
Fast Pyrolysis of Cellulose by Infrared Heating


Abstract
:1. Introduction
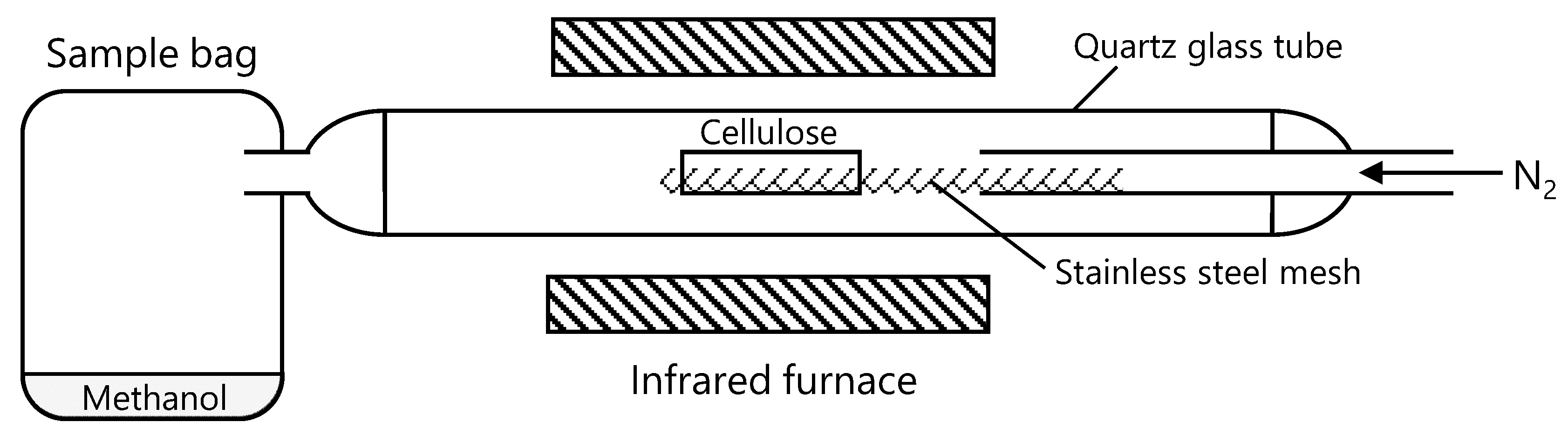
2. Materials and Methods
2.1. Cellulose Samples
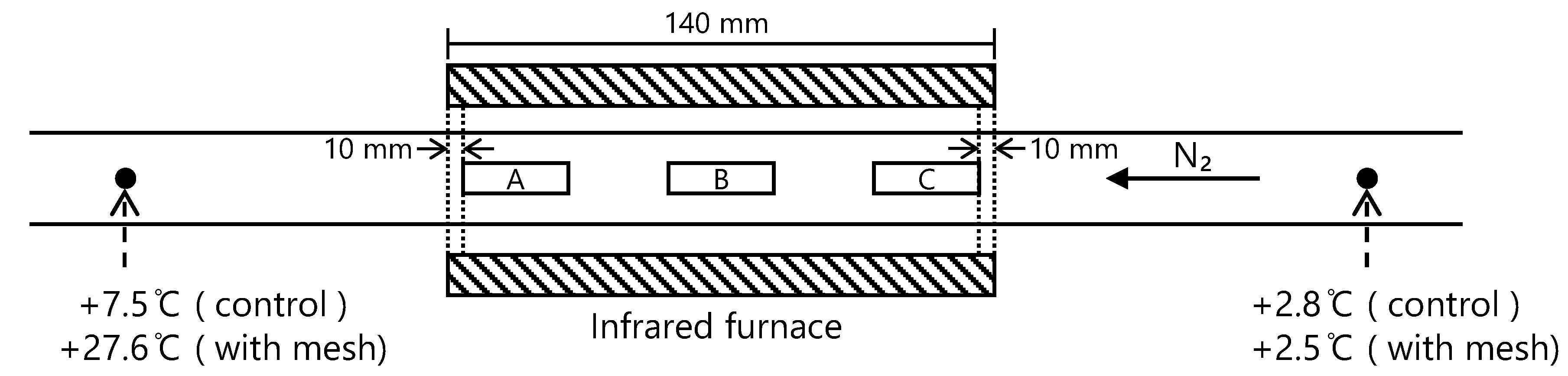
2.2. Pyrolysis and Product Analysis
3. Results and Discussion
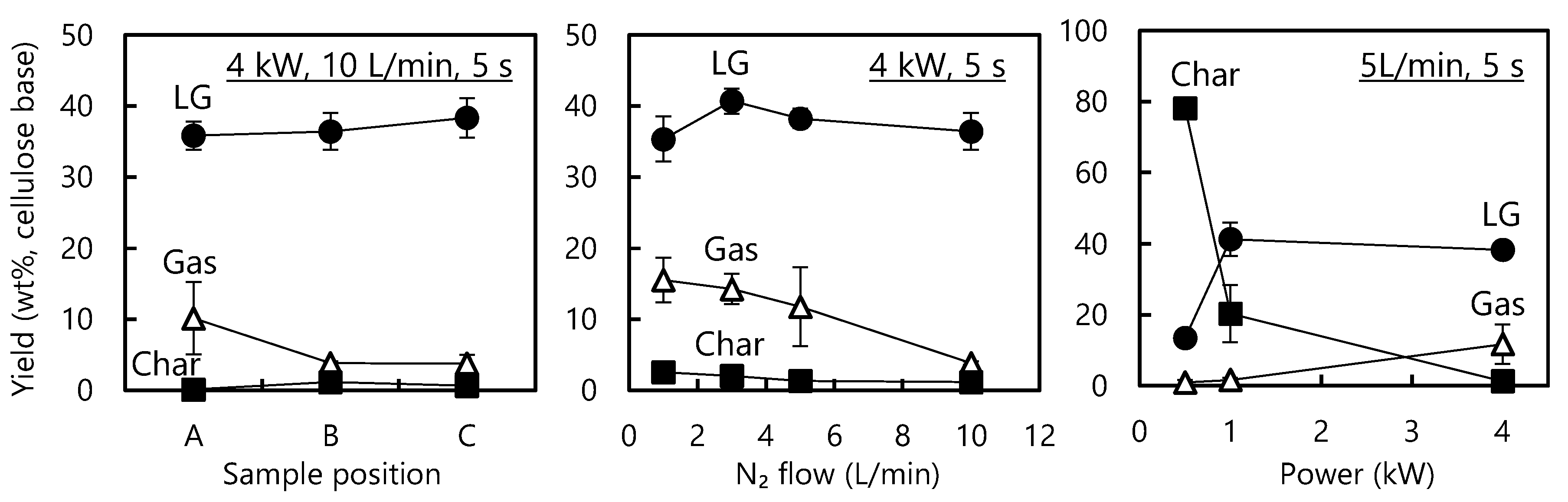
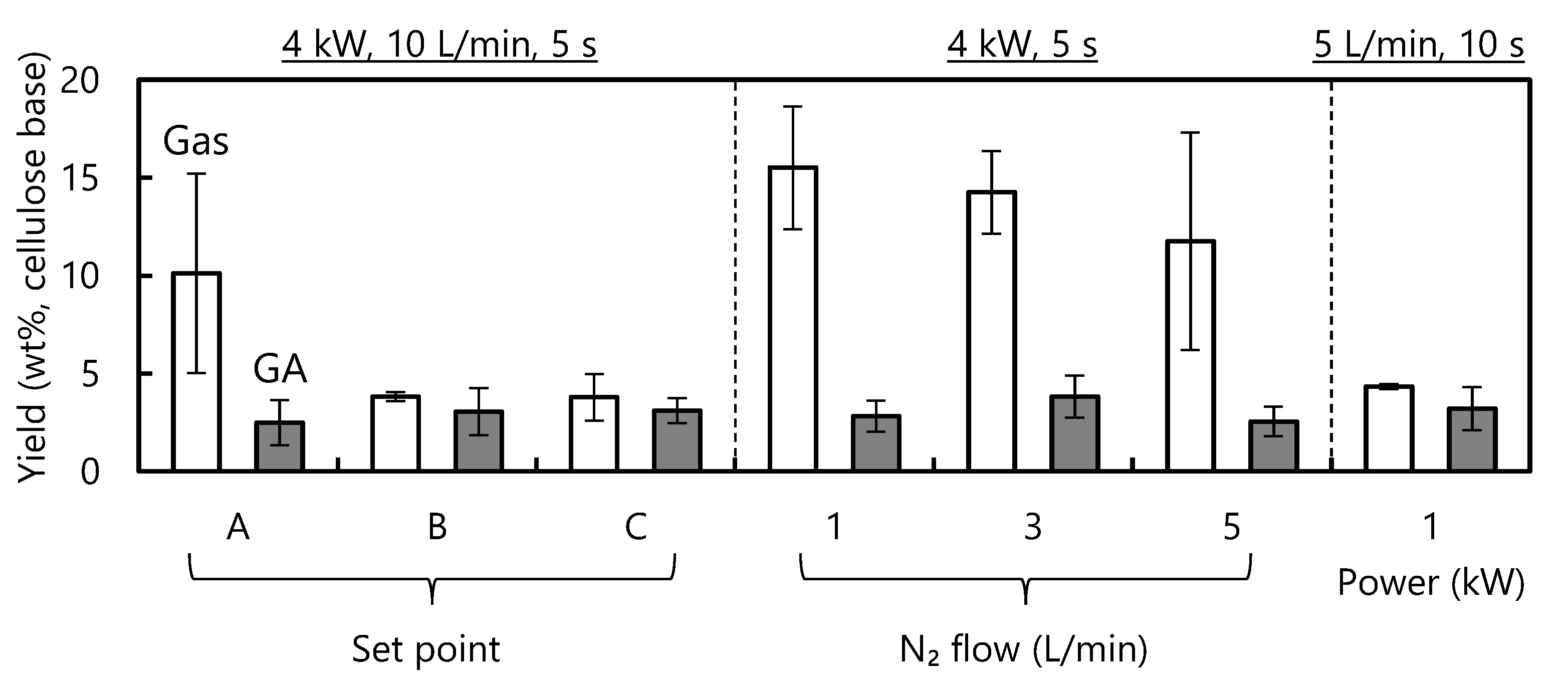
3.1. Effects of the Experimental Parameters on the Product Yield
3.2. The Mechanism for Cellulose Pyrolysis by Infrared Irradiation
4. Conclusions
- The sample position in the IR furnace, the IR power level and the nitrogen flow rate all affected the thermal degradation of cellulose by modifying the cellulose degradation temperature.
- Under the optimum conditions, LG was obtained in a 52.7 wt % yield from Whatman cellulose sheets (infrared power: 1.0 kW, nitrogen flow: 5 L/min, irradiation time: 10 s).
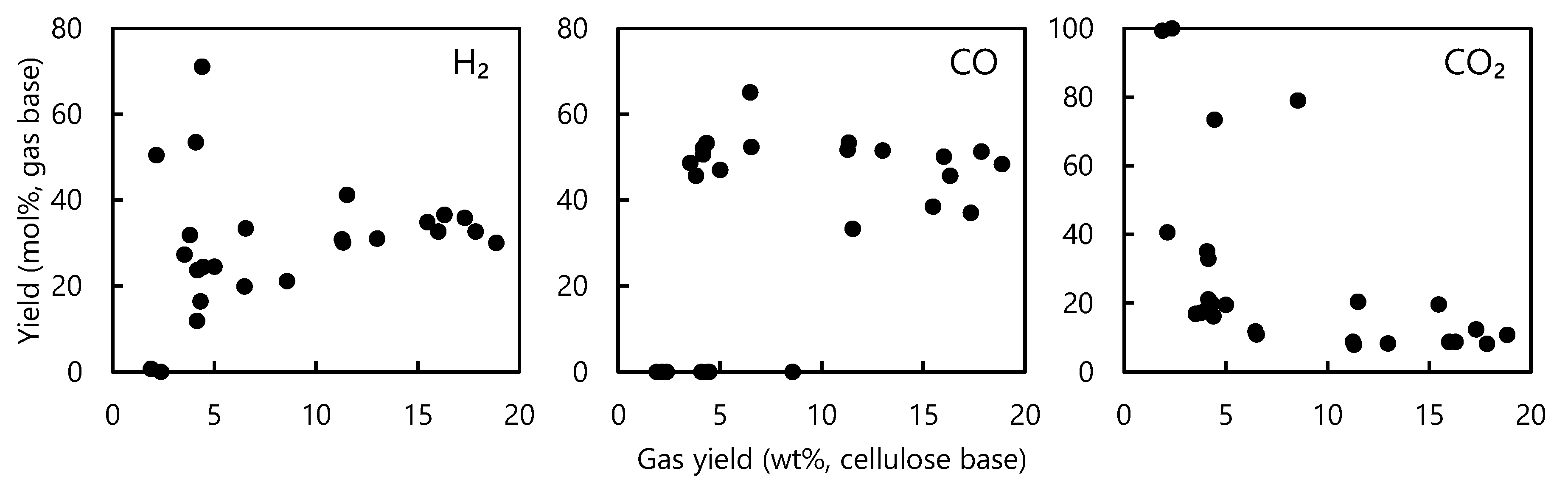
- The gas yield increased when the cellulose was overheated to above 600 °C as a result of the secondary degradation of LG. This effect could be monitored by tracking the formation of CO, although in situ temperature measurements were difficult due to the nonuniform progression of the cellulose thermal degradation.
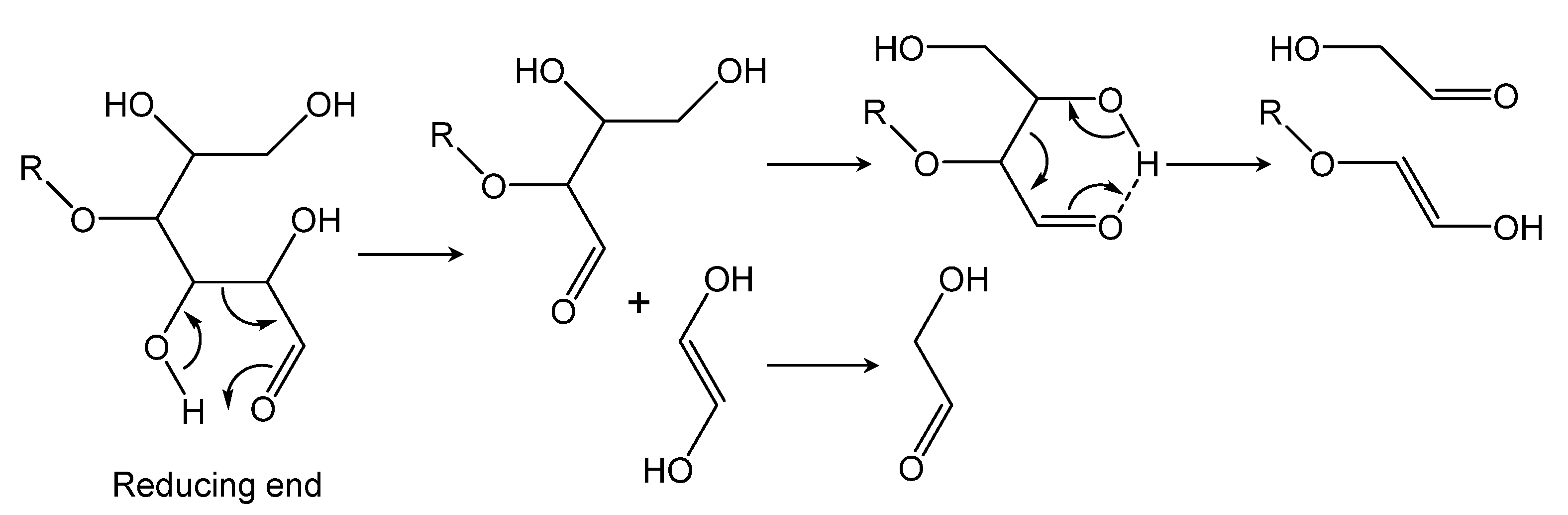
- GA was the other major product of cellulose degradation, and the yield of this compound was not correlated with the gas output, suggesting that it was not a secondary LG degradation product. GA was evidently produced during the primary cellulose pyrolysis stage via the retro-Aldol fragmentation of the reducing ends of cellulose and other intermediate carbohydrates.
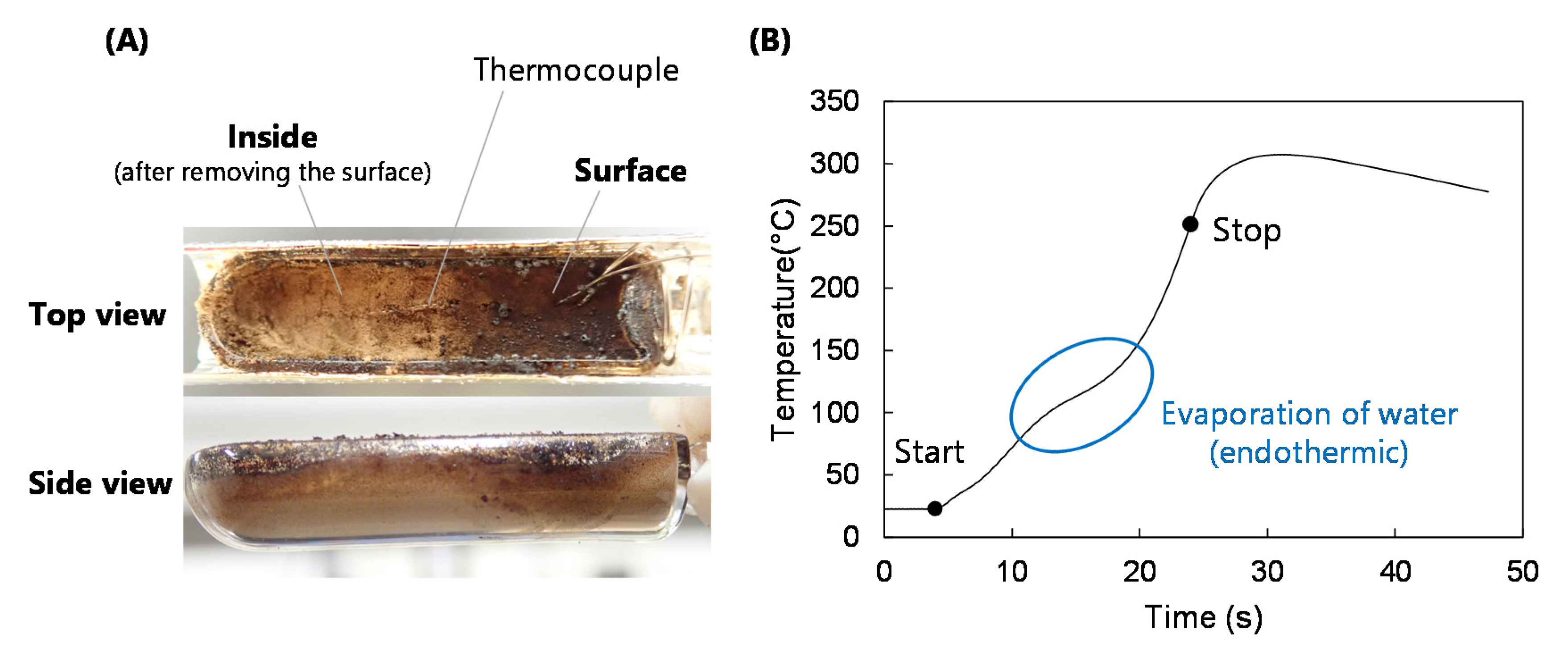
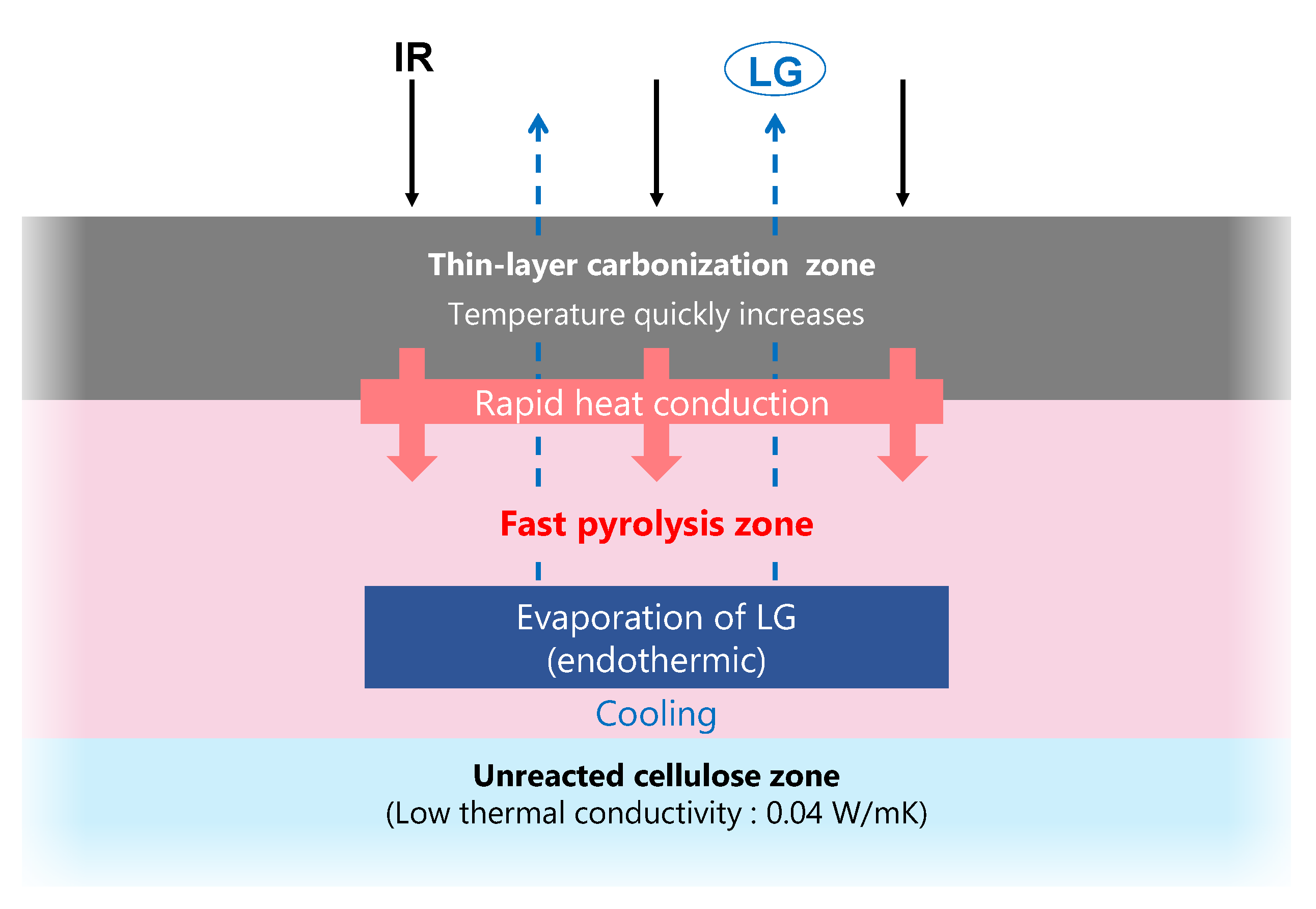
- The thermal degradation of cellulose occurred in a nonuniform manner in response to IR irradiation, with the formation of a narrow carbonization layer. This layer was rapidly heated by efficiently absorbing IR and, in turn, heated the adjacent cellulose. This process then propagated throughout the cellulose to maintain a high LG output rate.
- The use of IR heating during the production of LG from cellulose offers several advantages compared with other fast pyrolysis methods based on heat conduction. The latter methods require the cellulose to be ground and heated quickly to maintain a high sample temperature, while the IR heating methods allow the use of any cellulose, regardless of size. Infrared power can also be easily controlled by changing the electric power.
- These results give insights into the production of biochemicals and biofuels via LG and pyrolysis-based saccharification.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LG | levoglucosan |
| GA | glycolaldehyde |
References
- Jahirul, M.I.; Rasul, M.G.; Chowdhury, A.A.; Ashwath, N. Biofuels production through biomass pyrolysis-A technological review. Energies 2012, 5, 4952–5001. [Google Scholar] [CrossRef]
- Uddin, M.N.; Techato, K.; Taweekun, J.; Rahman, M.M.; Rasul, M.G.; Mahlia, T.M.I.; Ashrafur, S.M. An overview of recent developments in biomass pyrolysis technologies. Energies 2018, 11, 3115. [Google Scholar] [CrossRef] [Green Version]
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Kersten, S.; Garcia-Perez, M. Recent developments in fast pyrolysis of ligno-cellulosic materials. Curr. Opin. Biotechnol. 2013, 24, 414–420. [Google Scholar] [CrossRef]
- Sharifzadeh, M.; Sadeqzadeh, M.; Guo, M.; Borhani, T.N.; Murthy Konda, N.V.S.N.; Garcia, M.C.; Wang, L.; Hallett, J.; Shah, N. The multi-scale challenges of biomass fast pyrolysis and bio-oil upgrading: Review of the state of art and future research directions. Prog. Energy Combust. Sci. 2019, 71, 1–80. [Google Scholar] [CrossRef]
- Venderbosch, R.; Prins, W. Fast pyrolysis technology development. Biofuels Bioprod. Biorefin. 2010, 4, 178–208. [Google Scholar] [CrossRef]
- Perkins, G.; Bhaskar, T.; Konarova, M. Process development status of fast pyrolysis technologies for the manufacture of renewable transport fuels from biomass. Renew Sustain. Energy Rev. 2018, 90, 292–315. [Google Scholar] [CrossRef]
- Shafizadeh, F.; Furneaux, R.H.; Cochran, T.G.; Scholl, J.P.; Sakai, Y. Production of levoglucosan and glucose from pyrolysis of cellulosic materials. J. Appl. Polym. Sci. 1979, 23, 3525–3539. [Google Scholar] [CrossRef]
- Kwon, G.J.; Kim, D.-Y.; Kimura, S.; Kuga, S. Rapid-cooling, continuous-feed pyrolyzer for biomass processing: Preparation of levoglucosan from cellulose and starch. J. Anal. Appl. Pyrolysis 2007, 80, 1–5. [Google Scholar] [CrossRef]
- Itabaiana Junior, I.; Avelar Do Nascimento, M.; De Souza, R.O.M.A.; Dufour, A.; Wojcieszak, R. Levoglucosan: A promising platform molecule? Green Chem. 2020, 22, 5859–5880. [Google Scholar] [CrossRef]
- Boutin, O.; Ferrer, M.; Lédé, J. Flash pyrolysis of cellulose pellets submitted to a concentrated radiation: Experiments and modelling. Chem. Eng. Sci. 2002, 57, 15–25. [Google Scholar] [CrossRef]
- Boutin, O.; Ferrer, M.; Lédé, J. Radiant flash pyrolysis of cellulose—Evidence for the formation of short life time intermediate liquid species. J. Anal. Appl. Pyrolysis 1998, 47, 13–31. [Google Scholar] [CrossRef]
- Lédé, J.; Blanchard, F.; Boutin, O. Radiant flash pyrolysis of cellulose pellets: Products and mechanisms involved in transient and steady state conditions. Fuel 2002, 81, 1269–1279. [Google Scholar] [CrossRef]
- Shoji, T.; Kawamoto, H.; Saka, S. Boiling point of levoglucosan and devolatilization temperatures in cellulose pyrolysis measured at different heating area temperatures. J. Anal. Appl. Pyrolysis 2014, 109, 185–195. [Google Scholar] [CrossRef]
- Pictet, A. Sur la transformation de la lévoglucosane en dextrine. Helv. Chim. Acta 1918, 1, 226–230. [Google Scholar] [CrossRef]
- Fukutome, A.; Kawamoto, H.; Saka, S. Processes forming gas, tar, and coke in cellulose gasification from gas-phase reactions of levoglucosan as intermediate. ChemSusChem 2015, 8, 2240–2249. [Google Scholar] [CrossRef] [Green Version]
- Hosoya, T.; Kawamoto, H.; Saka, S. Different pyrolytic pathways of levoglucosan in vapor- and liquid/solid-phases. J. Anal. Appl. Pyrolysis 2008, 83, 64–70. [Google Scholar] [CrossRef]
- Kawamoto, H.; Murayama, M.; Saka, S. Pyrolysis behavior of levoglucosan as an intermediate in cellulose pyrolysis: Polymerization into polysaccharide as a key reaction to carbonized product formation. J. Wood Sci. 2003, 49, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, H.; Hosoya, T.; Ueno, Y.; Shoji, T.; Saka, S. Thermal stabilization and decomposition of simple glycosides in the presence of aromatic substances in closed ampoules: The role of OH π hydrogen bonding. J. Anal. Appl. Pyrolysis 2014, 109, 41–46. [Google Scholar] [CrossRef]
- Kawamoto, H.; Ueno, Y.; Saka, S. Thermal reactivities of non-reducing sugars in polyether—Role of intermolecular hydrogen bonding in pyrolysis. J. Anal. Appl. Pyrolysis 2013, 103, 287–292. [Google Scholar] [CrossRef]
- Suzuki, J.; Azuma, J.; Koshijima, T.; Okamura, K.; Okamoto, H. Characterization of mono- and oligosaccharides produed by CO2 laser irradiation on cellulose. Chem. Lett. 1983, 12, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Dauenhauer, P.J.; Colby, J.L.; Balonek, C.M.; Suszynski, W.J.; Schmidt, L.D. Reactive boiling of cellulose for integrated catalysis through an intermediate liquid. Green Chem. 2009, 11, 1555–1561. [Google Scholar] [CrossRef]
- Teixeira, A.R.; Mooney, K.G.; Kruger, J.S.; Williams, C.L.; Suszynski, W.J.; Schmidt, L.D.; Schmidt, D.P.; Dauenhauer, P.J. Aerosol generation by reactive boiling ejection of molten cellulose. Energy Environ. Sci. 2011, 4, 4306–4321. [Google Scholar] [CrossRef] [Green Version]
- Lédé, J.; Li, H.Z.; Villermaux, J.; Martin, H. Fusion-like behaviour of wood pyrolysis. J. Anal. Appl. Pyrolysis 1987, 10, 291–308. [Google Scholar] [CrossRef]
- Luo, G.; Chandler, D.S.; Anjos, L.C.A.; Eng, R.J.; Jia, P.; Resende, F.L.P. Pyrolysis of whole wood chips and rods in a novel ablative reactor. Fuel 2017, 194, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Weldekidan, H.; Strezov, V.; Town, G. Review of solar energy for biofuel extraction. Renew Sustain. Energy Rev. 2018, 88, 184–192. [Google Scholar] [CrossRef]
- Nordin, S.B.; Nyren, J.O.; Back, E.L. An indication of molten cellulose produced in a laser beam. Text. Res. J. 1974, 44, 152–154. [Google Scholar] [CrossRef]
- Schroeter, J.; Felix, F. Melting cellulose. Cellulose 2005, 12, 159–165. [Google Scholar] [CrossRef]
- Zeng, K.; Gauthier, D.; Soria, J.; Mazza, G.; Flamant, G. Solar pyrolysis of carbonaceous feedstocks: A review. Sol. Energy 2017, 156, 73–92. [Google Scholar] [CrossRef]
- Hopkins, M.W.; DeJenga, C.; Antal, M.J. The flash pyrolysis of cellulosic materials using concentrated visible light. Sol. Energy 1984, 32, 547–551. [Google Scholar] [CrossRef]
- Kwon, G.J.; Kuga, S.; Hori, K.; Yatagai, M.; Ando, K.; Hattori, N. Saccharification of cellulose by dry pyrolysis. J. Wood Sci. 2006, 52, 461–465. [Google Scholar] [CrossRef]
- Westerhof, R.J.M.; Oudenhoven, S.R.G.; Marathe, P.S.; Engelen, M.; Garcia-Perez, M.; Wang, Z.; Kersten, S.R.A. The interplay between chemistry and heat/mass transfer during the fast pyrolysis of cellulose. React. Chem. Eng. 2016, 1, 555–566. [Google Scholar] [CrossRef]
- Piskorz, J.; Radlein, D.S.A.G.; Scott, D.S.; Czernik, S. Pretreatment of wood and cellulose for production of sugars by fast pyrolysis. J. Anal. Appl. Pyrolysis 1989, 16, 127–142. [Google Scholar] [CrossRef]
- Piskorz, J.; Radlein, D.; Scott, D.S. On the mechanism of the rapid pyrolysis of cellulose. J. Anal. Appl. Pyrolysis 1986, 9, 121–137. [Google Scholar] [CrossRef]
- Richards, G.N. Glycolaldehyde from pyrolysis of cellulose. J. Anal. Appl. Pyrolysis 1987, 10, 251–255. [Google Scholar] [CrossRef]
- Banyasz, J.L.; Li, S.; Lyons-Hart, J.L.; Shafer, K.H. Cellulose pyrolysis: The kinetics of hydroxyacetaldehyde evolution. J. Anal. Appl. Pyrolysis 2001, 57, 223–248. [Google Scholar] [CrossRef]
- Degenstein, J.C.; Murria, P.; Easton, M.; Sheng, H.; Hurt, M.; Dow, A.R.; Gao, J.; Nash, J.J.; Agrawal, R.; Delgass, W.N.; et al. Fast pyrolysis of 13C-labeled cellobioses: Gaining insights into the mechanisms of fast pyrolysis of carbohydrates. J. Org. Chem. 2015, 80, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Milosavljevic, I.; Oja, V.; Suuberg, E.M. Thermal effects in cellulose pyrolysis: Relationship to char formation processes. Ind. Eng. Chem. Res. 1996, 35, 653–662. [Google Scholar] [CrossRef]
- Mok, W.S.L.; Antal, M.J. Effects of pressure on biomass pyrolysis. II. Heats of reaction of cellulose pyrolysis. Thermochim. Acta 1983, 68, 165–186. [Google Scholar] [CrossRef]
- Cho, J.; Davis, J.M.; Huber, G.W. The intrinsic kinetics and heats of reactions for cellulose pyrolysis and char formation. ChemSusChem 2010, 3, 1162–1165. [Google Scholar] [CrossRef]
- Di Blasi, C. Numerical simulation of cellulose pyrolysis. Biomass Bioenergy 1994, 7, 87–98. [Google Scholar] [CrossRef]
- Majumdar, A.; Mukhopadhyay, S.; Yadav, R. Thermal properties of knitted fabrics made from cotton and regenerated bamboo cellulosic fibres. Int. J. Therm. Sci. 2010, 49, 2042–2048. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yield (wt%, Cellulose Base) | ||||
|---|---|---|---|---|
| Infrared Power (kW) | Irradiation Time (s) | LG | Gas | Char |
| 0.5 | 5 | 4.9 ± 5.6 (48.1 ± 1.8) * | 0.9 ± 0.7 | 89.8 ± 7.9 |
| 10 | 35.0 ± 2.7 (51.3 ± 1.3) | 4.0 ± 3.3 | 31.7 ± 5.6 | |
| 1.0 | 5 | 41.3 ± 4.6 (51.9 ± 4.1) | 1.6 ± 0.6 | 20.4 ± 8.1 |
| 10 | 47.5 ± 3.8 (48.3 ± 3.7) | 4.3 ± 0.1 | 1.7 ± 0.8 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nomura, T.; Mizuno, H.; Minami, E.; Kawamoto, H. Fast Pyrolysis of Cellulose by Infrared Heating. Energies 2021, 14, 1842. https://doi.org/10.3390/en14071842
Nomura T, Mizuno H, Minami E, Kawamoto H. Fast Pyrolysis of Cellulose by Infrared Heating. Energies. 2021; 14(7):1842. https://doi.org/10.3390/en14071842
Chicago/Turabian StyleNomura, Takashi, Hinano Mizuno, Eiji Minami, and Haruo Kawamoto. 2021. "Fast Pyrolysis of Cellulose by Infrared Heating" Energies 14, no. 7: 1842. https://doi.org/10.3390/en14071842