Scholar] [CrossRef]
- Spadaro, L.; Arena, F.; Negro, P.; Palella, A. Sunfuels from CO2 exhaust emissions: Insights into the role of photoreactor configuration by the study in laboratory and industrial environment. J. CO2 Util. 2018, 26, 445–453. [Google Scholar] [CrossRef]
- Xiong, Z.; Zhao, X.S. Nitrogen-doped titanate-anatase core–shell nanobelts with exposed {101} anatase facets and enhanced visible light photocatalytic activity. J. Am. Chem. Soc. 2012, 134, 5754–5757. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Mari, B.; Sharma, S.; Kumari, V.; Maken, S.; Kumari, K.; Kumar, N. Non-metal modified TiO2: A step towards visible light photocatalysis. J. Mater. Sci. Mater. Electron. 2019, 30, 3186–3207. [Google Scholar] [CrossRef]
- Pelaez, M.; Nolan, N.T.; Pillai, S.C.; Seery, M.K.; Falaras, P.; Kontos, A.G.; Dunlop, P.S.M.; Hamilton, J.W.J.; Byrne, J.A.; O’Shea, K.; et al. A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl. Catal. B Environ. 2012, 125, 331–349. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, T.; Asahi, R.; Ohwaki, T.; Aoki, K.; Taga, Y. Band-gap narrowing of titanium dioxide by nitrogen doping. Jpn. J. Appl. Phys. 2001, 40, L561–L563. [Google Scholar] [CrossRef]
- Matějová, L.; Kočí, K.; Troppová, I.; Šihor, M.; Edelmannová, M.; Lang, J.; Čapek, L.; Matěj, Z.; Kuśtrowski, P.; Obalová, L. TiO2 and nitrogen doped TiO2 prepared by different methods; on the (micro)structure and photocatalytic activity in CO2 reduction and N2O decomposition. J. Nanosci. Nanotechnol. 2018, 18, 688–698. [Google Scholar] [CrossRef]
- Li, M.; Wang, M.; Zhu, L.; Li, Y.; Yan, Z.; Shen, Z.; Cao, X. Facile microwave assisted synthesis of n-rich carbon quantum dots/dual-phase TiO2 heterostructured nanocomposites with high activity in CO2 photoreduction. Appl. Catal. B Environ. 2018, 231, 269–276. [Google Scholar] [CrossRef]
- Zeng, S.; Zhang, X.; Bai, L.; Zhang, X.; Wang, H.; Wang, J.; Bao, D.; Li, M.; Liu, X.; Zhang, S. Ionic-liquid-based CO2 capture systems: Structure, interaction and process. Chem. Rev. 2017, 117, 9625–9673. [Google Scholar] [CrossRef]
- Sheridan, Q.R.; Schneider, W.F.; Maginn, E.J. Role of molecular modeling in the development of CO2–reactive ionic liquids. Chem. Rev. 2018, 118, 5242–5260. [Google Scholar] [CrossRef]
- Rebecca, S.; Miguel, T.; Katherine, B.; Nora, H. Reduction of carbon dioxide to formate at low overpotential using a superbase ionic liquid. Angew. Chem. Int. Ed. 2015, 127, 14370–14374. [Google Scholar]
- Lin, J.; Ding, Z.; Hou, Y.; Wang, X. Ionic liquid co-catalyzed artificial photosynthesis of CO. Sci. Rep. 2013, 3, 1056. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.; Yu, B.; Wu, Y.; Yu, X.; Guo, S.; Han, B.; Liu, Z. Visible light-driven photoreduction of CO2 to CH4 over TiO2 using a multiple-site ionic liquid as an absorbent and photosensitizer. ACS Sustain. Chem. Eng. 2020, 8, 9088–9094. [Google Scholar] [CrossRef]
- Banerjee, S.; Mohapatra, S.K.; Das, P.P.; Misra, M. Synthesis of coupled semiconductor by filling 1d TiO2 nanotubes with cds. Chem. Mater. 2008, 20, 6784–6791. [Google Scholar] [CrossRef]
- Shi, L.; Li, C.; Gu, H.; Fang, D. Morphology and properties of ultrafine SnO2–TiO2 coupled semiconductor particles. Mater. Chem. Phys. 2000, 62, 62–67. [Google Scholar] [CrossRef]
- Xu, Y.-F.; Wang, X.-D.; Liao, J.-F.; Chen, B.-X.; Chen, H.-Y.; Kuang, D.-B. Amorphous-TiO2-encapsulated CsPbBr3 nanocrystal composite photocatalyst with enhanced charge separation and CO2 fixation. Adv. Mater. Interfaces 2018, 5, 1801015. [Google Scholar] [CrossRef]
- Crake, A.; Christoforidis, K.C.; Godin, R.; Moss, B.; Kafizas, A.; Zafeiratos, S.; Durrant, J.R.; Petit, C. Titanium dioxide/carbon nitride nanosheet nanocomposites for gas phase CO2 photoreduction under uv-visible irradiation. Appl. Catal. B Environ. 2019, 242, 369–378. [Google Scholar] [CrossRef]
- Tan, J.Z.Y.; Xia, F.; Maroto-Valer, M.M. Raspberry-like microspheres of core–shell Cr2O3@TiO2 nanoparticles for CO2 photoreduction. ChemSusChem 2019, 12, 5246–5252. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, F.; Mumtaz, A.; Shahabuddin, S.; Abd Mutalib, M.I.; Shaharun, M.S.; Nguyen, T.D.; Khan, M.R.; Abdullah, B. Photocatalytic reduction of CO2 to methanol over ZnFe2O4/TiO2 (p–n) heterojunctions under visible light irradiation. J. Chem. Technol. Biotechnol. 2020, 95, 2208–2221. [Google Scholar] [CrossRef]
- Sadeghi, N.; Sillanpää, M. High selective photocatalytic CO2 conversion into liquid solar fuel over a cobalt porphyrin-based metal–organic framework. Photochem. Photobiol. Sci. 2021, 20, 391–399. [Google Scholar] [CrossRef]
- Li, R.; Zhang, W.; Zhou, K. Metal–organic-framework-based catalysts for photoreduction of CO2. Adv. Mater. 2018, 30, 1705512. [Google Scholar] [CrossRef]
- Llabrés i Xamena, F.X.; Corma, A.; Garcia, H. Applications for metal−organic frameworks (mofs) as quantum dot semiconductors. J. Phys. Chem. C 2007, 111, 80–85. [Google Scholar] [CrossRef]
- Khajavi, H.; Gascon, J.; Schins, J.M.; Siebbeles, L.D.A.; Kapteijn, F. Unraveling the optoelectronic and photochemical behavior of zn4o-based metal organic frameworks. J. Phys. Chem. C 2011, 115, 12487–12493. [Google Scholar] [CrossRef]
- Fu, Y.; Sun, D.; Chen, Y.; Huang, R.; Ding, Z.; Fu, X.; Li, Z. An amine-functionalized titanium metal–organic framework photocatalyst with visible-light-induced activity for CO2 reduction. Angew. Chem. Int. Ed. 2012, 51, 3364–3367. [Google Scholar] [CrossRef]
- Sun, D.; Fu, Y.; Liu, W.; Ye, L.; Wang, D.; Yang, L.; Fu, X.; Li, Z. Studies on photocatalytic CO2 reduction over nh2-uio-66(zr) and its derivatives: Towards a better understanding of photocatalysis on metal–organic frameworks. Chem. A Eur. J. 2013, 19, 14279–14285. [Google Scholar] [CrossRef]
- Wang, D.; Huang, R.; Liu, W.; Sun, D.; Li, Z. Fe-based mofs for photocatalytic CO2 reduction: Role of coordination unsaturated sites and dual excitation pathways. ACS Catal. 2014, 4, 4254–4260. [Google Scholar] [CrossRef]
- Li, S.; Huo, F. Metal–organic framework composites: From fundamentals to applications. Nanoscale 2015, 7, 7482–7501. [Google Scholar] [CrossRef]
- Blanco, L.A.; Gracia, F. 3—2d structures for CO2 utilization. In Nanomaterials for Sustainable Energy and Environmental Remediation; Naushad, M., Saravanan, R., Raju, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 47–58. [Google Scholar]
- Wu, J.; Wen, C.; Zou, X.; Jimenez, J.; Sun, J.; Xia, Y.; Fonseca Rodrigues, M.-T.; Vinod, S.; Zhong, J.; Chopra, N.; et al. Carbon dioxide hydrogenation over a metal-free carbon-based catalyst. ACS Catal. 2017, 7, 4497–4503. [Google Scholar] [CrossRef]
- Kumar, A.; Xu, Q. Two-dimensional layered materials as catalyst supports. ChemNanoMat 2018, 4, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Low, Z.-X.; Li, L.; Razmjou, A.; Wang, K.; Yao, J.; Wang, H. Zif-8/zn2geo4 nanorods with an enhanced CO2 adsorption property in an aqueous medium for photocatalytic synthesis of liquid fuel. J. Mater. Chem. A 2013, 1, 11563–11569. [Google Scholar] [CrossRef]
- Wang, S.; Lin, J.; Wang, X. Semiconductor–redox catalysis promoted by metal–organic frameworks for CO2 reduction. Phys. Chem. Chem. Phys. 2014, 16, 14656–14660. [Google Scholar] [CrossRef]
- Li, R.; Hu, J.; Deng, M.; Wang, H.; Wang, X.; Hu, Y.; Jiang, H.-L.; Jiang, J.; Zhang, Q.; Xie, Y.; et al. Integration of an inorganic semiconductor with a metal–organic framework: A platform for enhanced gaseous photocatalytic reactions. Adv. Mater. 2014, 26, 4783–4788. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y. Recent advances (2012–2015) in the photocatalytic conversion of carbon dioxide to fuels using solar energy: Feasibilty for a new energy. In Advances in CO2 Capture, Sequestration, and Conversion; American Chemical Society: Washington, DC, USA, 2015; Volume 1194, pp. 1–46. [Google Scholar]
- Lai, L.; Chen, L.; Zhan, D.; Sun, L.; Liu, J.; Lim, S.H.; Poh, C.K.; Shen, Z.; Lin, J. One-step synthesis of nh2-graphene from in situ graphene-oxide reduction and its improved electrochemical properties. Carbon 2011, 49, 3250–3257. [Google Scholar] [CrossRef]
- Dikin, D.A.; Stankovich, S.; Zimney, E.J.; Piner, R.D.; Dommett, G.H.B.; Evmenenko, G.; Nguyen, S.T.; Ruoff, R.S. Preparation and characterization of graphene oxide paper. Nature 2007, 448, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Jahan, M.; Bao, Q.; Loh, K.P. Electrocatalytically active graphene–porphyrin mof composite for oxygen reduction reaction. J. Am. Chem. Soc. 2012, 134, 6707–6713. [Google Scholar] [CrossRef]
- Sadeghi, N.; Sharifnia, S.; Do, T.-O. Enhanced CO2 photoreduction by a graphene–porphyrin metal–organic framework under visible light irradiation. J. Mater. Chem. A 2018, 6, 18031–18035. [Google Scholar] [CrossRef]
- Zhou, A.; Dou, Y.; Zhao, C.; Zhou, J.; Wu, X.-Q.; Li, J.-R. A leaf-branch TiO2/carbon@ mof composite for selective CO2 photoreduction. Appl. Catal. B Environ. 2020, 264, 118519. [Google Scholar] [CrossRef]
- Lohse, M.S.; Bein, T. Covalent organic frameworks: Structures, synthesis, and applications. Adv. Funct. Mater. 2018, 28, 1705553. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Lv, D.; Chen, Y.; Wu, H.; Liu, B.; Xia, Q.; Li, Z. Highly selective adsorption separation of light hydrocarbons with a porphyrinic zirconium metal-organic framework pcn-224. Sep. Purif. Technol. 2018, 207, 262–268. [Google Scholar] [CrossRef]
- Wang, L.; Jin, P.; Huang, J.; She, H.; Wang, Q. Integration of copper(ii)-porphyrin zirconium metal–organic framework and titanium dioxide to construct z-scheme system for highly improved photocatalytic CO2 reduction. ACS Sustain. Chem. Eng. 2019, 7, 15660–15670. [Google Scholar] [CrossRef]
- Ahmed, I.; Jhung, S.H. Composites of metal–organic frameworks: Preparation and application in adsorption. Mater. Today 2014, 17, 136–146. [Google Scholar] [CrossRef]
- Schröder, F.; Henke, S.; Zhang, X.; Fischer, R.A. Simultaneous gas-phase loading of mof-5 with two metal precursors: Towards bimetallics@mof. EurJIC 2009, 2009, 3131–3140. [Google Scholar] [CrossRef]
- Sun, D.; Liu, W.; Fu, Y.; Fang, Z.; Sun, F.; Fu, X.; Zhang, Y.; Li, Z. Noble metals can have different effects on photocatalysis over metal–organic frameworks (mofs): A case study on m/nh2-mil-125(ti) (m=pt and au). Chem. A Eur. J. 2014, 20, 4780–4788. [Google Scholar] [CrossRef]
- Call, A.; Cibian, M.; Yamamoto, K.; Nakazono, T.; Yamauchi, K.; Sakai, K. Highly efficient and selective photocatalytic CO2 reduction to co in water by a cobalt porphyrin molecular catalyst. ACS Catal. 2019, 9, 4867–4874. [Google Scholar] [CrossRef]
- Zhang, J.; Grzelczak, M.; Hou, Y.; Maeda, K.; Domen, K.; Fu, X.; Antonietti, M.; Wang, X. Photocatalytic oxidation of water by polymeric carbon nitride nanohybrids made of sustainable elements. Chem. Sci. 2012, 3, 443–446. [Google Scholar] [CrossRef]
- Wang, S.; Yao, W.; Lin, J.; Ding, Z.; Wang, X. Cobalt imidazolate metal–organic frameworks photosplit CO2 under mild reaction conditions. Angew. Chem. 2014, 126, 1052–1056. [Google Scholar] [CrossRef]
- Chambers, M.B.; Wang, X.; Elgrishi, N.; Hendon, C.H.; Walsh, A.; Bonnefoy, J.; Canivet, J.; Quadrelli, E.A.; Farrusseng, D.; Mellot-Draznieks, C.; et al. Photocatalytic carbon dioxide reduction with rhodium-based catalysts in solution and heterogenized within metal–organic frameworks. ChemSusChem 2015, 8, 603–608. [Google Scholar] [CrossRef]
- Sadeghi, N.; Sharifnia, S.; Sheikh Arabi, M. A porphyrin-based metal organic framework for high rate photoreduction of CO2 to CH4 in gas phase. J. CO2 Util. 2016, 16, 450–457. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, H.; Kang, M. Remarkable photoconversion of carbon dioxide into methane using bi-doped TiO2 nanoparticles prepared by a conventional sol–gel method. Mater. Lett. 2016, 178, 316–319. [Google Scholar] [CrossRef]
- Torres, J.A.; Nogueira, A.E.; da Silva, G.T.S.T.; Lopes, O.F.; Wang, Y.; He, T.; Ribeiro, C. Enhancing TiO2 activity for CO2 photoreduction through mgo decoration. J. CO2 Util. 2020, 35, 106–114. [Google Scholar] [CrossRef]
- Feng, S.; Zhao, J.; Bai, Y.; Liang, X.; Wang, T.; Wang, C. Facile synthesis of mo-doped TiO2 for selective photocatalytic CO2 reduction to methane: Promoted H2O dissociation by mo doping. J. CO2 Util. 2020, 38, 1–9. [Google Scholar] [CrossRef]
- Chen, M.; Wu, J.; Lu, C.; Luo, X.; Huang, Y.; Jin, B.; Gao, H.; Zhang, X.; Argyle, M.; Liang, Z. Photoreduction of CO2 in the presence of CH4 over g-c3n4 modified with TiO2 nanoparticles at room temperature. Green Energy Environ. 2020, 6, 938–951. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, S.; Kang, J.K.; Cohen, S.M. Photocatalytic CO2 reduction by a mixed metal (zr/ti), mixed ligand metal-organic framework under visible light irradiation. Chem. Commun. 2015, 51, 5735–5738. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Ouyang, S.; Xu, H.; Zhao, M.; Zhang, X.; Ye, J. Co-zif-9/tio 2 nanostructure for superior co 2 photoreduction activity. J. Mater. Chem. A 2016, 4, 15126–15133. [Google Scholar] [CrossRef]
- He, X.; Gan, Z.; Fisenko, S.; Wang, D.; El-Kaderi, H.M.; Wang, W.-N. Rapid formation of metal–organic frameworks (mofs) based nanocomposites in microdroplets and their applications for CO2 photoreduction. ACS Appl. Mater. Interfaces 2017, 9, 9688–9698. [Google Scholar] [CrossRef]
- Wang, M.; Wang, D.; Li, Z. Self-assembly of cpo-27-mg/TiO2 nanocomposite with enhanced performance for photocatalytic CO2 reduction. Appl. Catal. B Environ. 2016, 183, 47–52. [Google Scholar] [CrossRef]
- Shi, L.; Wang, T.; Zhang, H.; Chang, K.; Ye, J. Electrostatic self-assembly of nanosized carbon nitride nanosheet onto a zirconium metal–organic framework for enhanced photocatalytic CO2 reduction. Adv. Funct. Mater. 2015, 25, 5360–5367. [Google Scholar] [CrossRef]
- Crake, A.; Christoforidis, K.C.; Kafizas, A.; Zafeiratos, S.; Petit, C. CO2 capture and photocatalytic reduction using bifunctional TiO2/mof nanocomposites under uv–vis irradiation. Appl. Catal. B Environ. 2017, 210, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Ulagappan, N.; Frei, H. Mechanistic study of CO2 photoreduction in ti silicalite molecular sieve by ft-ir spectroscopy. J. Phys. Chem. A 2000, 104, 7834–7839. [Google Scholar] [CrossRef]
- Chen, J.; Xin, F.; Qin, S.; Yin, X. Photocatalytically reducing CO2 to methyl formate in methanol over zns and ni-doped zns photocatalysts. Chem. Eng. J. 2013, 230, 506–512. [Google Scholar] [CrossRef]
- Chen, J.; Xin, F.; Yin, X.; Xiang, T.; Wang, Y. Synthesis of hexagonal and cubic znin2s4 nanosheets for the photocatalytic reduction of CO2 with methanol. RSC Adv. 2015, 5, 3833–3839. [Google Scholar] [CrossRef]
- Sadeghi, N.; Sharifnia, S.; Do, T.-O. Optimization and modeling of CO2 photoconversion using a response surface methodology with porphyrin-based metal organic framework. React. Kinet. Mech. Catal. 2018, 125, 411–431. [Google Scholar] [CrossRef]
- Li, G.; Ciston, S.; Saponjic, Z.V.; Chen, L.; Dimitrijevic, N.M.; Rajh, T.; Gray, K.A. Synthesizing mixed-phase TiO2 nanocomposites using a hydrothermal method for photo-oxidation and photoreduction applications. J. Catal. 2008, 253, 105–110. [Google Scholar] [CrossRef]
- Sun, D.; Liu, W.; Qiu, M.; Zhang, Y.; Li, Z. Introduction of a mediator for enhancing photocatalytic performance via post-synthetic metal exchange in metal-organic frameworks (mofs). Chem. Commun. 2015, 51, 2056–2059. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Zhou, Y.; Zou, Z. Photocatalytic conversion of CO2 into renewable hydrocarbon fuels: State-of-the-art accomplishment, challenges, and prospects. Adv. Mater. 2014, 26, 4607–4626. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Jiang, J.; Sakata, Y.; Nakabayashi, M.; Shibata, N.; Nandal, V.; Seki, K.; Hisatomi, T.; Domen, K. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 2020, 581, 411–414. [Google Scholar] [CrossRef]
- Ham, Y.; Hisatomi, T.; Goto, Y.; Moriya, Y.; Sakata, Y.; Yamakata, A.; Kubota, J.; Domen, K. Flux-mediated doping of srtio3 photocatalysts for efficient overall water splitting. J. Mater. Chem. A 2016, 4, 3027–3033. [Google Scholar] [CrossRef] [Green Version]
- Kurashige, W.; Mori, Y.; Ozaki, S.; Kawachi, M.; Hossain, S.; Kawawaki, T.; Shearer, C.J.; Iwase, A.; Metha, G.F.; Yamazoe, S. Activation of water-splitting photocatalysts by loading with ultrafine rh–cr mixed-oxide cocatalyst nanoparticles. Angew. Chem. Int. Ed. 2020, 59, 7076–7082. [Google Scholar] [CrossRef]
- Fu, S.; Liu, X.; Ran, J.; Jiao, Y. Theoretical considerations on activity of the electrochemical CO2 reduction on metal single-atom catalysts with asymmetrical active sites. Catal. Today 2021. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Lin, H.; Zhu, J. Recent advances in metal–organic frameworks for photo-/electrocatalytic CO2 reduction. Chem. A Eur. J. 2019, 25, 14026–14035. [Google Scholar] [CrossRef]
- Li, J.; Chen, G.; Zhu, Y.; Liang, Z.; Pei, A.; Wu, C.-L.; Wang, H.; Lee, H.R.; Liu, K.; Chu, S. Efficient electrocatalytic CO2 reduction on a three-phase interface. Nat. Catal. 2018, 1, 592–600. [Google Scholar] [CrossRef]
- Trickett, C.A.; Helal, A.; Al-Maythalony, B.A.; Yamani, Z.H.; Cordova, K.E.; Yaghi, O.M. The chemistry of metal–organic frameworks for CO2 capture, regeneration and conversion. Nat. Rev. Mater. 2017, 2, 17045. [Google Scholar] [CrossRef]
- Loiudice, A.; Lobaccaro, P.; Kamali, E.A.; Thao, T.; Huang, B.H.; Ager, J.W.; Buonsanti, R. Tailoring copper nanocrystals towards C2 products in electrochemical CO2 reduction. Angew. Chem. Int. Ed. 2016, 55, 5789–5792. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.W.; Fritz, K.E.; Kim, J.; Suntivich, J.; Abruña, H.D.; Hanrath, T. Controlled selectivity of CO2 reduction on copper by pulsing the electrochemical potential. ChemSusChem 2018, 11, 1781–1786. [Google Scholar] [CrossRef]
- Park, S.; Wijaya, D.T.; Na, J.; Lee, C.W. Towards the large-scale electrochemical reduction of carbon dioxide. Catalysts 2021, 11, 253. [Google Scholar] [CrossRef]
- Gu, Z.-G.; Zhang, J. Epitaxial growth and applications of oriented metal–organic framework thin films. Coord. Chem. Rev. 2019, 378, 513–532. [Google Scholar] [CrossRef]
- Lei, Z.; Xue, Y.; Chen, W.; Qiu, W.; Zhang, Y.; Horike, S.; Tang, L. Mofs-based heterogeneous catalysts: New opportunities for energy-related CO2 conversion. Adv. Energy Mater. 2018, 8, 1801587. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Liu, M.; Wen, T.; Zhang, J. Synthetic design of functional boron imidazolate frameworks. Coord. Chem. Rev. 2016, 307, 255–266. [Google Scholar] [CrossRef]
- Yang, M.; Zhou, Y.-N.; Cao, Y.-N.; Tong, Z.; Dong, B.; Chai, Y.-M. Advances and challenges of fe-mofs based materials as electrocatalysts for water splitting. Appl. Mater. Today 2020, 20, 100692. [Google Scholar] [CrossRef]
- Hod, I.; Sampson, M.D.; Deria, P.; Kubiak, C.P.; Farha, O.K.; Hupp, J.T. Fe-porphyrin-based metal–organic framework films as high-surface concentration, heterogeneous catalysts for electrochemical reduction of CO2. ACS Catal. 2015, 5, 6302–6309. [Google Scholar] [CrossRef]
- Dong, B.-X.; Qian, S.-L.; Bu, F.-Y.; Wu, Y.-C.; Feng, L.-G.; Teng, Y.-L.; Liu, W.-L.; Li, Z.-W. Electrochemical reduction of CO2 to co by a heterogeneous catalyst of fe–porphyrin-based metal–organic framework. ACS Appl. Energy Mater. 2018, 1, 4662–4669. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Huang, Q.; He, C.-T.; Chen, Y.; Liu, J.; Shen, F.-C.; Lan, Y.-Q. Oriented electron transmission in polyoxometalate-metalloporphyrin organic framework for highly selective electroreduction of CO2. Nat. Commun. 2018, 9, 4466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, Y.; Murata, A.; Takahashi, R. Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1989, 85, 2309–2326. [Google Scholar] [CrossRef]
- Watanabe, M.; Shibata, M.; Kato, A.; Azuma, M.; Sakata, T. Design of alloy electrocatalysts for CO2 reduction: III. The selective and reversible reduction of on cu alloy electrodes. J. Electrochem. Soc. 1991, 138, 3382–3389. [Google Scholar] [CrossRef]
- Senthil Kumar, R.; Senthil Kumar, S.; Anbu Kulandainathan, M. Highly selective electrochemical reduction of carbon dioxide using cu based metal organic framework as an electrocatalyst. Electrochem. Commun. 2012, 25, 70–73. [Google Scholar] [CrossRef]
- Hinogami, R.; Yotsuhashi, S.; Deguchi, M.; Zenitani, Y.; Hashiba, H.; Yamada, Y. Electrochemical reduction of carbon dioxide using a copper rubeanate metal organic framework. ECS Electrochem. Lett. 2012, 1, H17. [Google Scholar] [CrossRef]
- Shao, P.; Yi, L.; Chen, S.; Zhou, T.; Zhang, J. Metal-organic frameworks for electrochemical reduction of carbon dioxide: The role of metal centers. J. Energy Chem. 2020, 40, 156–170. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Gurau, G.; Rogers, R.D. Ionic liquid processing of cellulose. Chem. Soc. Rev. 2012, 41, 1519–1537. [Google Scholar] [CrossRef]
- Lei, Z.; Dai, C.; Chen, B. Gas solubility in ionic liquids. Chem. Rev. 2014, 114, 1289–1326. [Google Scholar] [CrossRef]
- Lui, M.Y.; Crowhurst, L.; Hallett, J.P.; Hunt, P.A.; Niedermeyer, H.; Welton, T. Salts dissolved in salts: Ionic liquid mixtures. Chem. Sci. 2011, 2, 1491–1496. [Google Scholar] [CrossRef]
- Kang, X.; Zhu, Q.; Sun, X.; Hu, J.; Zhang, J.; Liu, Z.; Han, B. Highly efficient electrochemical reduction of CO2 to CH4 in an ionic liquid using a metal–organic framework cathode. Chem. Sci. 2016, 7, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.-F.; Tao, K.; Li, G.-C.; Wu, M.-K.; Zhu, S.-R.; Yi, F.-Y.; Zhao, W.-N.; Han, L. In situ growth of zif-8 nanocrystals on layered double hydroxide nanosheets for enhanced CO2 capture. Dalton Trans. 2016, 45, 12632–12635. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, P.; Wang, Z.; Kang, P. Zinc imidazolate metal–organic frameworks (zif-8) for electrochemical reduction of CO2 to co. ChemPhysChem 2017, 18, 3142–3147. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Liu, J.; Gao, Y.; Gong, C.; Addicoat, M.; Heine, T.; Wöll, C.; Sun, L. Highly oriented mof thin film-based electrocatalytic device for the reduction of CO2 to co exhibiting high faradaic efficiency. J. Mater. Chem. A 2016, 4, 15320–15326. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, H.; Chang, S.; Si, R.; Miao, S.; Huang, W.; Li, Y.; Wang, G.; Bao, X. Boosting CO2 electroreduction over layered zeolitic imidazolate frameworks decorated with ag2o nanoparticles. J. Mater. Chem. A 2017, 5, 19371–19377. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, J. Rational design of cu-based electrocatalysts for electrochemical reduction of carbon dioxide. J. Energy Chem. 2017, 26, 1050–1066. [Google Scholar] [CrossRef] [Green Version]
- Yoshio, H.; Katsuhei, K.; Shin, S. Production of co and CH4 in electrochemical reduction of CO2 at metal electrodes in aqueous hydrogencarbonate solution. Chem. Lett. 1985, 14, 1695–1698. [Google Scholar]
- Hori, Y.; Murata, A.; Takahashi, R.; Suzuki, S. Electroreduction of carbon monoxide to methane and ethylene at a copper electrode in aqueous solutions at ambient temperature and pressure. J. Am. Chem. Soc. 1987, 109, 5022–5023. [Google Scholar] [CrossRef]
- Zhao, C.; Dai, X.; Yao, T.; Chen, W.; Wang, X.; Wang, J.; Yang, J.; Wei, S.; Wu, Y.; Li, Y. Ionic exchange of metal–organic frameworks to access single nickel sites for efficient electroreduction of CO2. J. Am. Chem. Soc. 2017, 139, 8078–8081. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.-X.; Zijlstra, B.; Filot, I.A.W.; Zhou, Z.; Sun, S.; Hensen, E.J.M. Optimum cu nanoparticle catalysts for CO2 hydrogenation towards methanol. Nano Energy 2018, 43, 200–209. [Google Scholar] [CrossRef]
- Reske, R.; Mistry, H.; Behafarid, F.; Roldan Cuenya, B.; Strasser, P. Particle size effects in the catalytic electroreduction of CO2 on cu nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. [Google Scholar] [CrossRef]
- Nam, D.-H.; Bushuyev, O.S.; Li, J.; De Luna, P.; Seifitokaldani, A.; Dinh, C.-T.; García de Arquer, F.P.; Wang, Y.; Liang, Z.; Proppe, A.H.; et al. Metal–organic frameworks mediate cu coordination for selective CO2 electroreduction. J. Am. Chem. Soc. 2018, 140, 11378–11386. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-M.; Choi, S.Y.; Youn, M.H.; Lee, W.; Park, K.T.; Gothandapani, K.; Grace, A.N.; Jeong, S.K. Investigation on electroreduction of CO2 to formic acid using cu3(btc)2 metal–organic framework (cu-mof) and graphene oxide. ACS Omega 2020, 5, 23919–23930. [Google Scholar] [CrossRef] [PubMed]
- Rayer, A.V.; Reid, E.; Kataria, A.; Luz, I.; Thompson, S.J.; Lail, M.; Zhou, J.; Soukri, M. Electrochemical carbon dioxide reduction to isopropanol using novel carbonized copper metal organic framework derived electrodes. J. CO2 Util. 2020, 39, 101159. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Z.; Zhao, X.; Yao, T.; Chen, W.; You, R.; Zhao, C.; Wu, G.; Wang, J.; Huang, W. Regulation of coordination number over single co sites: Triggering the efficient electroreduction of CO2. Angew. Chem. 2018, 130, 1962–1966. [Google Scholar] [CrossRef]
- Ye, Y.; Cai, F.; Li, H.; Wu, H.; Wang, G.; Li, Y.; Miao, S.; Xie, S.; Si, R.; Wang, J.; et al. Surface functionalization of zif-8 with ammonium ferric citrate toward high exposure of fe-n active sites for efficient oxygen and carbon dioxide electroreduction. Nano Energy 2017, 38, 281–289. [Google Scholar] [CrossRef]
- Zhu, M.; Chen, J.; Huang, L.; Ye, R.; Xu, J.; Han, Y.-F. Covalently grafting cobalt porphyrin onto carbon nanotubes for efficient CO2 electroreduction. Angew. Chem. Int. Ed. 2019, 58, 6595–6599. [Google Scholar] [CrossRef]
- Kim, M.K.; Kim, H.J.; Lim, H.; Kwon, Y.; Jeong, H.M. Metal–organic framework-mediated strategy for enhanced methane production on copper nanoparticles in electrochemical CO2 reduction. Electrochim. Acta 2019, 306, 28–34. [Google Scholar] [CrossRef]
- Albo, J.; Perfecto-Irigaray, M.; Beobide, G.; Irabien, A. Cu/bi metal-organic framework-based systems for an enhanced electrochemical transformation of CO2 to alcohols. J. CO2 Util. 2019, 33, 157–165. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Xi, S.; Du, Y.; Hai, X.; Wang, J.; Xu, H.; Wu, G.; Zhang, J.; Lu, J. A graphene-supported single-atom fen5 catalytic site for efficient electrochemical CO2 reduction. Angew. Chem. 2019, 131, 15013–15018. [Google Scholar] [CrossRef]
- Huang, J.; Mensi, M.; Oveisi, E.; Mantella, V.; Buonsanti, R. Structural sensitivities in bimetallic catalysts for electrochemical CO2 reduction revealed by ag–cu nanodimers. J. Am. Chem. Soc. 2019, 141, 2490–2499. [Google Scholar] [CrossRef]
- Dou, S.; Song, J.; Xi, S.; Du, Y.; Wang, J.; Huang, Z.F.; Xu, Z.J.; Wang, X. Boosting electrochemical CO2 reduction on metal–organic frameworks via ligand doping. Angew. Chem. 2019, 131, 4081–4085. [Google Scholar] [CrossRef]
- Vasileff, A.; Zhi, X.; Xu, C.; Ge, L.; Jiao, Y.; Zheng, Y.; Qiao, S.-Z. Selectivity control for electrochemical CO2 reduction by charge redistribution on the surface of copper alloys. ACS Catal. 2019, 9, 9411–9417. [Google Scholar] [CrossRef]
- Deng, W.; Zhang, L.; Li, L.; Chen, S.; Hu, C.; Zhao, Z.-J.; Wang, T.; Gong, J. Crucial role of surface hydroxyls on the activity and stability in electrochemical CO2 reduction. J. Am. Chem. Soc. 2019, 141, 2911–2915. [Google Scholar] [CrossRef]
- Daiyan, R.; Lu, X.; Tan, X.; Zhu, X.; Chen, R.; Smith, S.C.; Amal, R. Antipoisoning nickel–carbon electrocatalyst for practical electrochemical CO2 reduction to co. ACS Appl. Energy Mater. 2019, 2, 8002–8009. [Google Scholar] [CrossRef]
- Choi, J.; Wagner, P.; Gambhir, S.; Jalili, R.; MacFarlane, D.R.; Wallace, G.G.; Officer, D.L. Steric modification of a cobalt phthalocyanine/graphene catalyst to give enhanced and stable electrochemical CO2 reduction to co. ACS Energy Lett. 2019, 4, 666–672. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, R.; Zhu, X.; Han, N.; Song, B.; Liu, T.; Hu, G.; Li, Y.; Lu, J.; Li, Y. Mesoporous pdag nanospheres for stable electrochemical CO2 reduction to formate. Adv. Mater. 2020, 32, 2000992. [Google Scholar] [CrossRef]
- Lee, M.-Y.; Ringe, S.; Kim, H.; Kang, S.; Kwon, Y. Electric field mediated selectivity switching of electrochemical CO2 reduction from formate to co on carbon supported sn. ACS Energy Lett. 2020, 5, 2987–2994. [Google Scholar] [CrossRef]
- Sun, Z.; Ma, T.; Tao, H.; Fan, Q.; Han, B. Fundamentals and challenges of electrochemical CO2 reduction using two-dimensional materials. Chem 2017, 3, 560–587. [Google Scholar] [CrossRef] [Green Version]
- Jouny, M.; Luc, W.; Jiao, F. Correction to “general techno-economic analysis of CO2 electrolysis systems”. Ind. Eng. Chem. Res. 2020, 59, 8121–8123. [Google Scholar] [CrossRef] [Green Version]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
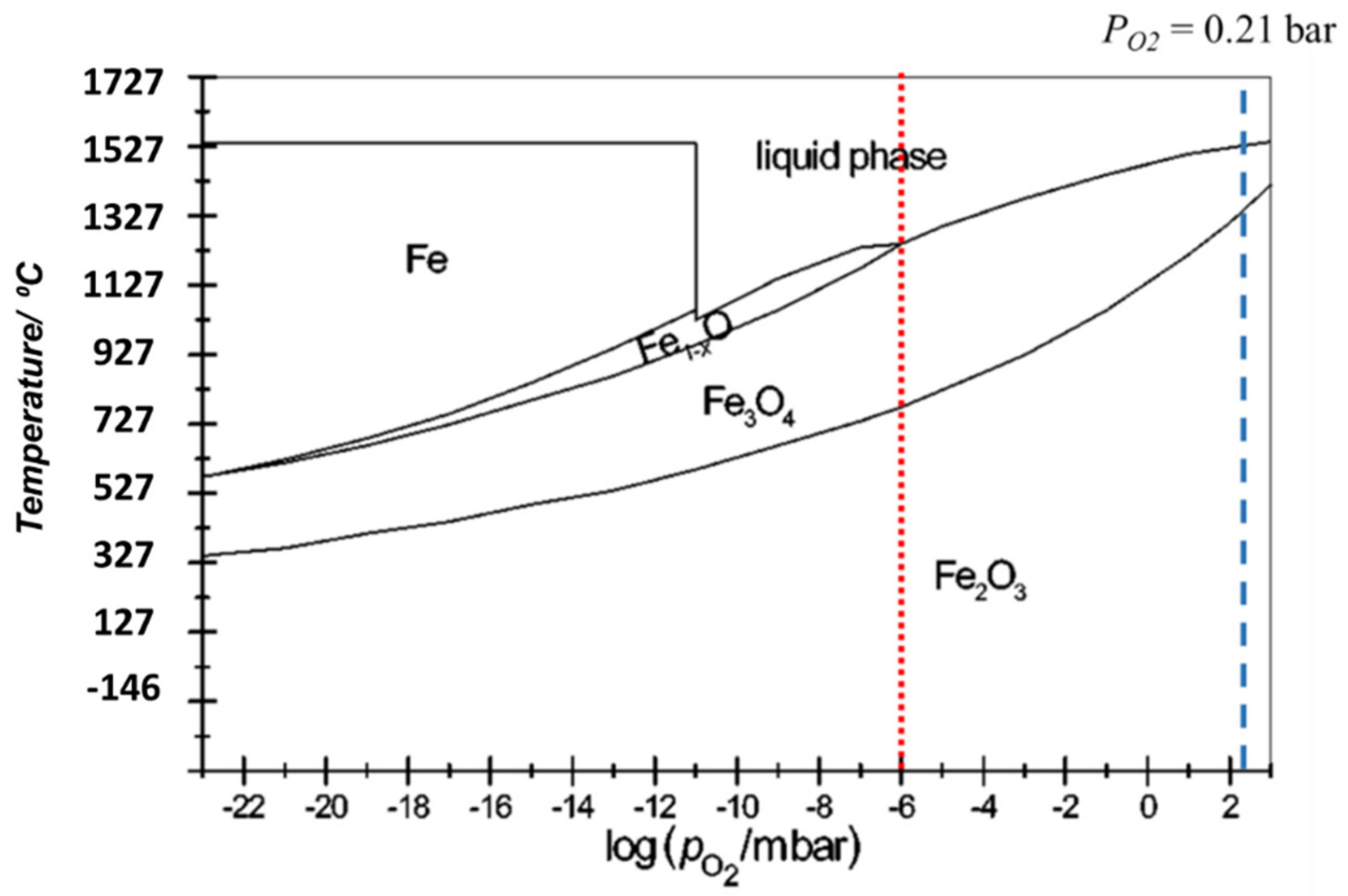{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
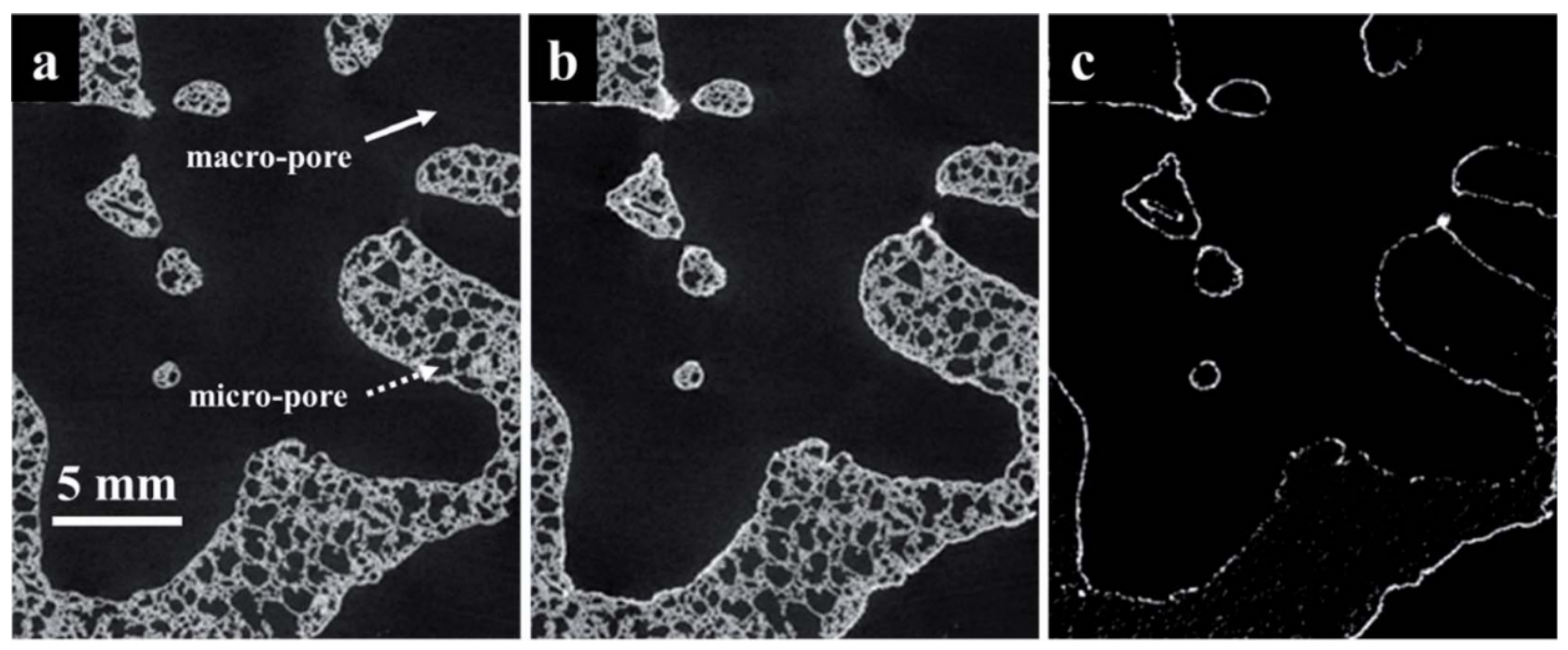{kind=link}
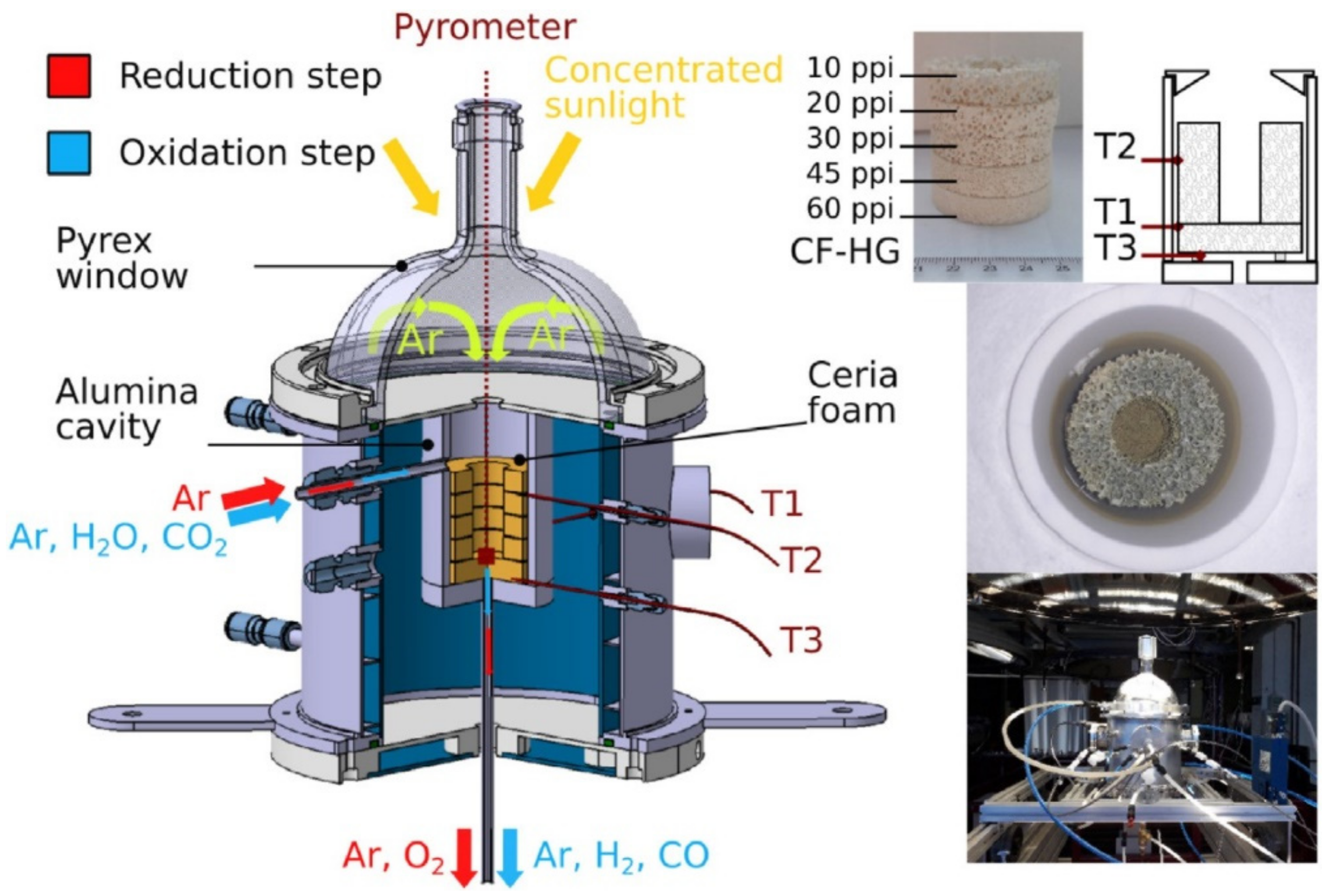{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
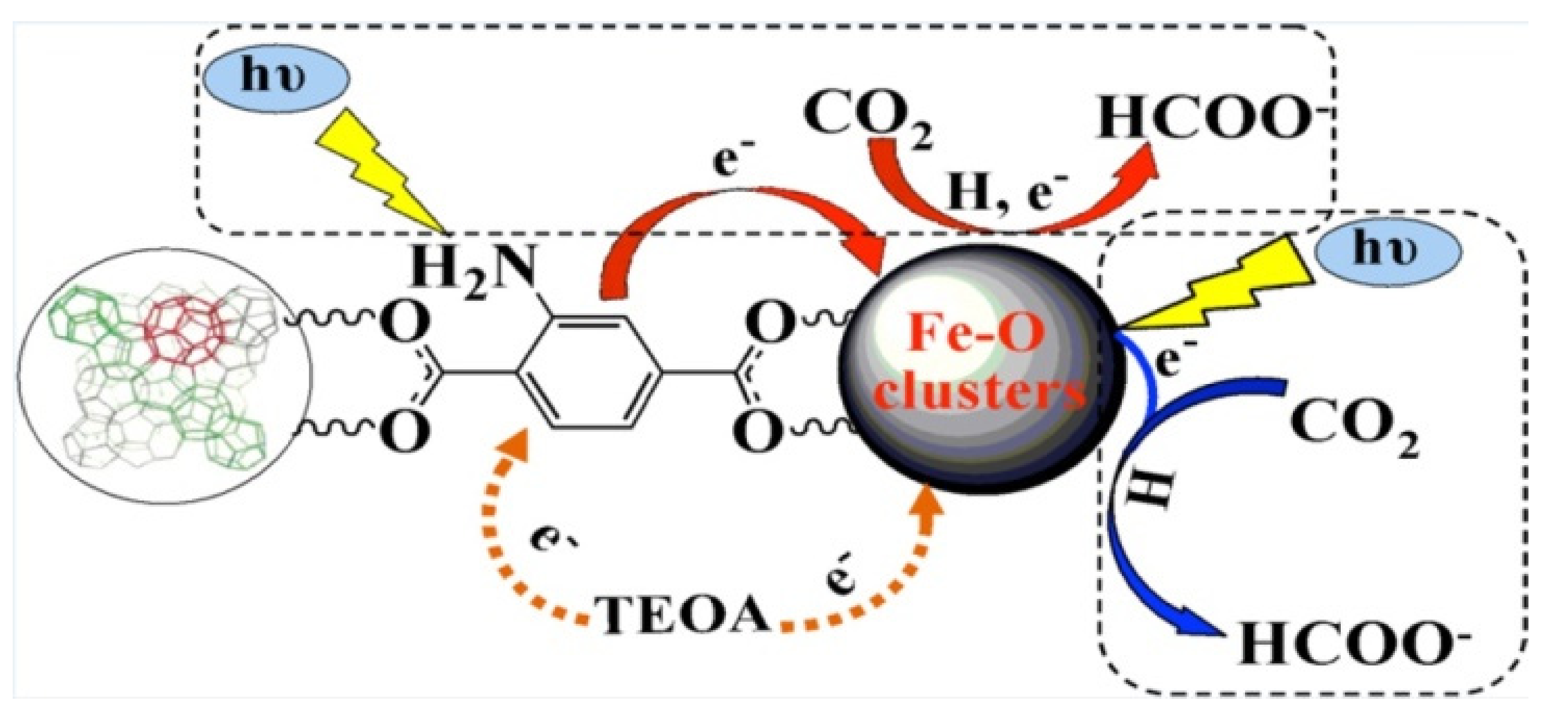{kind=link}
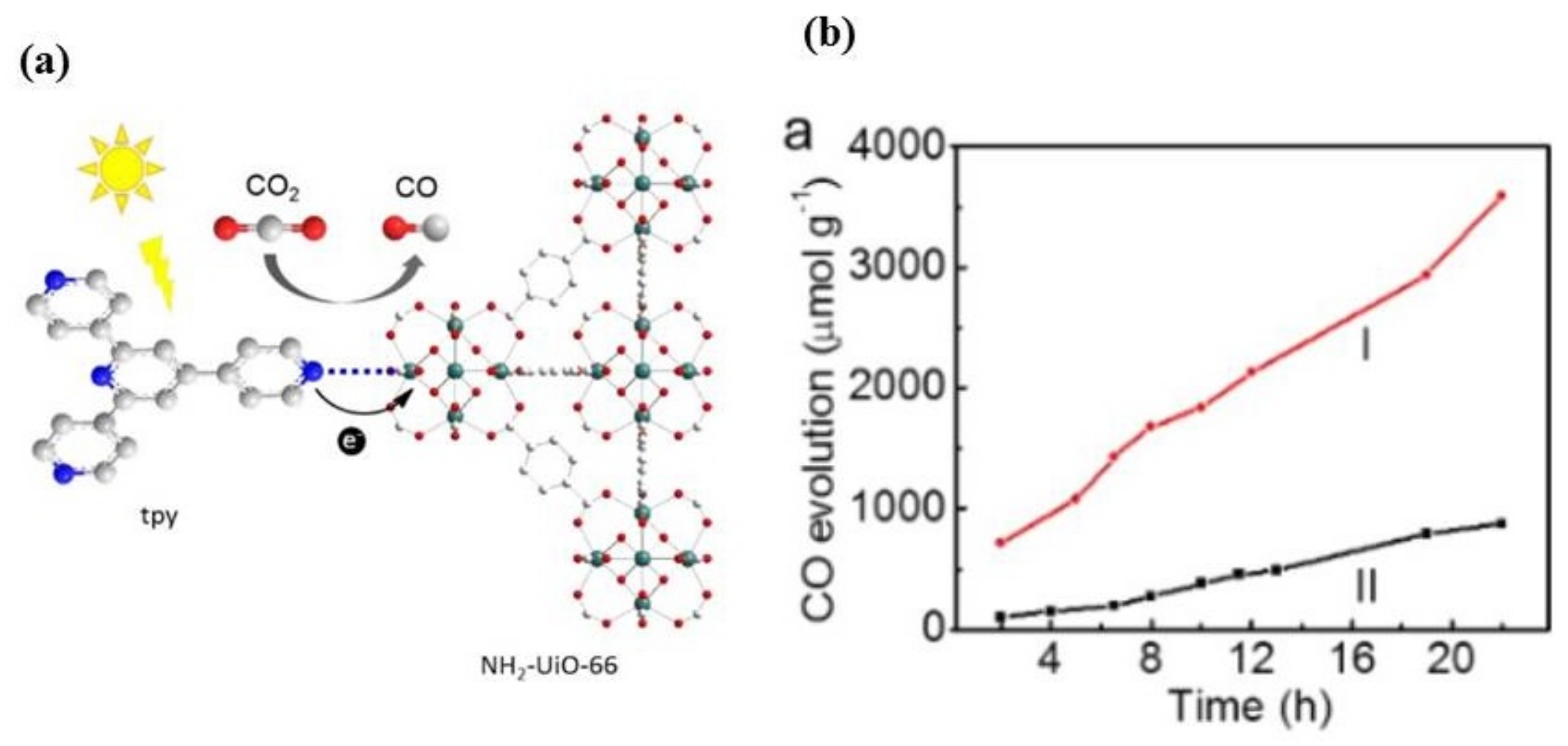{kind=link}
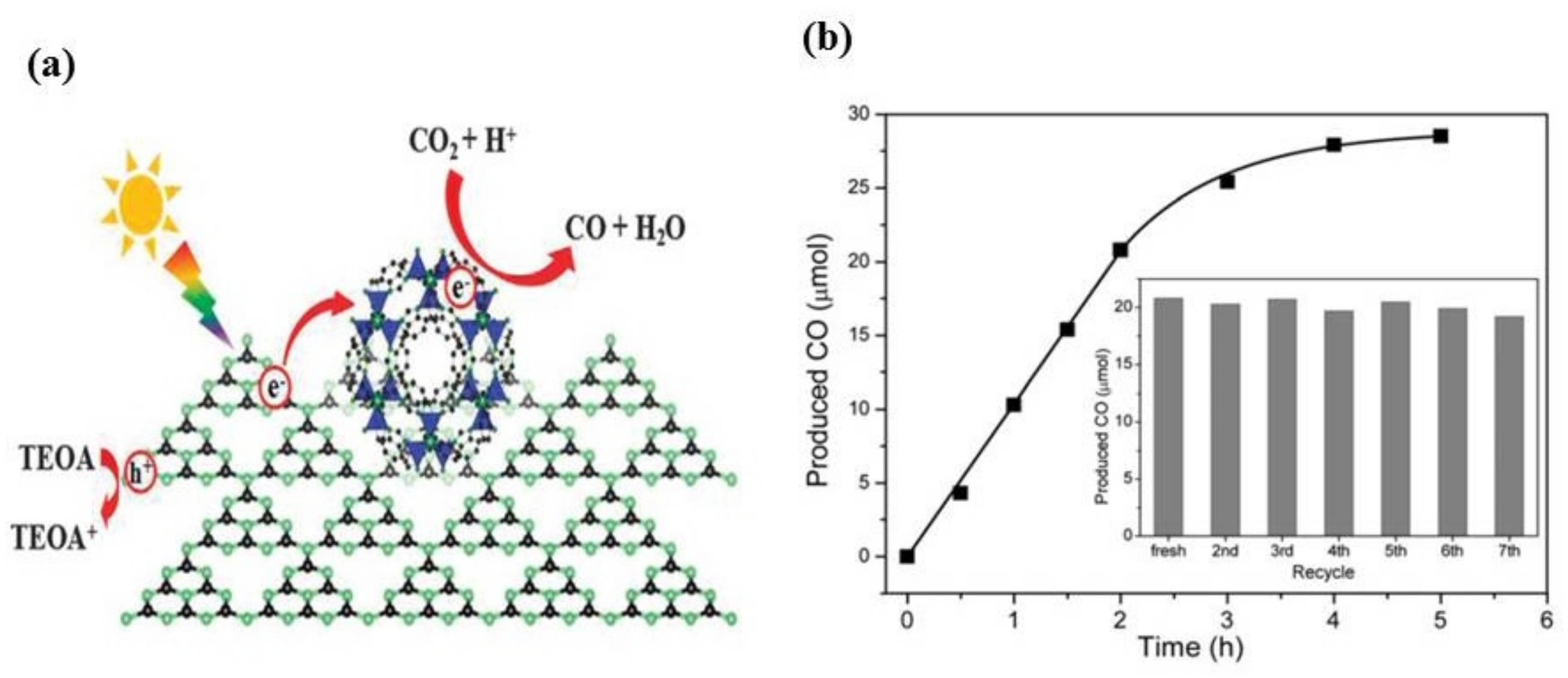{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CDR Route | Main Chemical Reactions | Comments | References |
|---|---|---|---|
| Dry reforming for syngas production | CO2 + CH4 ⇄ 2CO + 2H2 CO + H2O ⇄ CO2 + H2 | Commercially available | [12,31,32,33,34,35,36] |
| CO2 hydrogenation for methanol production | CO + 2H2 ⇄ CH3OH CO2 + 3H2 ⇄ CH3OH + H2O CO + H2O ⇄ CO2 + H2 | Commercially available | [37,38,39,40,41,42] |
| CO2 hydrogenation to DME | CO2 + 3H2 ⇄ CH3OH + H2O CO + 2H2 ⇄ CH3OH CO + H2O ⇄ CO2 + H2 2CH3OH ⇄ CH3OCH3 + H2O | - | [40,43,44,45] |
| Urea production | 2NH3 + CO2 ⇄ H2N-COONH4 H2N-COONH4 ⇄ (NH2)2CO + H2O | Commercially available | [46,47,48] |
| Polyethercarbonate polyols | Propylene oxide (C3H6O) + CO2 → Polyethercarbonate polyols | Commercially available | [49] |
| Fischer–Tropsch (FT) synthesis by dry reforming of natural gas | n CO + 2nH2→(−CH2-) + nH2O | Commercially available | [12,36,50,51,52] |
| Solar thermochemical | MOoxidized + (∆H) → MOreduced + 1⁄2 O2 (g) MOreduced + H2O/CO2(g)→MOoxidized + H2/CO(g) + (∆H) | Under development | [19,53,54] |
| Photochemical (During light irradiation, the energy of photons is absorbed. Excite electron/hole pairs are produced, which reduce and oxidize the chemical species over the surface of the photocatalyst.) | CO2 + 2H+ + 2e−→HCO2H | Under development | [55,56,57,58] |
| CO2 + 2H+ + 2e−→CO + H2O | |||
| CO2 + 2H+ + 4e−→HCHO + H2O | |||
| CO2 + 6H+ + 6e−→CH3OH + H2O | |||
| CO2 + 8H+ + 8e−→CH4 + 2H2O | |||
| 2CO2 + 12H+ + 12e−→C2H5OH + 3H2O | |||
| Electrochemical (In this process, a chemical reaction occurs by the applied electrical current. It involves oxidation—reduction reactions where CO2 is reduced on the cathode and oxygen evolves at the anode.) | CO2 + 2H+ + 2e−→HCO2H | Under development | [59,60,61] |
| CO2 + 2H+ + 2e−→CO + H2O | |||
| CO2 + 2H+ + 4e−→HCHO + H2O | |||
| CO2 + 6H+ + 6e−→CH3OH + H2O | |||
| CO2 + 8H+ + 8e−→CH4 + 2H2O | |||
| 2CO2 + 12H+ + 12e−→C2H4 + 4H2O |
| Year | Porous Support | RedOx Coating Material | Chemical Process | Performance | References |
|---|---|---|---|---|---|
| 1989 | Alumina honeycomb/foam | Rh | CO2 methane reforming | more efficiency for honeycomb structure compared to foams | [145,146] |
| 2005 | re-crystallized SiC honeycomb | Mn/Zn ferrites | water splitting | conversion efficiency ~80% and hydrogen yield >90% at low oxidation temperatures (800 °C) | [141,147] |
| 2008 | c-YSZ / MPSZ foam | Fe3O4 | water splitting | ferrite conversion of 20–27% for a 10.5 wt% Fe3O4-coated porous MPSZ | [137] |
| 2009 | MPSZ foam | m-ZrO2 supported NiFe2O4, Fe3O4 | water splitting | ferrite conversion of 24–76% for a 25 wt% NiFe2O4 coating on porous MPSZ | [97] |
| 2008 1 | 1:3 Co0.67Fe2.33O4/YSZ, Al2O3 and TiO2 | - | water splitting | unfavorable side reactions of ferrite with the YSZ supports and, thus, weak performance of the porous RedOx material | [127,148] |
| 2010 | cerium oxide (CeO2) monolith | - | water splitting/carbon dioxide splitting | with ƞsolar-to-fuel = 0.7–0.8% and the possibility of improvement through upscaling and removing the heat losses’ effects | [70] |
| 2011 | siliconized SiC monoliths | Fe/Zn mixed oxide | water splitting | conversion efficiency ~30% and RedOx materials’ degradation due to zinc content volatilization and inhomogeneous temperature distribution | [149] |
| MPSZ foam | zirconia supported Fe3O4 or NiFe2O4 | water splitting | maximum ferrite conversion of 60% for NiFe2O4/m-ZrO2/MPSZ foam device | [143] | |
| 2012 2 | 75 vol% YSZ and 25 vol% Fe2O3 | - | carbon dioxide splitting | yttria addition led to the oxygen conductivity improvement and Iron oxide conversion (max: 58%) and the stability of the CO production in consecutive RedOx cycles. The increasing of co-extruded honeycomb substrates’ surface area from ~2.6 to ~8.5 cm2 did not lead to a notable improvement in CO generation per unit volume | [142] |
| porous ceria felt | - | water splitting/carbon dioxide splitting | ceria sublimation and deposition on reactor components were detected as the main technical challenges, which eventually were responsible for deterioration of the active material and, thus, reactor yield | [150] | |
| ceria RPC | - | carbon dioxide splitting | with mean ƞsolar-to-fuel = 1.73% and max ƞsolar-to-fuel = 3.53%, a 17 times’ improvement in the fuel yield per cycle compared to ceria felt in previous study | [114] | |
| 2013 | 3DOM 3 CeO2, NOM 4 CeO2 | - | carbon dioxide splitting | more structural stability and CO production rate (10-fold) of porous structures over non-porous ones in over 55 cycles | [151] |
| 2014 | ceria RPC with dual porosities | - | carbon dioxide splitting | a 10 times higher yield for samples with porous struts (44% porosity) compared to samples with non-porous solid struts. The mean ηsolar-to-fuel of 1.72% was also detected in a 3.8-kW solar cavity receiver | [118] |
| MPSZ | NiFe2O4/m-ZrO2 and CeO2 | water splitting | lower yields of NiFe2O4/m-ZrO2/MPSZ compared to CeO2/MPSZ as a result of some sintering effects | [138] | |
| SiC, Ni, Cu foams | ZrO2-supported CeO2 | methane reforming/water splitting | higher gas yields for the Ni and Cu foams than for SiC. The poor thermal conductivity of SiC foam was also responsible for CeO2 particle sintering and, thus, an overall efficiency decrement. | [112] | |
| 2015 | porous ceria | - | carbon dioxide splitting | high degree of reactivity (even after 2000 cycles) was reported | [152] |
| 2017 | ceria RPC with dual porosities | - | carbon dioxide splitting | molar CO2 conversion of 83% and ƞsolar-to-fuel = 5.25% | [139,153] |
| 2019 | Ceria RPC | - | water splitting/carbon dioxide splitting | max. ƞsolar-to-fuel = 5.22% while increasing methane flow rate and decreasing the reduction temperature will enhance the nonstoichiometry value and, thus, syngas yield | [154] |
| 2020 | SiC RPC with dual porosities | La0.6Ca0.4Mn0..6Al0.4O3±δ (LCMA) | carbon dioxide splitting | CO2 conversion with [CO%] = 3.2. A coagulation of smaller pores because of reaction between LCMA coating and SiC substrate was reported. The smallest pore size of 75 ppi delivered the highest CO yield of ca. 0.07 molg−1 LCMA and δ = 0.4 | [18,136] |
| ceria RPC (with gradient porosities 10–60 ppi) | - | water splitting/carbon dioxide splitting | max. ηsolar-to-fuel of ~7.5% after 64 cycles was measured with a high stability of the porous RedOx structures | [73] | |
| ceria RPC | La0.5Sr0.5Mn0.9Mg0.1O3 (LSMMg) | water splitting/carbon dioxide splitting | max. ηsolar-to-fuel = 5.3 and the perovskite coating had just a positive effect on the reduction extent, which hindered oxidant gas (H2O or CO2) accessing the reactive ceria and can result in the poor re-oxidation compared to the pure, uncoated ceria RPC | [14] |
| Photoreactor. | Photocatalyst. | Product Formation Rate (µmol gcat.−1 h−1) | Photoreactor Configuration | |||
|---|---|---|---|---|---|---|
| CH3OH | CH4 | CO | Energy (Wh/m2) | AQEmax (%) | ||
| SB | TiO2 | 4.3 | - | - | 4.6 | 1.19 |
| Cu-TiO2 | 22 | - | - | |||
| PB | TiO2 | 5 | 9 | - | 315.2 | 5.7 |
| Cu-TiO2 | 47 | 3 | - | |||
| MT | Cu-TiO2 | - | 37.5 | 40 | 5.3 | 0.063 |
| Photocatalyst | Production Yield (µmol g−1) | Sel. for CO2 Reduction (%) | Particle Size (nm) | ||
|---|---|---|---|---|---|
| H2 | CO | CH4 | |||
| CsPbBr3NC | 1.64 | 3.35 | 2.06 | 90.3 | 16.4 |
| CsPbBr3NC/a-TiO2(10) | 5.08 | 7.73 | 10.12 | 90.5 | 13.4 |
| CsPbBr3NC/a-TiO2(20) | 4.38 | 11.71 | 20.15 | 95.5 | 8.5 |
| CsPbBr3NC/a-TiO2(30) | 4.42 | 8.05 | 14.40 | 93.7 | 7.8 |
| CsPbBr3NC/a-TiO2(50) | 5.01 | 8.72 | 6.47 | 87.4 | 5.9 |
| Reaction Media | Light Source | Product | Product Formation Rate [µmol h−1g−1] | Comment | References | ||
|---|---|---|---|---|---|---|---|
| TiO2-Based Photocatalyst | |||||||
| Metals and Non-metals Doping | 10 wt.% In/TiO2 | H2O vapor | UV light | CH4 | 244 | [182] | |
| CO | 81 | ||||||
| C2H4 | 0.06 | ||||||
| C2H6 | 2.78 | ||||||
| C3H6 | 0.02 | ||||||
| C3H8 | 0.02 | ||||||
| TiO2 | CH4 | 31 | |||||
| CO | 46 | ||||||
| C2H4 | 0 | ||||||
| C2H6 | 0 | ||||||
| C3H6 | 0 | ||||||
| C3H8 | 0 | ||||||
| 5 mol% Bi-TiO2 | H2O vapor | UV light | CH4 | 34,000 | The Bi ion has appropriate conductivity and remarkable CO2 adsorption. Therefore, the presence of Bi in the Bi-TiO2 structure enhanced the number of adsorbed CO2 and H2O molecules. | [251] | |
| N3/TiO2 | water | UV light | CH3COCH3 | 52.6 | [184] | ||
| Cu0.6N4/TiO2 | 33.2 | ||||||
| N/TiO2-SG-PFE | 0.2 M NaOH | UV light | CO | 0.2 | [207] | ||
| CH4 | 0.0014 | ||||||
| H2 | 0.012 | ||||||
| TiO2/MgO-1% | 0.1 M NaOH | UVC light (λmax = 253.7 nm) | HCOOH | 0.8125 | With increasing the amount of MgO, the catalytic efficiency decreased due to the full coverage of the TiO2 by MgO layers, consequently hindering the transferring of the photogenerated charge carriers at the TiO2 surface and thereby photoactivity was reduced. | [252] | |
| CH3COOH | 1.037 | ||||||
| CH4 | 1.437 | ||||||
| CO | 2.946 | ||||||
| TNi1Bi3 | H2O vapor | Visible light | CH4 | 2.113 | [186] | ||
| Pt-TiO2/MWCNT | H2O vapor | Visible light | CH4 | 1.9 | [194] | ||
| Pt-TiO2/SWCNT | 0.7 | ||||||
| Pt-TiO2/rGO | 0.5 | ||||||
| Pt-TiO2/AC | 0.16 | ||||||
| 0.3% Mo- doped TiO2 | H2O vapor | Xe lamp (300 W) | CO | 13.67 | CH4 selectivity increased with increasing Mo concentration to 0.3 wt.%. Thereafter, CH4 selectivity decreased with increasing Mo concentration. This was attributed to the decrease in electron-hole separation efficiency. | [253] | |
| CH4 | 16.33 | ||||||
| Surface Sensitizer | NCQDs/TiO2 | H2O vapor | Solar | CO | 0.769 | [208] | |
| CH4 | 1.153 | ||||||
| TiO2-[P4444]3[p-2,6-O-4-COO] | TEOA | Visible light (λ > 420 nm) | CH4 | 3.52 | [213] | ||
| H2 | 0.14 | ||||||
| TiO2-[P4444]2[p-2-O-4-COO] | CH4 | 0.19 | |||||
| H2 | 0.17 | ||||||
| TiO2-[P4444] [p-2-O] | CH4 | 0.1 | |||||
| H2 | 0.07 | ||||||
| TiO2-[P4444] [p-4-COO] | CH4 | 0.1 | |||||
| H2 | 0.06 | ||||||
| Other Semiconductors | CsPbBr3NC/a-TiO2(20) | CH4 | 6.72 | [216] | |||
| CO | 3.9 | ||||||
| H2 | 1.46 | ||||||
| H2 | 0.55 | ||||||
| Ti-NS/CN | H2 | UV-Vis. light | CO | 2.04 | [217] | ||
| Ti/ISO/CN | CO | 1.55 | |||||
| Ti-NS/CN | H2O | CO | 0.8 | ||||
| H2 | 2.67 | ||||||
| 700-Cr2O3@TiO2 | H2O | UV light | CH4 | 167.69 | [218] | ||
| CO | 0.488 | ||||||
| ZnFe2O4/TiO2 (1:1) | Na2S, Na2SO3, KOH in water | Visible light | CH3OH | 693.31 | [219] | ||
| ZnFe2O4/TiO2 (1:2) | 519.69 | ||||||
| ZnFe2O4/TiO2 (2:1) | 33.53 | ||||||
| (20)TNPs-CN/450 | CH4 | Solar simulator (λ = 320–780 nm) | CO | 4.71 | The sensitivity factors were investigated and implied the order of FC (Catalyst amount) > FE (Calcination temperature) > FB (CH4 to CO2 ratio) > FD (TNPs loading) > FA (Absolute pressure). | [254] | |
| (20)P25-CN/450 | 2.77 | ||||||
| Bulk g-C3N4 | 2.275 | ||||||
| MOF-Based Photocatalyst | |||||||
| NH2-Modified MOFs | MIL-125 (Ti) | MeCN/TEOA (5:1) | Visible light | HCOO− | 0 | [224] | |
| NH2-MIL-125 (Ti) | 16.28 | ||||||
| UiO-66(Zr) | MeCN/TEOA (5:1) | Visible light | HCOO− | 0 | In addition to TEOA, benzyl alcohol, ethylenediaminetetraacetic, and methanol were utilized as reaction media and no HCOO− was produced | [225] | |
| NH2-UiO-66(zr) | 26.4 | ||||||
| MIL-101 (Fe) | MeCN/TEOA (5:1) | Visible light | HCOO− | 147.5 | MIL-101 (Fe) demonstrated the best photocatalytic performance due to the presence of coordination unsaturated Fe metal sites (CUSs) in its structure. | [248] | |
| NH2- MIL-101 (Fe) | 445 | ||||||
| MIL-53 (Fe) | 74.25 | ||||||
| NH2-MIL-53 (Fe) | 116.25 | ||||||
| MIL-88(Fe) | 22.5 | ||||||
| NH2-MIL-88(Fe) | 75 | ||||||
| NH2-UiO-66 (Zr) | MeCN/TEOA (4:1) BNAH (0.1 M) | Visible light | HCOO− | 0 | Introducing Ti ions into UiO-66 structure created new energy levels and broader light absorption and also improved charge transfer, which increased photoactivity of the MOF structure. | [255] | |
| NH2-UiO-66 (Zr/Ti) | 741 | ||||||
| (NH2)2-UiO-66 (Zr/Ti) | 1052.3 ± 54.7 | ||||||
| NH2-UiO-66 | MeCN/TEOA (11:1) | Simulate sunlight (350 < λ < 780 nm) | CO | 34 | [22] | ||
| NH2-UiO-66-tpy | 209.2 | ||||||
| Semiconductor-MOF composite | g-C3N4-Co-ZIF-9 | MeCN/H2O/TEOA (3:2:1)20 mg g-C3N4, 10 mg bpy, 1 mg MOF cocatalyst | Visible light (λ > 420 nm) | CO | 495 | [248] | |
| H2 | 78.6 | ||||||
| TiO2-Co-ZIF-9 | H2O vapor | Visible light (200 < λ < 900) | CO | 8.8 | Cocatalyst Co-ZIF-9 can facilitate CO2 adsorption and activation and also improve charge transfer. | [256] | |
| CH4 | 2.0 | ||||||
| H2 | 2.6 | ||||||
| Zn2GeO4-Zn-ZIF-8 | Na2SO3 (0.1 M) | 500 W Xe arc lamp | CH3OH | 0.22 | [231] | ||
| TiO2@Cu3(BTC)2 | H2O vapor | UV light | CH4 | 2.64 a | [233] | ||
| HKUST-1/TiO2 | CO | 256.3 a | HKUST-1 was selected because of the high porosity and surface area, about 40.7% and over 600 m2/g, respectively. Introducing TiO2 into the HKUST-1 matrix increased CO2 photoreduction efficiency. Additionally, the hydrophilicity of HKUST-1 led to simultaneous adsorption of water (H2O), resulting in improving CO2 photoreduction processes. | [257] | |||
| TiO2 | CO | 11.48 a | |||||
| TiO2-Mg-CPO-27 | H2O vapor | UV lamps (λ = 365) | CO | 4.09 | CPO-27-Mg was selected due to the superior CO2 adsorption capacity (about 35.2 wt.%) and consisted of great concentration of alkaline metal sites (Mg2+), which led to improved CO2 activation. | [258] | |
| CH4 | 2.35 | ||||||
| CNNS-UiO-66 (Zr) | MeCN/TEOA (4:1) | Visible light (400 < λ < 900) | CO | 2.9 | [259] | ||
| CNNS | 0.99 | ||||||
| UiO-66 (Zr) | 0 | ||||||
| TiO2-NH2-UiO-66 (Zr) (18.5%) b | CO2/H2 (1.5:1) | Visible light λ > 325 | CO | 4.24 | [260] | ||
| TiO2 | 2.85 | ||||||
| NH2-UiO-66 (Zr) | 1.5 | ||||||
| NH2-rGO (5 wt%)/Al-PMOF | MeCN/TEOA (5:1) | Visible light | HCOO− | 685.6 | [238] | ||
| Al-PMOF | 165.3 | ||||||
| TiO2/C@ZnCo-ZIF-L | - | Visible light | CO | 28.6 | [239] | ||
| TiO2/C@Co-ZIF-L | 22.7 | ||||||
| TiO2/C@Zn-ZIF-L | 18.7 | ||||||
| TiO2/C | 7.8 | ||||||
| ZIF-L | 0.36 | ||||||
| PCN-224 (Cu) | Water | Xe lamp (λ > 300) | CO | 3.72 | [242] | ||
| 6% PCN-224 (Cu)/TiO2 | 19.35 | ||||||
| 7.5% PCN-224 (Cu)/TiO2 | 26.78 | ||||||
| 10% PCN-224 (Cu)/TiO2 | 31.67 | ||||||
| 15% PCN-224 (Cu)/TiO2 | 37.21 | ||||||
| 30% PCN-224 (Cu)/TiO2 | 26.04 | ||||||
| TiO2 | 0.82 | ||||||
| Metal-MOF composite | Pt/NH2-MIL-125(Ti) | MeCN/TEOA (5:1) | Visible light | HCOO− | 32.4 | [245] | |
| Au/NH2-MIL-125(Ti) | 16.3 | ||||||
| Ni0.87Mg0.13-MOF-74 | MeCN/TEOA (5:1) [Ru(bpy c)3] Cl2 as photosensitizer | Visible light | HCOO− | 540 | [20] | ||
| CO | 520 | ||||||
| H2 | 2240 | ||||||
| Ni0.75Mg0.25-MOF-74 | HCOO− | 640 | |||||
| CO | 460 | ||||||
| H2 | 2610 | ||||||
| Other form of MOF-based photocatalyst | Zn/PMOF | H2O vapor | UV/Visible light | CH4 | 8.7 | [250] | |
| UiO-67-Cp*Rh d (5,5′-dcbpy) Cl2 (10%) | MeCN/TEOA (5:1) | Visible light (λ > 415) | HCOO− | 271 | [249] | ||
| H2 | 457 | ||||||
| Al-PMOF embedded Cu2+ | H2O/TEA (99:1) | Visible light | CH3OH | 262.6 (ppm h−1g−1) | CO2 adsorbed chemically on the prepared photocatalyst, which led to bending the linear CO2 molecule, resulting in lower photoreduction barrier and subsequently improving the photocatalytic efficiency. | [21] | |
| Al-PMOF | 37.5 (ppm h−1g−1) | ||||||
| Co/PMOF | MeCN/TEOA (5:1) | Visible light | HCOO− | 23.21 | [220] | ||
| Electrocatalyst | FECO2RR (%) | FEisopropanol/FEother |
|---|---|---|
| Cu Mesh | 43.3 | 1.5 |
| Cu Foil | 37.1 | 1.6 |
| Cu-PCN62 800 °C | 8.2 | 1.9 |
| Cu-PCN62 600 °C | 7.3 | 2 |
| Ni-PCN62 800 °C | 29.1 | 0.4 |
| Cu-HKUST-1 600 °C | 22.5 | 2.7 |
| Ni-HKUST-1 600 °C | 75.1 | 0.1 |
| Ni-HKUST-1 400 °C | 7.9 | 1.9 |
| Electrocatalyst | Potential (V vs. RHE) | Electrolyte | Product | FE (%) | References |
|---|---|---|---|---|---|
| Co-PP@CNT | −0.49 | 0.5 M NaHCO3 | CO | 98.3 | [309] |
| Cu-MOF-74 NPs | −1.3 | 0.1 M KHCO3 | CH4 | 50 | [310] |
| CuBi12 | −0.21 | 0.5 M KHCO3 | CH3OH | 8.6 | [311] |
| C2H5OH | 28.3 | ||||
| alcohols | 36.9 | ||||
| H-M-G | −0.46 | 0.1 M KHCO3 | CO | 96.9 | [312] |
| Ag1-Cu1.1 NDs | −1.2 | 0.1 M KHCO3 | C2H3O2− | 1.25 | [313] |
| (CH2OH)2 | 0.34 | ||||
| C2H5OH | 4.3 | ||||
| C3H8O | 0.83 | ||||
| ZIF-A-LD/CB | −1.1 | 0.1 M KHCO3 | CO | 90.57 | [314] |
| ZIF-7-A-LD/CB | 53.7 | ||||
| Cu12Sn a | −0.76 | 0.1 M KHCO3 | CO | 66.5 | [315] |
| Cu4Sn | −0.97 | HCOO− | 56.77 | ||
| Cu3Sn2 | −0.67 | H2 | 50.33 | ||
| Sn-OH-5.9 branches | −1.6 V vs. Ag/AgCl | 0.1 M KCl | HCOOH | 82 | [316] |
| C1 | 93 | ||||
| Ni@NC-900 | −1 | 0.1 M KHCO3 | CO | 96 | [317] |
| w-CCG/CoPc-A hybrid | −0.69 | 0.1 M KHCO3 | H2 | 11.4 | [318] |
| CO | 90.9 | ||||
| −0.79 | H2 | 8.8 | |||
| CO | 91.5 | ||||
| Cu2O/Cu@NC-800 | −6.8 | 0.1 M KHCO3 | HCOO− | 70.5 | [16] |
| PdAg_2 | −0.27 | 0.1 M KHCO3 | HCOO− | 94 | [319] |
| Sn-CHF (10 wt.%) | −0.78 b | 0.1 M KHCO3 | CO | 8.91 | [320] |
| −0.98 b | HCOO− | 0.88 | |||
| −0.48 b | H2 | 87.72 | |||
| −0.84 c | CO | 48.36 | |||
| −0.96 c | HCOO− | 8.94 | |||
| −0.32 c | H2 | 92.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parvanian, A.M.; Sadeghi, N.; Rafiee, A.; Shearer, C.J.; Jafarian, M. Application of Porous Materials for CO2 Reutilization: A Review. Energies 2022, 15, 63. https://doi.org/10.3390/en15010063
Parvanian AM, Sadeghi N, Rafiee A, Shearer CJ, Jafarian M. Application of Porous Materials for CO2 Reutilization: A Review. Energies. 2022; 15(1):63. https://doi.org/10.3390/en15010063
Chicago/Turabian StyleParvanian, Amir Masoud, Nasrin Sadeghi, Ahmad Rafiee, Cameron J. Shearer, and Mehdi Jafarian. 2022. "Application of Porous Materials for CO2 Reutilization: A Review" Energies 15, no. 1: 63. https://doi.org/10.3390/en15010063