
A Neural Network-Inspired Matrix Formulation of Chemical Kinetics for Acceleration on GPUs

Abstract
:1. Introduction
2. Methodology
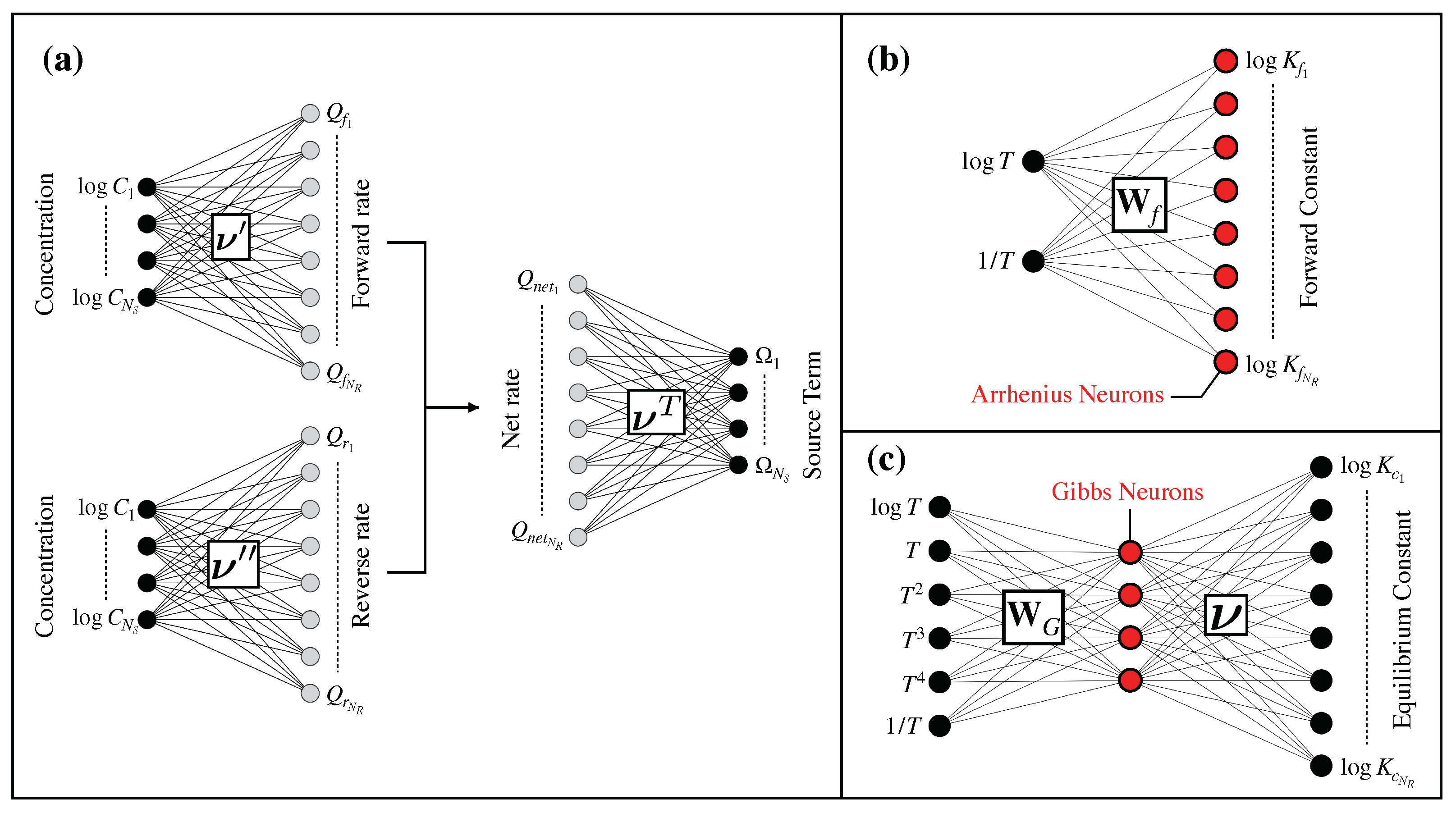
2.1. Matrix-Based Kinetics Equations
- Let the GPU handle the zero concentrations natively. For example, CUDA supports the operation of exp(log(0))=0 through the Inf floating point placeholder. That is, CUDA ensures that the operation log(0)=-Inf, and that exp(-Inf)=0.
- Replace either the zero concentrations or mass fractions with an extremely small positive number and then proceed with the computations. This number should be small enough (i.e., 1.0e-300) to effectively produce a huge negative number upon taking the logarithm.
- Replace the logarithm of the zero concentrations directly with a huge negative number, e.g., through a re-definition as log(0)=-1.0e300.
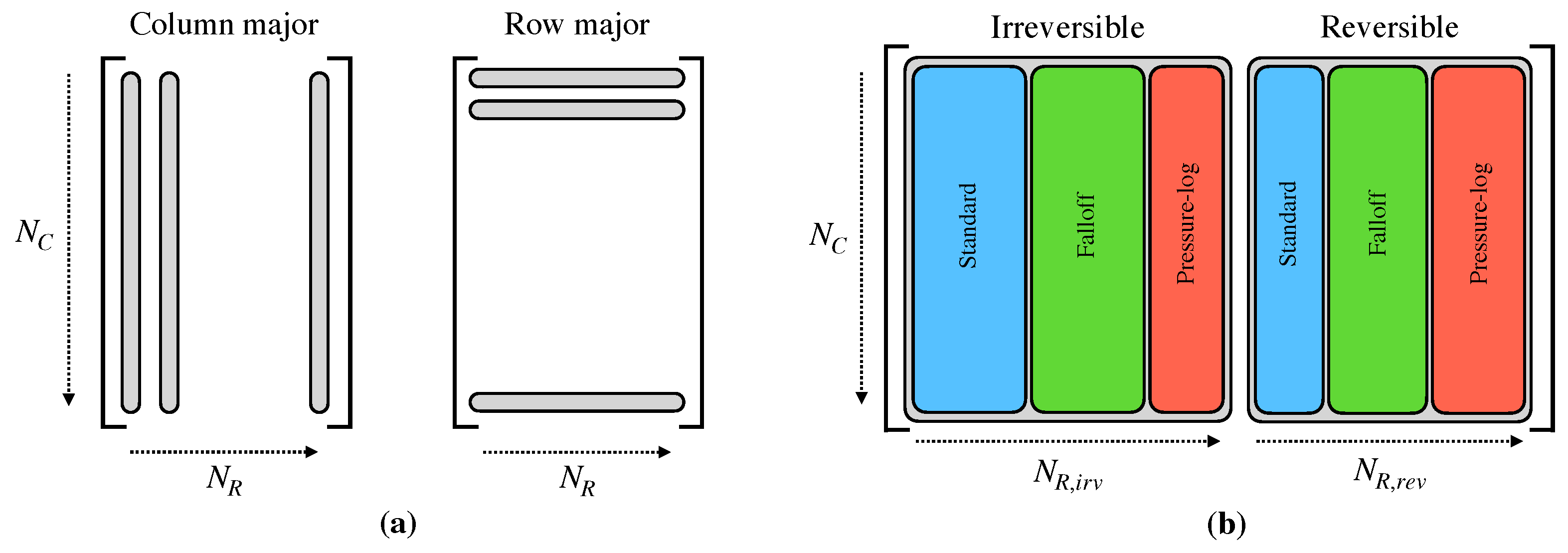
2.2. Organization of Data
2.3. Reaction Decomposition and Classification
- Standard reactions utilize the standard Arrhenius equation for the forward rate constant (Equation (6)). They may or may not include third body contributions.
- Falloff reactions employ significantly more complex expressions of the forward rate constant computation for reactions involving third bodies. Falloff reactions can be Lindemann, Troe, or SRI type, each with slightly different implementations. The reader is referred to the Cantera [26] or Chemkin [27] documentation for mathematical details.
- Pressure-log reactions add pressure dependence into the computation of the forward rate constant through an interpolation routine. They may or may not include third body contributions. The reader is referred to the Cantera or Chemkin documentation for mathematical details.
3. GPU Performance Analysis
3.1. Compute Times and Throughput
3.2. Speedup
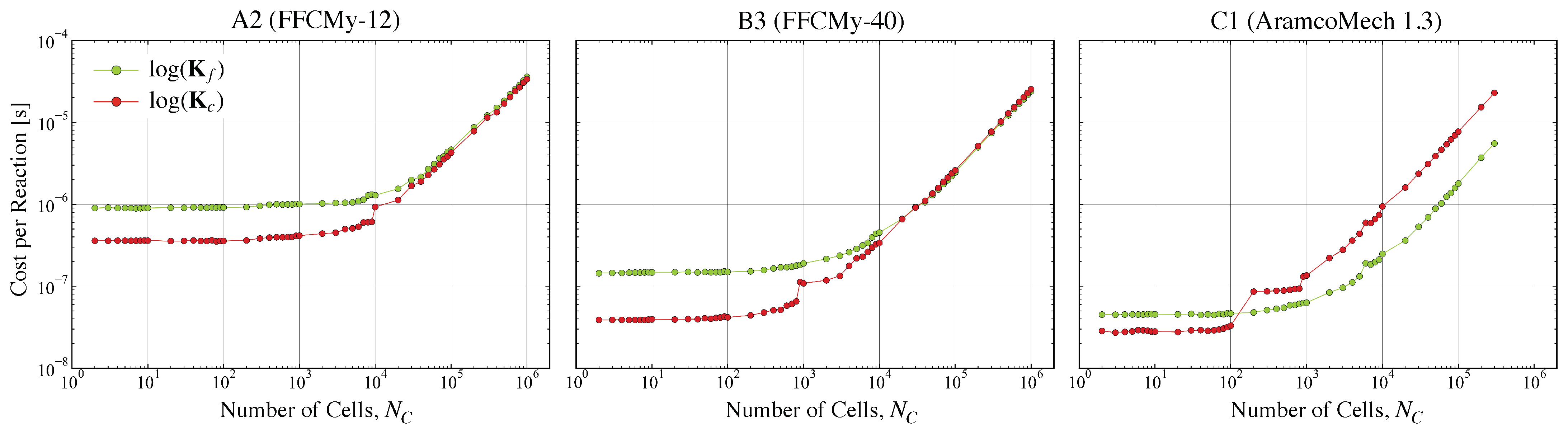
3.3. Cost of Reaction Types
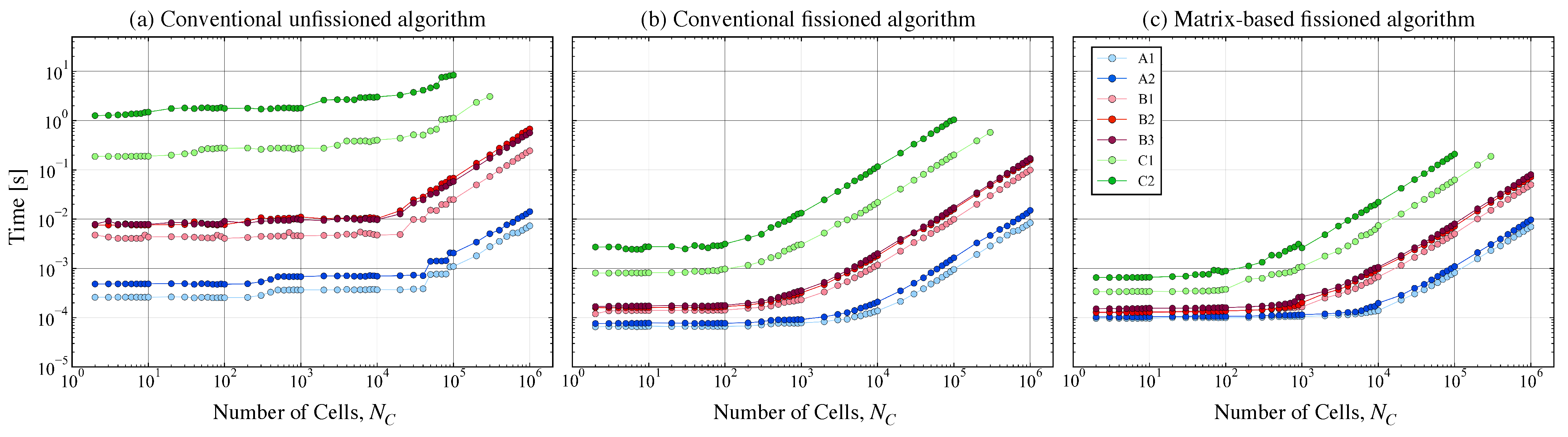
3.4. Improving Speedup for Large Mechanisms
- The preprocessing routine consists of kernels that: (a) recover the primitive species mass fractions from conserved values; (b) normalize the species mass fractions in each cell to sum to unity; and (c) obtain molar concentrations from the mass fractions.
- The forward rate constant routine for computes Equation (6) along with any other non-standard reaction types that modify the forward rate (three-body, pressure-log, and falloff).
- The equilibrium rate constant routine for is outlined in Equation (9).
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Substitution with Approximate Artificial Neural Networks

Appendix B. Verification of GPU Implementation

Appendix C. Performance Comparison with Non-Matrix Approaches



References
- Hochgreb, S. Mind the gap: Turbulent combustion model validation and future needs. Proc. Combust. Inst. 2019, 37, 2091–2107. [Google Scholar] [CrossRef] [Green Version]
- Raman, V.; Hassanaly, M. Emerging trends in numerical simulations of combustion systems. Proc. Combust. Inst. 2019, 37, 2073–2089. [Google Scholar] [CrossRef] [Green Version]
- Pitsch, H. Large-eddy simulation of turbulent combustion. Annu. Rev. Fluid Mech. 2006, 38, 453–482. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.H. Petascale direct numerical simulation of turbulent combustion—Fundamental insights towards predictive models. Proc. Combust. Inst. 2011, 33, 99–123. [Google Scholar] [CrossRef]
- Jaravel, T.; Riber, E.; Cuenot, B.; Pepiot, P. Prediction of flame structure and pollutant formation of Sandia flame D using Large Eddy Simulation with direct integration of chemical kinetics. Combust. Flame 2018, 188, 180–198. [Google Scholar] [CrossRef]
- Mueller, M.E. A computationally efficient turnkey approach to turbulent combustion modeling: From elusive fantasy to impending reality. In Proceedings of the AIAA Scitech 2019 Forum, San Diego, CA, USA, 7–11 January 2019; p. 0994. [Google Scholar]
- Pope, S.B. Turbulent Flows; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Raman, V.; Pitsch, H. A consistent LES/filtered-density function formulation for the simulation of turbulent flames with detailed chemistry. Proc. Combust. Inst. 2007, 31, 1711–1719. [Google Scholar] [CrossRef]
- Menon, S.; Kerstein, A.R. The linear-eddy model. In Turbulent Combustion Modeling; Springer: Dordrecht, The Netherlands, 2011; pp. 221–247. [Google Scholar]
- Pope, S. Computationally efficient implementation of combustion chemistry using in situ adaptive tabulation. Combust. Theory Model. 1997, 1, 41–63. [Google Scholar] [CrossRef]
- Tonse, S.R.; Moriarty, N.W.; Brown, N.J.; Frenklach, M. PRISM: Piecewise reusable implementation of solution mapping. An economical strategy for chemical kinetics. Isr. J. Chem. 1999, 39, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Christo, F.; Masri, A.; Nebot, E.; Pope, S. An integrated PDF/neural network approach for simulating turbulent reacting systems. Symp. (Int.) Combust. 1996, 26, 43–48. [Google Scholar] [CrossRef]
- Sen, B.A.; Menon, S. Turbulent premixed flame modeling using artificial neural networks based chemical kinetics. Proc. Combust. Inst. 2009, 32, 1605–1611. [Google Scholar] [CrossRef]
- Kempf, A.; Flemming, F.; Janicka, J. Investigation of lengthscales, scalar dissipation, and flame orientation in a piloted diffusion flame by LES. Proc. Combust. Inst. 2005, 30, 557–565. [Google Scholar] [CrossRef]
- Owoyele, O.; Kundu, P.; Ameen, M.M.; Echekki, T.; Som, S. Application of deep artificial neural networks to multi-dimensional flamelet libraries and spray flames. Int. J. Engine Res. 2020, 21, 151–168. [Google Scholar] [CrossRef]
- Barwey, S.; Prakash, S.; Hassanaly, M.; Raman, V. Data-driven Classification and Modeling of Combustion Regimes in Detonation Waves. Flow Turbul. Combust. 2020, 106, 1065–1089. [Google Scholar] [CrossRef]
- Reed, D.A.; Dongarra, J. Exascale computing and big data. Commun. ACM 2015, 58, 56–68. [Google Scholar] [CrossRef]
- Nickolls, J.; Dally, W.J. The GPU computing era. IEEE Micro 2010, 30, 56–69. [Google Scholar] [CrossRef]
- Niemeyer, K.E.; Sung, C.J. Recent progress and challenges in exploiting graphics processors in computational fluid dynamics. J. Supercomput. 2014, 67, 528–564. [Google Scholar] [CrossRef] [Green Version]
- Niemeyer, K.E.; Sung, C.J. Accelerating moderately stiff chemical kinetics in reactive-flow simulations using GPUs. J. Comput. Phys. 2014, 256, 854–871. [Google Scholar] [CrossRef] [Green Version]
- Curtis, N.J.; Niemeyer, K.E.; Sung, C.J. Using SIMD and SIMT vectorization to evaluate sparse chemical kinetic Jacobian matrices and thermochemical source terms. Combust. Flame 2018, 198, 186–204. [Google Scholar] [CrossRef] [Green Version]
- Sewerin, F.; Rigopoulos, S. A methodology for the integration of stiff chemical kinetics on GPUs. Combust. Flame 2015, 162, 1375–1394. [Google Scholar] [CrossRef]
- Pérez, F.E.H.; Mukhadiyev, N.; Xu, X.; Sow, A.; Lee, B.J.; Sankaran, R.; Im, H.G. Direct numerical simulations of reacting flows with detailed chemistry using many-core/GPU acceleration. Comput. Fluids 2018, 173, 73–79. [Google Scholar] [CrossRef] [Green Version]
- cuBLAS, The CUDA Basic Linear Algebra Subroutine Library. NVIDIA Corporation. 2021. Available online: https://docs.nvidia.com/cuda/cublas/index.html (accessed on March 2021).
- Poinsot, T.; Veynante, D. Theoretical and Numerical Combustion; RT Edwards, Inc.: Philadelphia, PA, USA, 2005. [Google Scholar]
- Goodwin, D.G.; Moffat, H.K.; Speth, R.L. Cantera: An Object-Oriented Software Toolkit for Chemical Kinetics, Thermodynamics, and Transport Processes. 2017. Available online: https://www.cantera.org (accessed on March 2021).
- Kee, R.J.; Rupley, F.M.; Miller, J.A. Chemkin-II: A Fortran Chemical Kinetics Package for the Analysis of Gas-Phase Chemical Kinetics; Technical Report; Sandia National Lab.(SNL-CA): Livermore, CA, USA, 1989. [Google Scholar]
- Mueller, M.; Kim, T.; Yetter, R.; Dryer, F. Flow reactor studies and kinetic modeling of the H2/O2 reaction. Int. J. Chem. Kinet. 1999, 31, 113–125. [Google Scholar] [CrossRef]
- Xu, R.; (Stanford University, 440 Escondido Mall, Stanford, CA, USA); Wang, H.; (Stanford University, 440 Escondido Mall, Stanford, CA, USA). Reduced Reaction Models for Methane and Ethylene Combustion. Personal communication, 2020. [Google Scholar]
- Smith, G.; Tao, Y.; Wang, H. Foundational Fuel Chemistry Model Version 1.0 (FFCM-1). 2016. Available online: http://nanoenergy.stanford.edu/ffcm1 (accessed on August 2020).
- Chemical-Kinetic Mechanisms for Combustion Applications. San Diego Mechanism web page, Mechanical and Aerospace Engineering (Combustion Research), University of California at San Diego. 2016. Available online: http://web.eng.ucsd.edu/mae/groups/combustion/mechanism.html (accessed on August 2020).
- Metcalfe, W.K.; Burke, S.M.; Ahmed, S.S.; Curran, H.J. A hierarchical and comparative kinetic modeling study of C1–C2 hydrocarbon and oxygenated fuels. Int. J. Chem. Kinet. 2013, 45, 638–675. [Google Scholar] [CrossRef]
- Mehl, M.; Pitz, W.J.; Westbrook, C.K.; Curran, H.J. Kinetic modeling of gasoline surrogate components and mixtures under engine conditions. Proc. Combust. Inst. 2011, 33, 193–200. [Google Scholar] [CrossRef] [Green Version]
- The Nvprof Profiling Tool. NVIDIA Corporation. 2021. Available online: https://docs.nvidia.com/cuda/profiler-users-guide (accessed on March 2021).
- Williams, S.; Waterman, A.; Patterson, D. Roofline: An insightful visual performance model for multicore architectures. Commun. ACM 2009, 52, 65–76. [Google Scholar] [CrossRef]
- cuSPARSE, The CUDA Sparse Matrix Library. NVIDIA Corporation. 2021. Available online: https://docs.nvidia.com/cuda/cusparse/index.html (accessed on March 2021).
- Clevert, D.A.; Unterthiner, T.; Hochreiter, S. Fast and accurate deep network learning by exponential linear units (elus). arXiv 2015, arXiv:1511.07289. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Reference | Description | ||
|---|---|---|---|---|
| A1 | Mueller et al. [28] | Hydrogen/Air | 9 | 21 |
| A2 | FFCMy-12 [29,30] | Methane/Oxygen | 12 | 38 |
| B1 | FFCMy-30 [29] | Ethylene/Air | 30 | 231 |
| B2 | UCSD [31] | Hydrogen/Air | 57 | 268 |
| B3 | FFCMy-40 [29] | Ethylene/Air | 41 | 361 |
| C1 | AramcoMech 1.3 [32] | – | 253 | 1542 |
| C2 | LLNL [33] | – | 654 | 4846 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barwey, S.; Raman, V. A Neural Network-Inspired Matrix Formulation of Chemical Kinetics for Acceleration on GPUs. Energies 2021, 14, 2710. https://doi.org/10.3390/en14092710
Barwey S, Raman V. A Neural Network-Inspired Matrix Formulation of Chemical Kinetics for Acceleration on GPUs. Energies. 2021; 14(9):2710. https://doi.org/10.3390/en14092710
Chicago/Turabian StyleBarwey, Shivam, and Venkat Raman. 2021. "A Neural Network-Inspired Matrix Formulation of Chemical Kinetics for Acceleration on GPUs" Energies 14, no. 9: 2710. https://doi.org/10.3390/en14092710