Bio-Based Carbon Materials from Potato Waste as Electrode Materials in Supercapacitors


Abstract
:1. Introduction
- high electric conductivity
- high specific surface (>2000 m2 g−1)
- high corrosion resistance
- good thermal stability
- homogeneous pore structure
- high material purity
- low cost
- microporous carbon structure positively influences the electric conductivity [4]
2. Materials and Methods
2.1. Biomass
2.2. Hydrothermal Carbonization
2.3. Pyrolysis
2.4. Elemental Analysis
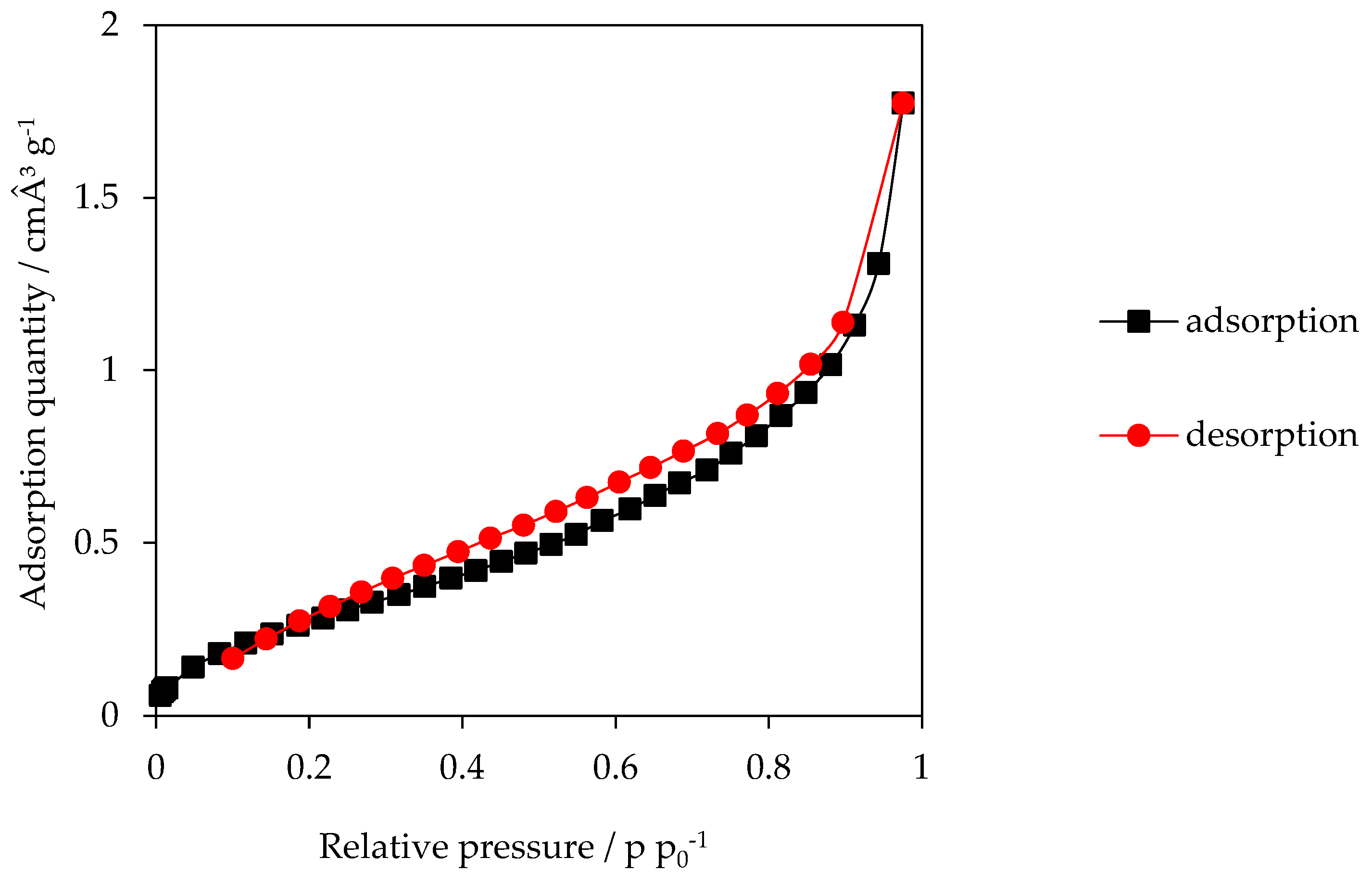
2.5. Specific Surface Area (BET)
- q—loading of the adsorbent (mass of adsorbate relative to mass of adsorbent)
- K—adsorption coefficient
- qmax—maximum concentration of adsorbate in a layer on the surface of the carbon
- Ceq—concentration of adsorbate in solution
- Csat—solubility of adsorbate
2.6. Electric Conductivity (EC)
- EC—electrical conductivity [S m−1]
- H—height of the carbon in the measuring cylinder [cm]
- R—measured resistance of carbon [Ω]
- A—area of the measuring cylinder (7.9 × 10−5 m2)
- ρ—density of the coal [kg m−3]
- m—mass of coal [g]
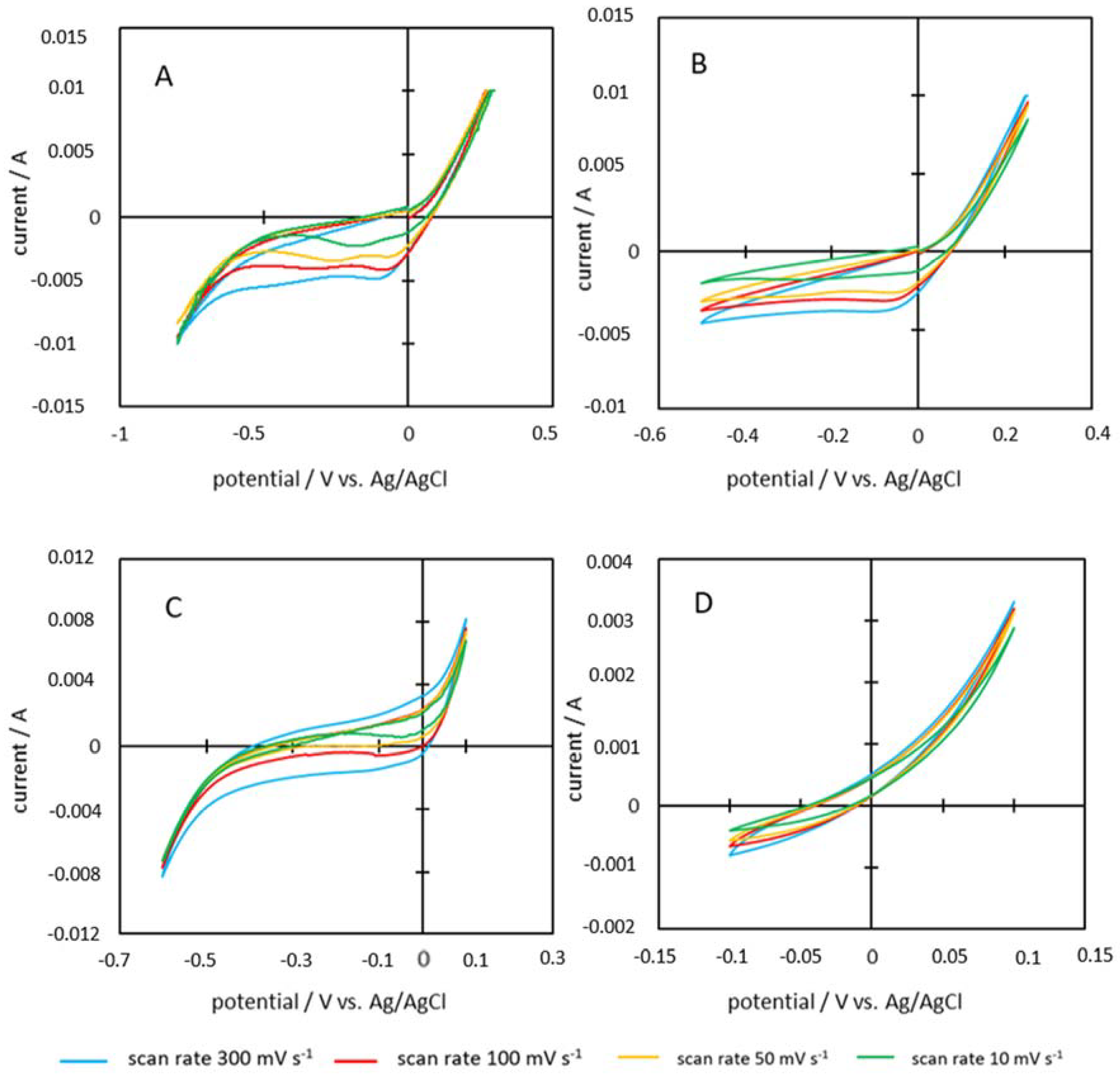
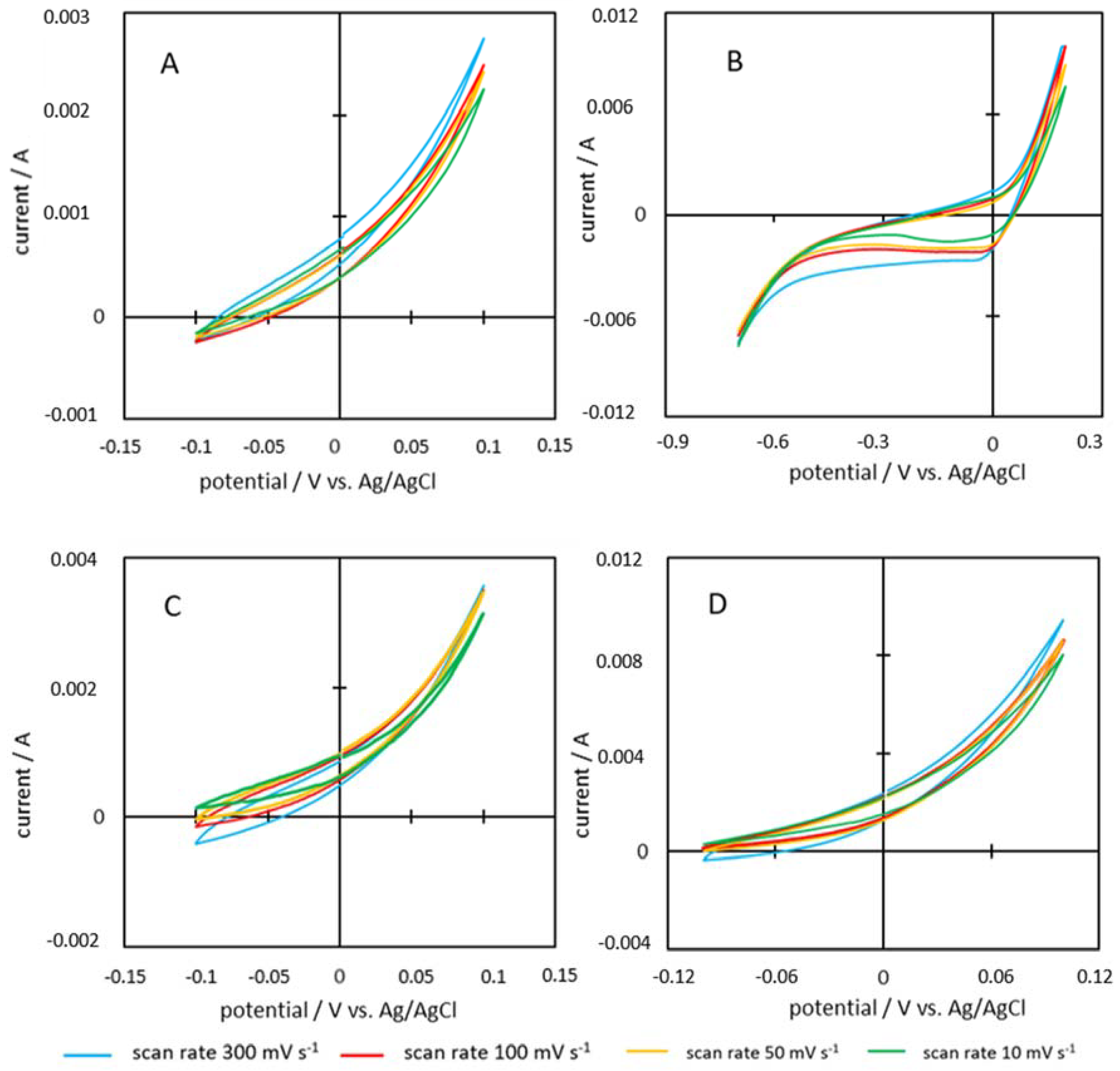
2.7. Cyclic Voltammetry
- m—masse of carbon on the working electrode [g]
- V—scan rate [Vs−1]
- ΔV—voltage range [V]
- —current [A]
2.8. Ash and Oxygen Content
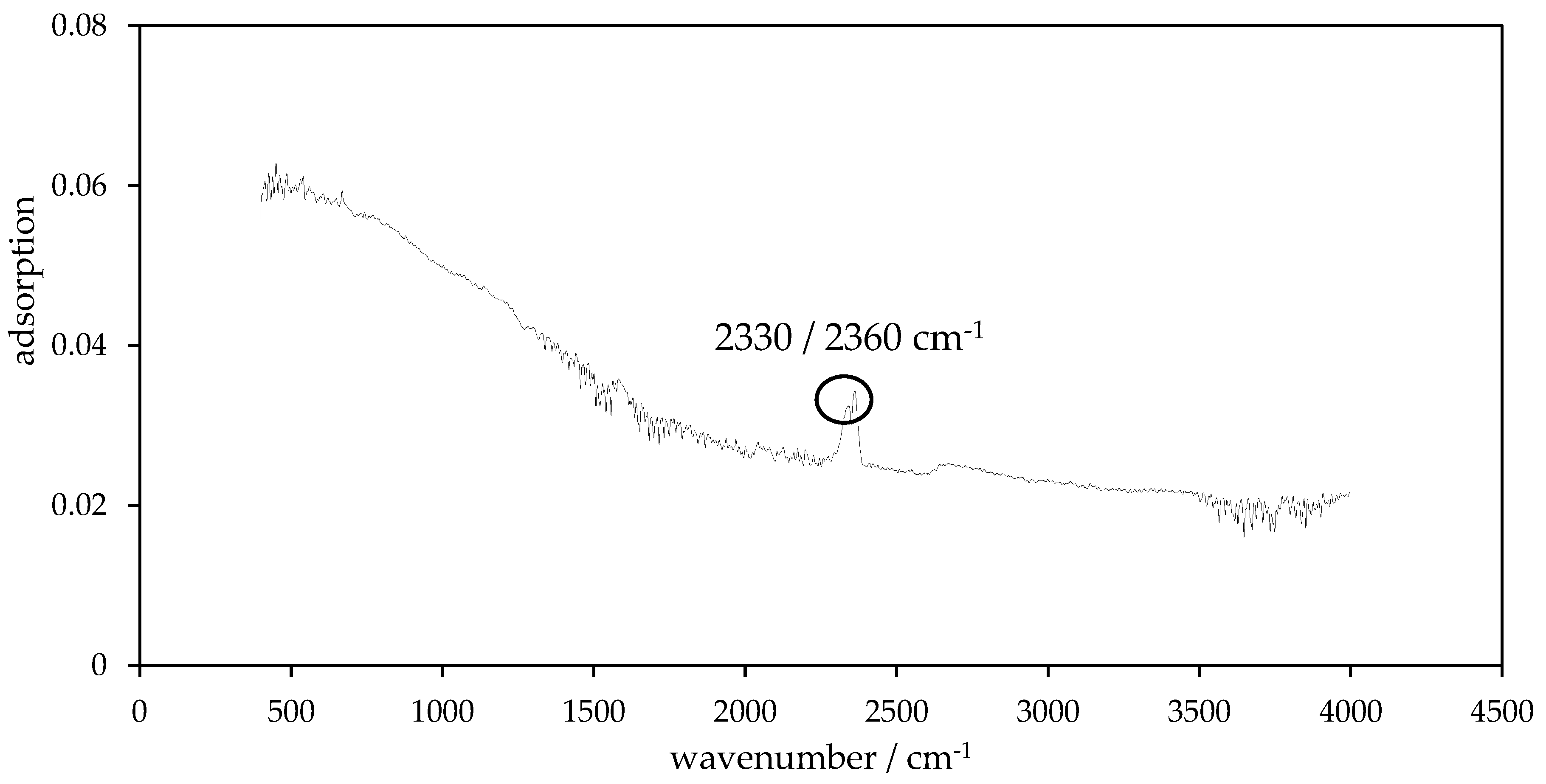
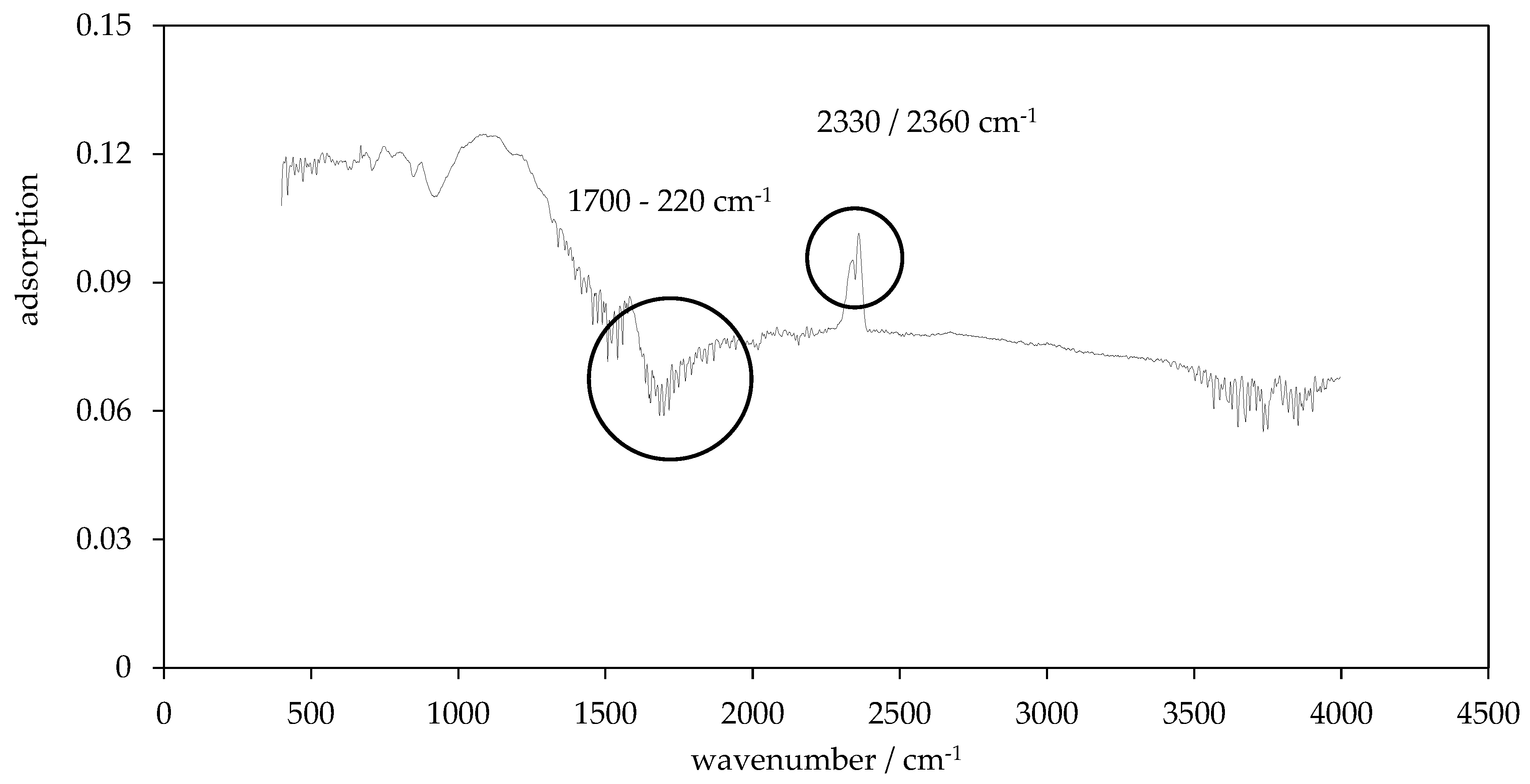
2.9. Fourier Transform Infrared Spectroscopy (FT-IR)
2.10. Experimental Error
3. Results and Discussions
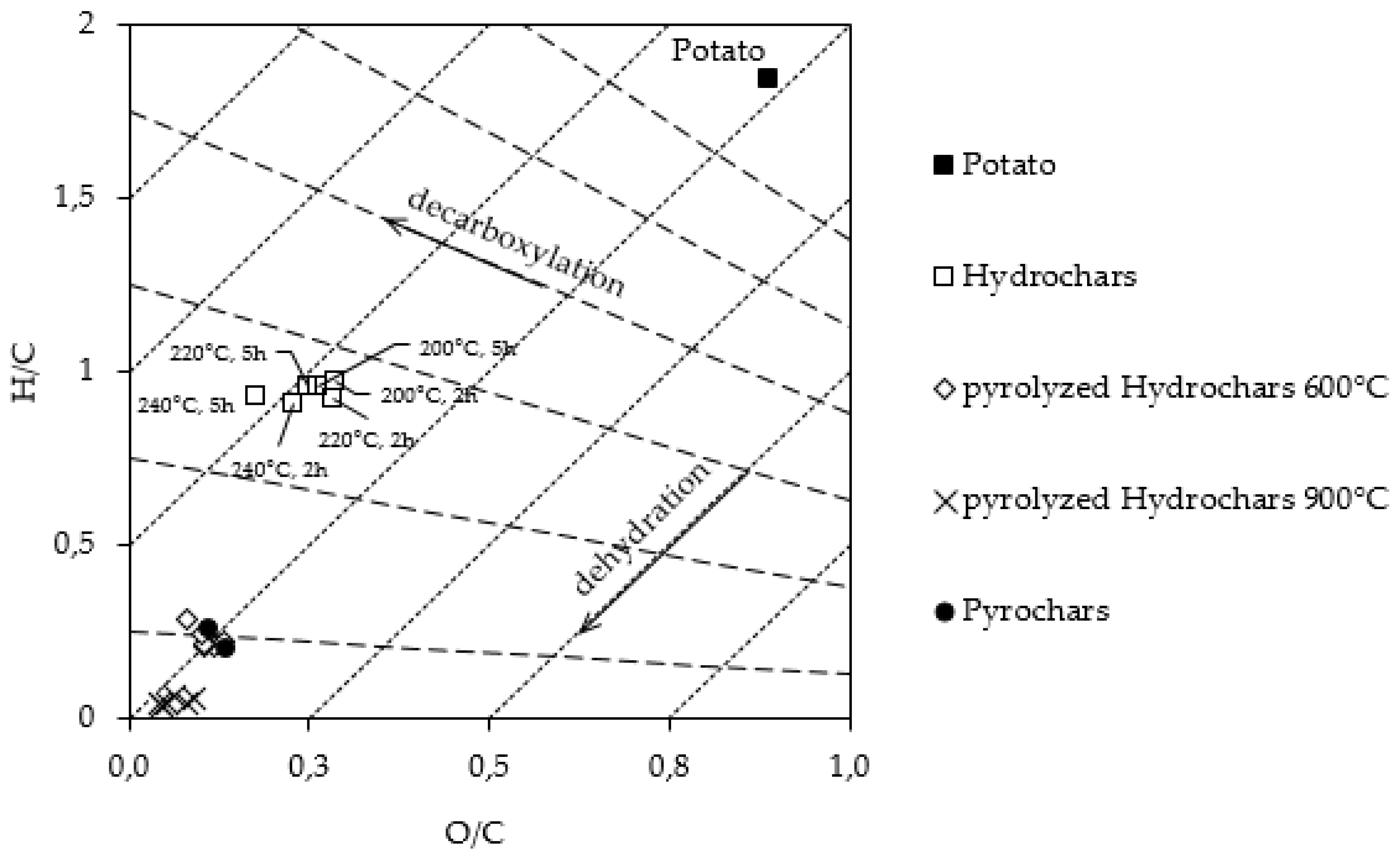
3.1. Thermochemical Conversion
3.1.1. Hydrochars
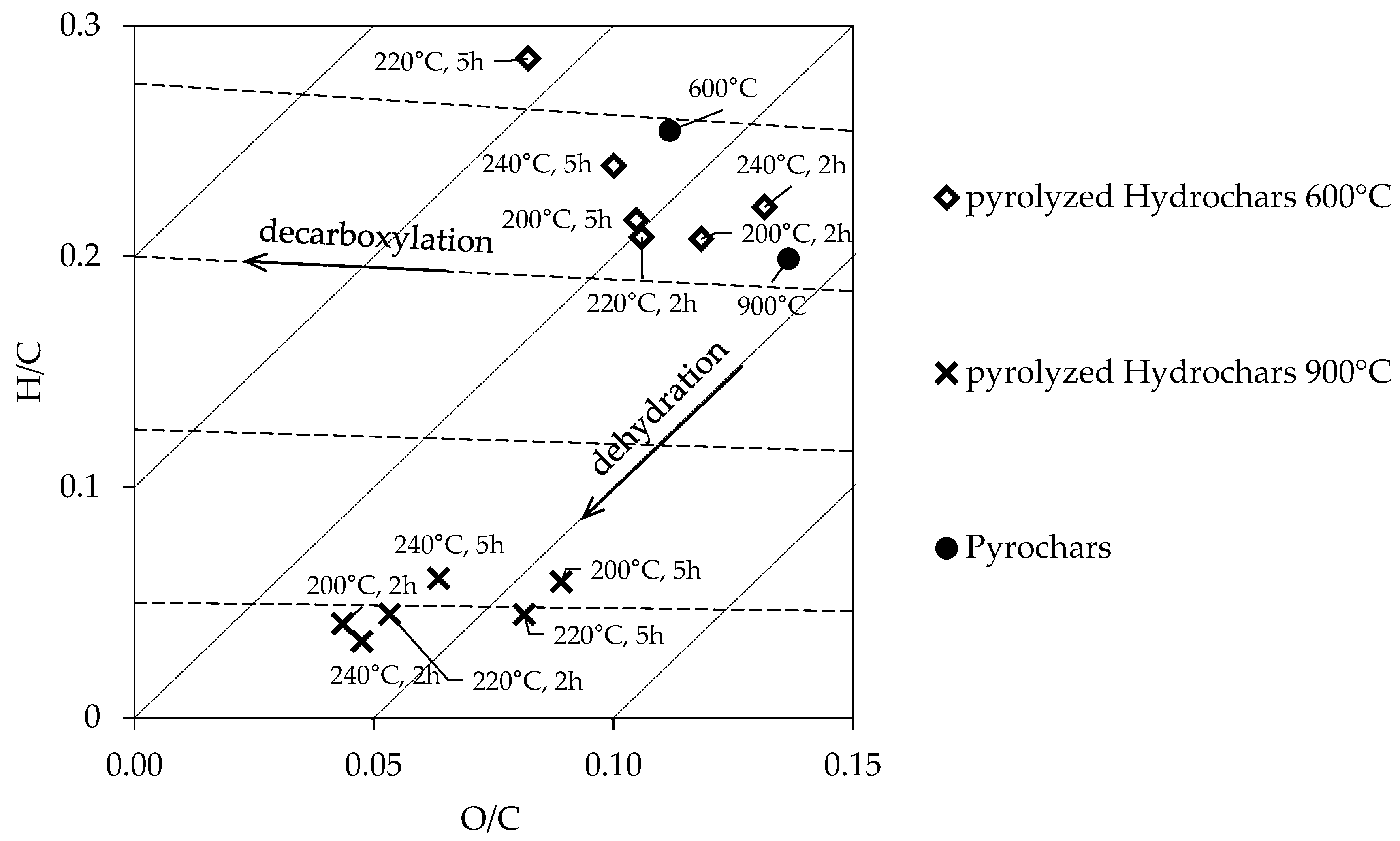
3.1.2. Pyrochars and Pyrolyzed Hydrochars
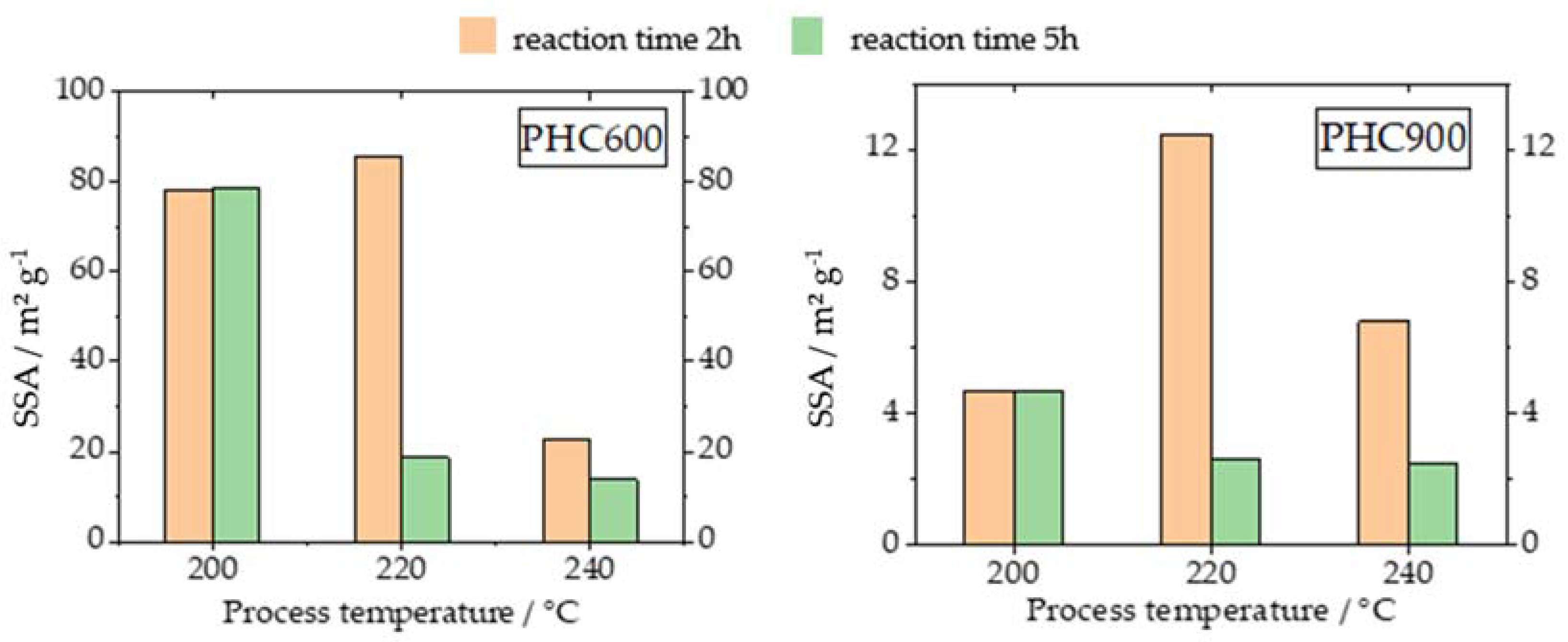
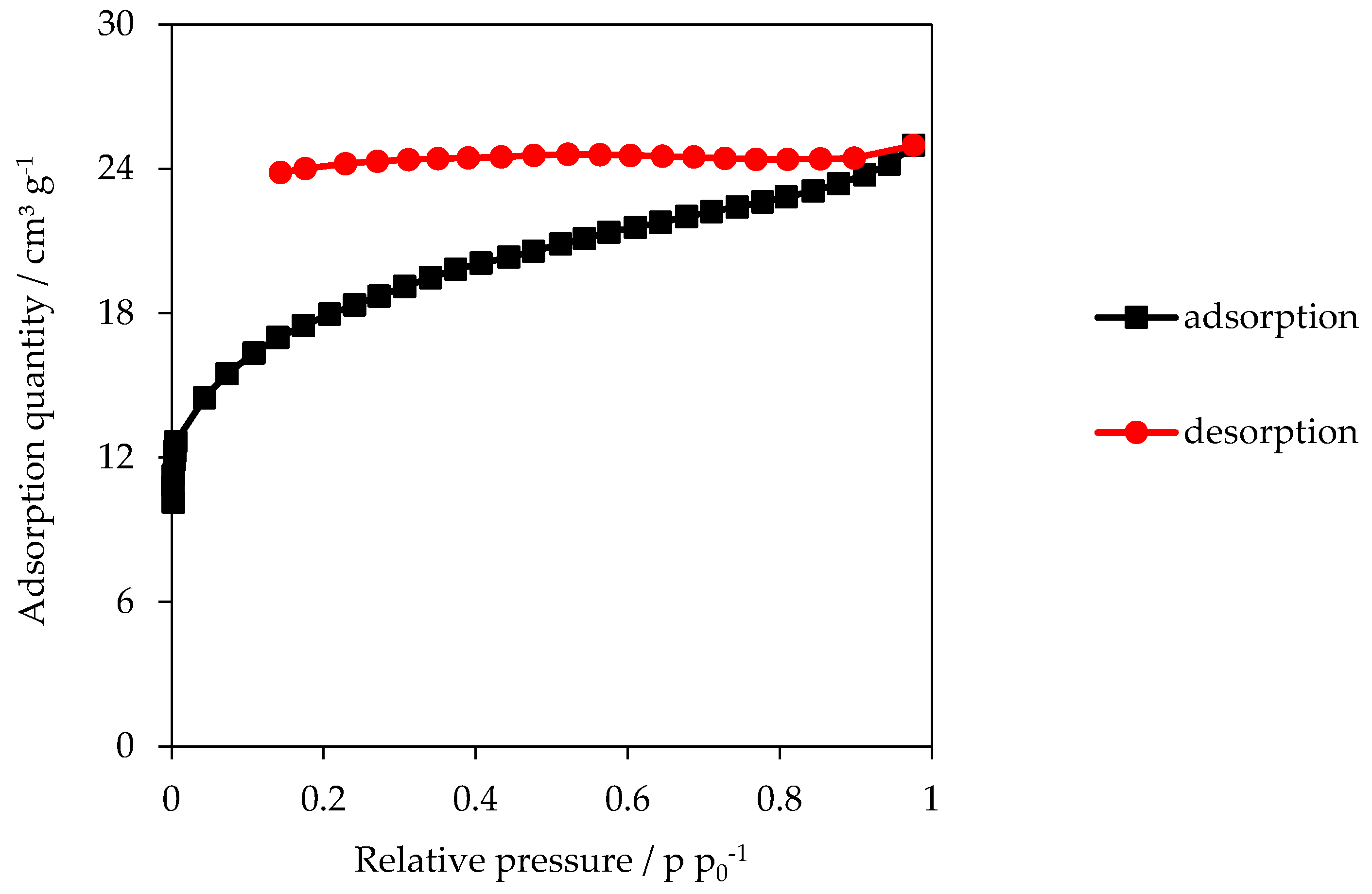
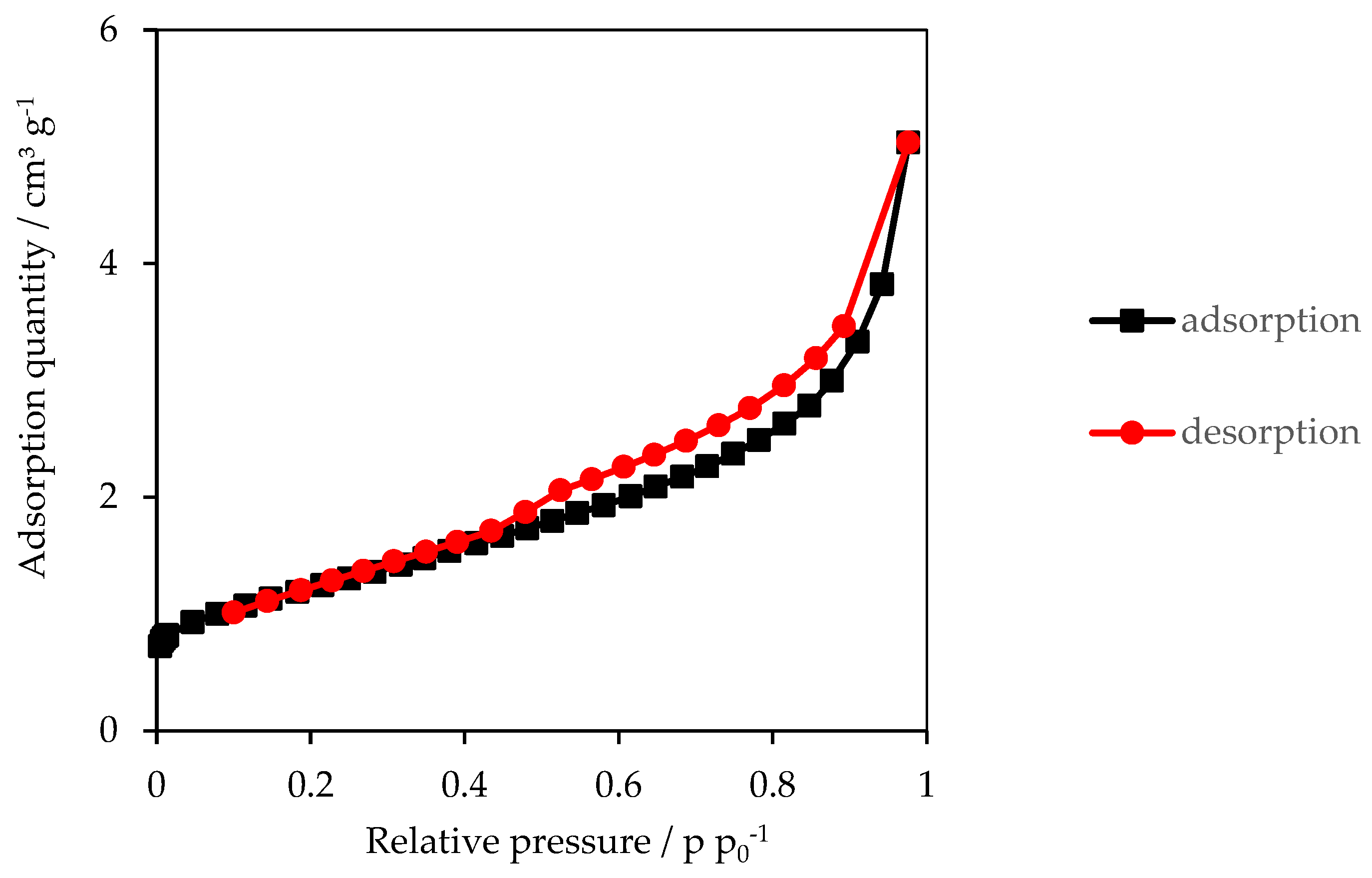
3.2. Specific Surface Area (SSA)
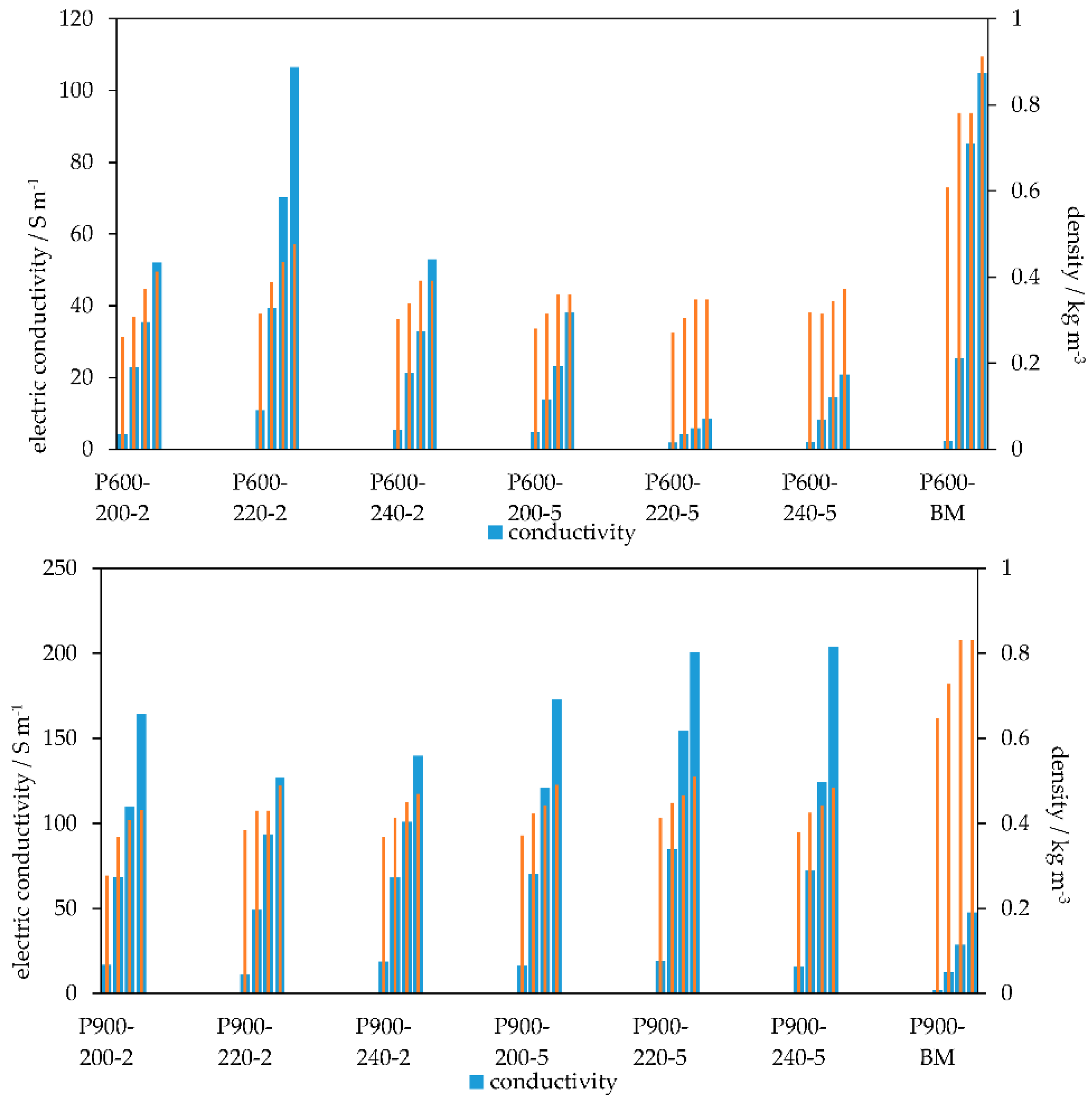
3.3. Electric Conductivity and Bulk Density
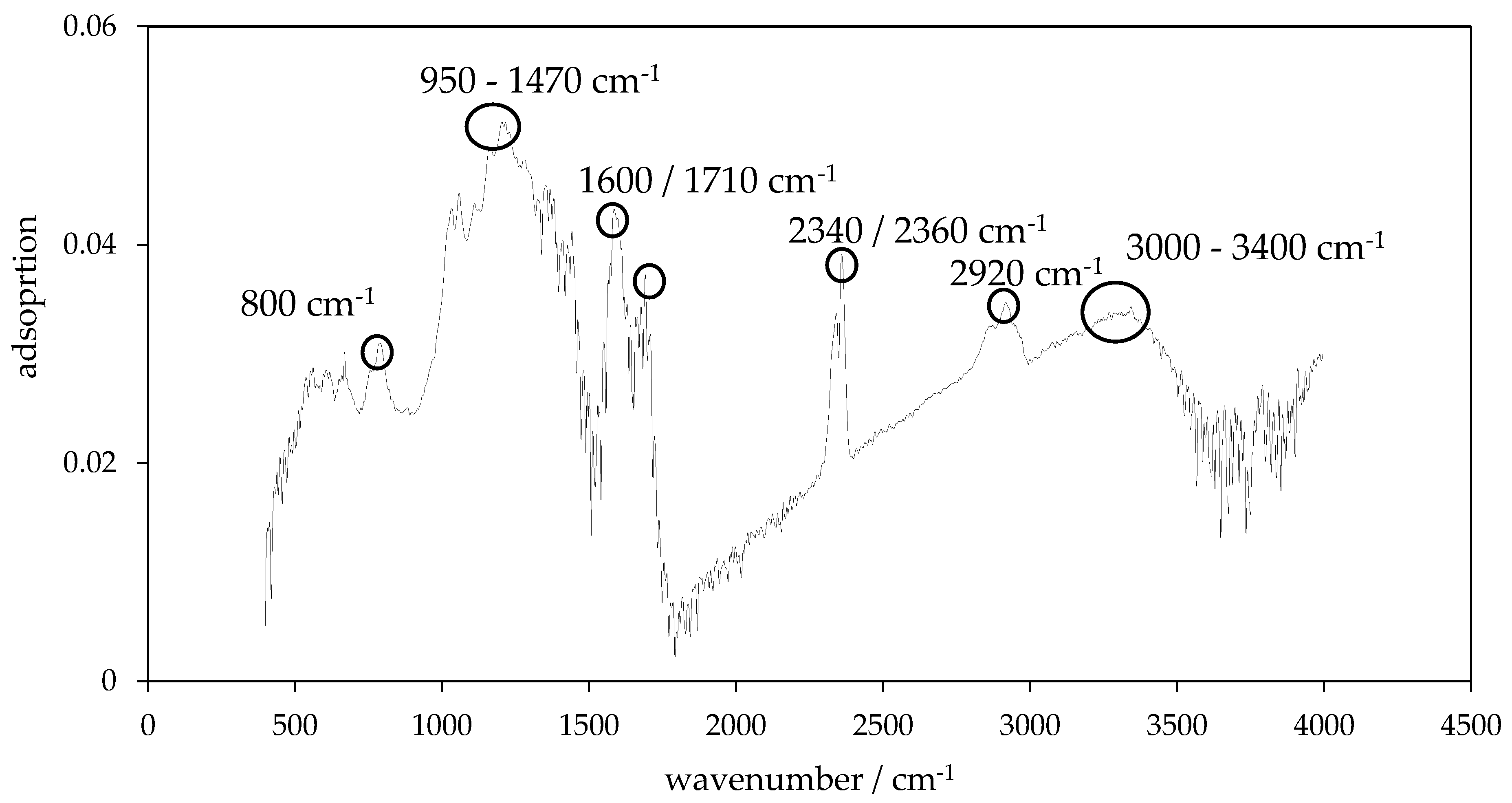
3.4. Surface Functionality
3.5. Capacitance
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HTC Yield % | ||||
|---|---|---|---|---|
| Sample | Rep1 | Rep2 | Mean | STD |
| HTC-200-2 | 42.1 | 40.7 | 41.4 | 0.700 |
| HTC-220-2 | 41.1 | 41.7 | 41.4 | 0.300 |
| HTC-240-2 | 37.4 | 37.6 | 37.5 | 0.120 |
| HTC-200-5 | 42.8 | 43.1 | 43.0 | 0.140 |
| HTC-220-5 | 38.6 | 38.5 | 38.6 | 0.080 |
| HTC-240-5 | 36.6 | 36.6 | 36.6 | 0.020 |
| Average Standard Deviation | 0.227 |
| Nitrogen % | Carbon % | Hydrogen % | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Rep1 | Rep2 | Mean | STD | Rep1 | Rep2 | Mean | STD | Rep1 | Rep2 | Mean | STD |
| Potatoe | 1.89 | 1.87 | 1.88 | 0.012 | 40.05 | 40.01 | 40.03 | 0.021 | 6.20 | 6.07 | 6.13 | 0.062 |
| HTC-200-2 | 3.34 | 3.45 | 3.39 | 0.059 | 63.33 | 64.96 | 64.15 | 0.816 | 5.16 | 5.18 | 5.17 | 0.010 |
| HTC220-2 | 3.37 | 3.40 | 3.39 | 0.015 | 64.32 | 64.12 | 64.22 | 0.100 | 4.84 | 5.00 | 4.92 | 0.079 |
| HTC240-2 | 3.51 | 3.53 | 3.52 | 0.007 | 67.80 | 67.90 | 67.85 | 0.049 | 5.07 | 5.17 | 5.12 | 0.051 |
| HTC-200-5 | 3.31 | 3.30 | 3.30 | 0.004 | 65.12 | 65.16 | 65.14 | 0.020 | 5.29 | 5.07 | 5.18 | 0.110 |
| HTC-220-5 | 3.43 | 3.48 | 3.45 | 0.028 | 65.96 | 67.33 | 66.65 | 0.687 | 5.33 | 5.27 | 5.30 | 0.031 |
| HTC-240-5 | 3.71 | 3.71 | 3.71 | 0.002 | 72.07 | 71.63 | 71.85 | 0.221 | 5.55 | 5.54 | 5.55 | 0.004 |
| BM-P600 | 2.69 | 2.66 | 2.67 | 0.015 | 69.21 | 69.04 | 69.13 | 0.084 | 1.54 | 1.40 | 1.47 | 0.069 |
| BM-P900 | 2.50 | 2.48 | 2.49 | 0.011 | 67.11 | 66.87 | 66.99 | 0.120 | 1.00 | 1.22 | 1.11 | 0.111 |
| P600-200-2 | 3.85 | 3.88 | 3.87 | 0.016 | 75.38 | 77.68 | 76.53 | 1.149 | 1.26 | 1.39 | 1.32 | 0.067 |
| P600-220-2 | 4.02 | 4.01 | 4.02 | 0.006 | 77.93 | 78.81 | 78.37 | 0.439 | 1.23 | 1.49 | 1.36 | 0.127 |
| P600-240-2 | 3.62 | 3.99 | 3.80 | 0.186 | 73.06 | 81.23 | 77.14 | 4.087 | 1.30 | 1.54 | 1.42 | 0.120 |
| P600-200-5 | 3.95 | 3.99 | 3.97 | 0.022 | 78.19 | 77.59 | 77.89 | 0.300 | 1.31 | 1.49 | 1.40 | 0.090 |
| P600-220-5 | 3.69 | 3.89 | 3.79 | 0.104 | 81.10 | 81.04 | 81.07 | 0.031 | 2.04 | 1.82 | 1.93 | 0.107 |
| P600-240-5 | 4.03 | 4.01 | 4.02 | 0.008 | 80.67 | 80.00 | 80.33 | 0.334 | 1.58 | 1.63 | 1.60 | 0.025 |
| P900-200-2 | 3.48 | 3.50 | 3.49 | 0.011 | 85.22 | 85.47 | 85.34 | 0.122 | 0.37 | 0.21 | 0.29 | 0.084 |
| P900-220-2 | 3.65 | 3.78 | 3.71 | 0.065 | 84.38 | 86.25 | 85.32 | 0.935 | 0.37 | 0.27 | 0.32 | 0.051 |
| P900-240-2 | 3.75 | 3.74 | 3.75 | 0.004 | 86.96 | 87.00 | 86.98 | 0.018 | 0.29 | 0.20 | 0.24 | 0.045 |
| P900-200-5 | 3.39 | 3.36 | 3.38 | 0.015 | 81.15 | 80.79 | 80.97 | 0.179 | 0.44 | 0.36 | 0.40 | 0.038 |
| P900-220-5 | 3.53 | 3.51 | 3.52 | 0.008 | 82.64 | 82.75 | 82.69 | 0.053 | 0.31 | 0.30 | 0.31 | 0.005 |
| P900-240-5 | 3.51 | 3.53 | 3.52 | 0.010 | 83.64 | 83.91 | 83.77 | 0.134 | 0.45 | 0.39 | 0.42 | 0.032 |
| Average Standard Deviation | 0.029 | 0.471 | 0.063 |
References
- Tititrici, M.-M. Sustainable Carbon Materials from Hydrothermal Processes; John Wiley & Sons, Ltd.: London, UK, 2013. [Google Scholar]
- Reif, K.; Noreikat, K.-E.; Borgeest, K. Kraftfahrzeug-Hybridantriebe; Vieweg+Teubner Verlag: Wiesbaden, Germany, 2012. [Google Scholar]
- Mahon, P.J.; Drummond, C.J. Essay: Supercapacitors—Nanostructured materials and nanoscale processes contributing to the next mobile generation. Aust. J. Chem. 2001, 54, 473. [Google Scholar] [CrossRef]
- Pandolfo, A.G.; Hollenkamp, A. Carbon properties and their role in supercapacitors. J. Power Sources 2006, 157, 11–27. [Google Scholar] [CrossRef]
- Shukla, A.K.; Sampath, S.; Vijayamohanan, K. Electrochemical supercapacitors: Energy storage beyond batteries. Curr. Sci. 2000, 79, 1656–1661. [Google Scholar]
- Frackowiak, E. Electrode materials with pseudocapacitive properties. In Supercapacitors; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 207–237. [Google Scholar]
- Frackowiak, E.; Beguin, F. Carbon materials for the electrochemical storage of energy in capacitors. Carbon 2001, 39, 937–950. [Google Scholar] [CrossRef]
- Hsieh, C.-T.; Teng, H. Influence of oxygen treatment on electric double-layer capacitance of activated carbon fabrics. Carbon 2002, 40, 667–674. [Google Scholar] [CrossRef]
- Jung, D.; Zimmermann, M.; Kruse, A. Hydrothermal carbonization of fructose: Growth mechanism and kinetic model. ACS Sustain. Chem. Eng. 2018, 6, 13877–13887. [Google Scholar] [CrossRef]
- Inyang, M.; Gao, B.; Pullammanappallil, P.; Ding, W.; Zimmerman, A.R. Biochar from anaerobically digested sugarcane bagasse. Bioresour. Technol. 2010, 101, 8868–8872. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ma, X.; Peng, X.; Lin, Y.; Yao, Z. Conversion of sweet potato waste to solid fuel via hydrothermal carbonization. Bioresour. Technol. 2018, 249, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Suwelack, K.; Wüst, D.; Zeller, M.; Kruse, A.; Krümpel, J. Hydrothermal carbonization of wheat straw—Prediction of product mass yields and degree of carbonization by severity parameter. Biomass Convers. Biorefin. 2015, 6, 347–354. [Google Scholar] [CrossRef]
- Hoffmann, V.; Correa, C.R.; Sautter, D.; Maringolo, E.; Kruse, A.; Hoffman, V. Study of the electrical conductivity of biobased carbonaceous powder materials under moderate pressure for the application as electrode materials in energy storage technologies. GCB Bioenergy 2018, 11, 230–248. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, V.; Jung, D.; Zimmermann, J.; Correa, C.R.; Elleuch, A.; Halouani, K.; Kruse, A. Conductive carbon materials from the hydrothermal carbonization of vineyard residues for the application in electrochemical double-layer capacitors (EDLCs) and direct carbon fuel cells (DCFCs). Materials 2019, 12, 1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.-W.; Jung, D.-W.; Jeong, J.-H.; Oh, E.-S. Effect of pyrolysis temperature on carbon obtained from green tea biomass for superior lithium ion battery anodes. Chem. Eng. J. 2014, 254, 597–604. [Google Scholar] [CrossRef]
- Lozano-Castello, D.; Marco-Lozar, J.P.; Bleda-Martínez, M.J.; Montilla, F.; Morallòn, E.; Linares-Solano, A.; Cazorla-Amorós, D. Relevance of porosity and surface chemistry of superactivated carbons in capacitors. Carbon N. Y. 2013, 57, 539. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Gonzalez, J.; Macias-Garcia, A.; Franco, M.A.; Gómez-Serrano, V. Electrical conductivity of carbon blacks under compression. Carbon 2005, 43, 741–747. [Google Scholar] [CrossRef]
- Hoffmann, V.; Alhnidi, M.J.; Correa, C.R.; Kruse, A. In-Situ functionalizing of HTC chars for the production of biobased electrode materials for electromobility. In Proceedings of the 2nd International Symposium on Hydrothermal Carbonization, Berlin, Germany, 14–16 May 2019; p. 160. [Google Scholar]
- Priyanga, K.; Reji, A.; Bhagat, J.k.; Anbuselvi, S. Production of organic manure from potato peel waste. Int. J. Chem.Tech. Res. 2016, 9, 845–847. [Google Scholar]
- Brendel, F. Kleine Makel-Grosse Folgen: Nahrungsmittelverschwendung am Beispiel Kartoffel; WWF Deutschland: Frankfurt am Main, Germany, 2017. [Google Scholar]
- Willersinn, C.; Mack, G.; Mouron, P.; Keiser, A.; Siegrist, M. Quantity and quality of food losses along the Swiss potato supply chain: Stepwise investigation and the influence of quality standards on losses. Waste Manag. 2015, 46, 120–132. [Google Scholar] [CrossRef]
- Bächtle, C.; Winkler, P.; Stellbrink, B. Eine frage der richtigen stärke. Chem. Unserer Zeit 2011, 45, 250–255. [Google Scholar] [CrossRef]
- Bleda-Martínez, M.; Maciá-Agulló, J.; Lozano-Castello, D.; Morallón, E.; Cazorla-Amorós, D.; Linares-Solano, A. Role of surface chemistry on electric double layer capacitance of carbon materials. Carbon 2005, 43, 2677–2684. [Google Scholar] [CrossRef] [Green Version]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Holm, R. Elektrische Kontakte/Electric Contacts Handbook; Springer: Berlin/Heidelberg, Germany, 1958. [Google Scholar]
- Celzard, A.; Marêché, J.; Payot, F.; Furdin, G. Electrical conductivity of carbonaceous powders. Carbon 2002, 40, 2801–2815. [Google Scholar] [CrossRef]
- Heinze, J. Cyclovoltammetrie—Die ”Spektroskopie” des Elektrochemikers. Angew. Chem. 1984, 96, 823–840. [Google Scholar] [CrossRef]
- Pröbstle, H.; Wiener, M.; Fricke, J. Carbon aerogels for electrochemical double layer capacitors. J. Porous Mater. 2003, 10, 213–222. [Google Scholar] [CrossRef]
- Choi, J.-H. Fabrication of a carbon electrode using activated carbon powder and application to the capacitive deionization process. Sep. Purif. Technol. 2010, 70, 362–366. [Google Scholar] [CrossRef]
- Falco, C.; Baccile, N.; Titirici, M.-M. Morphological and structural differences between glucose, cellulose and lignocellulosic biomass derived hydrothermal carbons. Green Chem. 2011, 13, 3273. [Google Scholar] [CrossRef] [Green Version]
- Karayıldırım, T.; Sınağ, A.; Kruse, A. Char and coke formation as unwanted side reaction of the hydrothermal biomass gasification. Chem. Eng. Technol. 2008, 31, 1561–1568. [Google Scholar] [CrossRef]
- Correa, C.R.; Kruse, A. Biobased functional carbon materials: Production, characterization, and applications—A review. Materials 2018, 11, 1568. [Google Scholar] [CrossRef] [Green Version]
- Antal, M.J.; Mok, W.S.; Richards, G.N. Mechanism of formation of 5-(hydroxymethyl)-2-furaldehyde from d-fructose and sucrose. Carbohydr. Res. 1990, 199, 91–109. [Google Scholar] [CrossRef]
- Kruse, A.; Badoux, F.; Grandl, R.; Wüst, D. Hydrothermal carbonization: 2. kinetics of draff conversion. Chem. Ing. Tech. 2012, 84, 509–512. [Google Scholar] [CrossRef]
- Yu, Q.-Z.; Brage, C.; Chen, G.-X.; Sjöström, K. The fate of fuel-nitrogen during gasification of biomass in a pressurised fluidised bed gasifier. Fuel 2007, 86, 611–618. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Barroso-Bogeat, A.; Franco, M.A.; Fernández-González, C.; Macias-Garcia, A.; Gómez-Serrano, V. Electrical conductivity of activated carbon–metal oxide nanocomposites under compression: A comparison study. Phys. Chem. Chem. Phys. 2014, 16, 25161–25175. [Google Scholar] [CrossRef] [PubMed]
- Pakuła, M.; Swiatkowski, A.; Biniak, S. Electrochemical behaviour of modified activated carbons in aqueous and nonaqueous solutions. J. Appl. Electrochem. 1995, 25, 1038–1044. [Google Scholar] [CrossRef]
- Sevilla, M.; Fuertes, A.B. The production of carbon materials by hydrothermal carbonization of cellulose. Carbon 2009, 47, 2281–2289. [Google Scholar] [CrossRef] [Green Version]
- Garlapalli, R.K.; Wirth, B.; Reza, M.T. Pyrolysis of hydrochar from digestate: Effect of hydrothermal carbonization and pyrolysis temperatures on pyrochar formation. Bioresour. Technol. 2016, 220, 168–174. [Google Scholar] [CrossRef]
- Sing, K.S.W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Rouquerol, F.; Rouquerol, J.; Sing, K. Adsorption by Powders and Porous Solids: Principles, Methodology and Applications; Academic Press: London, 1999. [Google Scholar]
- Thommes, M. Physical adsorption characterization of nanoporous materials. Chem. Ing. Tech. 2010, 82, 1059–1073. [Google Scholar] [CrossRef]
- Dieguez-Alonso, A.; Funke, A.; Anca-Couce, A.; Rombola’, A.G.; Ojeda, G.; Bachmann, J.; Behrendt, F. Towards biochar and hydrochar engineering—Influence of process conditions on surface physical and chemical properties, thermal stability, nutrient availability, toxicity and wettability. Energies 2018, 11, 496. [Google Scholar] [CrossRef] [Green Version]
- Lua, A.C.; Yang, T. Effect of activation temperature on the textural and chemical properties ofpotassium hydroxide activated carbon prepared from pistachio-nut shell. J. Colloid Interface Sci. 2004, 274, 594–601. [Google Scholar] [CrossRef]
- Sun, X.; Li, Y. Colloidal carbon spheres and their core/shell structures with noble-metal nanoparticles. Angew. Chem. 2004, 43, 597–601. [Google Scholar] [CrossRef]
- Günzler, H.; Gremlich, H.U. IR-Spektroskopie: Eine Einführung; Wiley: Weinheim, Germany, 2012. [Google Scholar]
- Araujo-Andrade, C.; Ruiz, F.; Martínez-Mendoza, J.; Terrones, H. Infrared and Raman spectra, conformational stability, ab initio calculations of structure, and vibrational assignment of α and β glucose. J. Mol. Struct. THEOCHEM 2005, 714, 143–146. [Google Scholar] [CrossRef]
- Ibarra, J.; Muñoz, E.; Moliner, R. FTIR study of the evolution of coal structure during the coalification process. Org. Geochem. 1996, 24, 725–735. [Google Scholar] [CrossRef]
- Biniak, S.; Swiatkowski, A.; Pakula, M.; Radovic, R. Chemistry and Physics of Carbon; Marcel Dekker: New York, NY, USA; Basel, Switzerland, 2017; Volume 27. [Google Scholar]
- Stiny, L. Aktive Elektronische Bauelemente Aufbau, Struktur, Wirkungsweise, Eigenschaften und Praktischer Einsatz Diskreter und Integrierter Halbleiter-Bauteile; Springer: Heidelberg, Germany, 2016. [Google Scholar]
- Bandosz, T.; Ania, C. Chapter 4 surface chemistry of activated carbons and its characterization. In Interface Science and Technology; Academic Press Inc.: San Diego, CA, USA, 2006; Volume 7, pp. 159–229. [Google Scholar]
- Kötz, R.; Carlen, M. Principles and applications of electrochemical capacitors. Electrochim. Acta 2000, 45, 2483–2498. [Google Scholar] [CrossRef]
- Lozano-Castello, D.; Cazorla-Amorós, D.; Linares-Solano, A.; Shiraishi, S.; Kurihara, H.; Oya, A. Influence of pore structure and surface chemistry on electric double layer capacitance in non-aqueous electrolyte. Carbon 2003, 41, 1765–1775. [Google Scholar] [CrossRef]
- Salitra, G.; Soffer, A.; Eliad, L.; Cohen, Y.; Aurbach, D. Carbon electrodes for double-layer capacitors I. Relations between ion and pore dimensions. J. Electrochem. Soc. 2000, 147, 2486–2493. [Google Scholar] [CrossRef]
- Qu, D.; Shi, H. Studies of activated carbons used in double-layer capacitors. J. Power Sources 1998, 74, 99–107. [Google Scholar] [CrossRef]
- Iro, Z.S. A brief review on electrode materials for supercapacitor. Int. J. Electrochem. Sci. 2016, 11, 10628–10643. [Google Scholar] [CrossRef]
- Wei, L.; Sevilla, M.; Fuertes, A.B.; Mokaya, R.; Yushin, G. Hydrothermal carbonization of abundant renewable natural organic chemicals for high-performance supercapacitor electrodes. Adv. Energy Mater. 2011, 1, 356–361. [Google Scholar] [CrossRef] [Green Version]
- Hulicova-Jurcakova, D.; Kodama, M.; Shiraishi, S.; Hatori, H.; Zhu, Z.; Lu, G. Nitrogen-enriched nonporous carbon electrodes with extraordinary supercapacitance. Adv. Funct. Mater. 2009, 19, 1800–1809. [Google Scholar] [CrossRef]
- Yan, L.; Yu, J.; Houston, J.; Flores, N.; Luo, H. Biomass derived porous nitrogen doped carbon for electrochemical devices. Green Energy Environ. 2017, 2, 84–99. [Google Scholar] [CrossRef]
- Okajima, K.; Ohta, K.; Sudoh, M. Capacitance behavior of activated carbon fibers with oxygen-plasma treatment. Electrochim. Acta 2005, 50, 2227–2231. [Google Scholar] [CrossRef]
- Raymundo-Piñero, E.; Čadek, M.; Beguin, F. Tuning carbon materials for supercapacitors by direct pyrolysis of seaweeds. Adv. Funct. Mater. 2009, 19, 1032–1039. [Google Scholar] [CrossRef]
- Rafiee, J.; Mi, X.; Gullapalli, H.; Thomas, A.V.; Yavari, F.; Shi, Y.; Ajayan, P.M.; Koratkar, N.A. Wetting transparency of graphene. Nat. Mater. 2012, 11, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Benedix, R. Redoxreaktionen—Grundlagen der elektrochemie. In Bauchemie; Vieweg+Teubner: Wiesbaden, Germany, 2008; pp. 206–230. [Google Scholar]













| Sample | HTC | Pyrolysis | Yield | C 1 | H | N | O | Carbon Yield 2 | Carbon Yield 3 | Ash | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| °C | h | °C | wt.% | wt.% | wt.% | wt.% | wt.% | wt.% | wt.% | wt.% | |
| Potato | 40.0 (42) | 6.1 | 1.9 | 47.4 | 4.4 | ||||||
| HTC-200-2 | 200 | 2 | - | 41.4 | 64.1 (66) | 5.2 | 3.4 | 24.5 | 66.3 | 2.7 | |
| HTC-220-2 | 220 | 2 | - | 41.4 | 64.2 (66) | 4.9 | 3.4 | 24.3 | 66.4 | 3.1 | |
| HTC-240-2 | 240 | 2 | - | 37.5 | 67.9 (70) | 5.1 | 3.5 | 20.8 | 63.5 | 2.7 | |
| HTC-200-5 | 200 | 5 | - | 43.0 | 65.1 (67) | 5.2 | 3.3 | 22.9 | 69.9 | 3.4 | |
| HTC-220-5 | 220 | 5 | - | 38.6 | 66.6 (68) | 5.3 | 3.5 | 22.0 | 64.2 | 2.5 | |
| HTC-240-5 | 240 | 5 | - | 36.6 | 71.9 (73) | 5.5 | 3.7 | 16.8 | 65.7 | 2.1 | |
| P600-200-2 | 200 | 2 | 600 | 50.4 | 76.5 (82) | 1.3 | 3.9 | 12.1 | 60.2 | 39.9 | 6.1 |
| P600-220-2 | 220 | 2 | 600 | 51.0 | 78.4 (83) | 1.4 | 4.0 | 11.1 | 62.2 | 41.3 | 5.1 |
| P600-240-2 | 240 | 2 | 600 | 60.6 | 77.1 (80) | 1.4 | 3.8 | 13.5 | 68.9 | 43.7 | 4.1 |
| P600-200-5 | 200 | 5 | 600 | 51.5 | 77.9 (83) | 1.4 | 4.0 | 10.9 | 61.6 | 43.1 | 5.8 |
| P600-220-5 | 220 | 5 | 600 | 59.5 | 81.1 (85) | 1.9 | 3.8 | 8.9 | 72.4 | 46.5 | 4.3 |
| P600-240-5 | 240 | 5 | 600 | 60.6 | 80.3 (83) | 1.6 | 4.0 | 10.7 | 67.8 | 44.5 | 3.3 |
| P900-200-2 | 200 | 2 | 900 | 48.7 | 85.3 (91) | 0.3 | 3.5 | 4.9 | 64.7 | 42.9 | 5.8 |
| P900-220-2 | 220 | 2 | 900 | 50.7 | 85.3 (89) | 0.3 | 3.7 | 6.1 | 67.4 | 44.8 | 4.5 |
| P900-240-2 | 240 | 2 | 900 | 53.4 | 87.0 (90) | 0.2 | 3.7 | 5.5 | 68.5 | 43.5 | 3.5 |
| P900-200-5 | 200 | 5 | 900 | 49.0 | 81.0 (86) | 0.4 | 3.4 | 9.6 | 60.9 | 42.6 | 5.6 |
| P900-220-5 | 220 | 5 | 900 | 52.8 | 82.7 (87) | 0.3 | 3.5 | 9.0 | 65.5 | 42.0 | 4.5 |
| P900-240-5 | 240 | 5 | 900 | 53.0 | 83.8 (88) | 0.4 | 3.5 | 7.1 | 61.8 | 40.6 | 5.1 |
| P600 | 600 | 26.5 | 69.1 (83) | 1.5 | 2.7 | 10.3 | 45.8 | 16.3 | |||
| P900 | 900 | 27.1 | 67.0 (81) | 1.1 | 2.5 | 12.2 | 45.4 | 17.2 | |||
| Sample | N2 | CO2 | EC | Bulk Density |
|---|---|---|---|---|
| (m2 g−1) | (m2 g−1) | (S m−1) | (kg m−3) | |
| HTC-220-2 | 1.8 | - | 0 | 0.55 |
| HTC-220-2 | 1.3 | - | 0 | 0.41 |
| HTC-240-2 | 1.6 | - | 0 | 0.33 |
| HTC-200-5 | 2.0 | - | 0 | 0.34 |
| HTC-220-5 | 1.7 | - | 0 | 0.32 |
| HTC-240-5 | 1.4 | - | 0 | 0.31 |
| P600-200-2 | 78 | 293 | 52 | 0.41 |
| P600-220-2 | 86 | 294 | 106 | 0.48 |
| P600-240-2 | 23 | - | 53 | 0.39 |
| P600-200-5 | 79 | 290 | 38 | 0.36 |
| P600-220-5 | 19 | 202 | 9 | 0.35 |
| P600-240-5 | 14 | - | 21 | 0.37 |
| P600 | 274 | 105 | 0.91 | |
| P900-200-2 | 4.7 | 288 | 165 | 0.43 |
| P900-220-2 | 12.5 | 89 | 127 | 0.49 |
| P900-240-2 | 6.8 | - | 140 | 0.47 |
| P900-200-5 | 4.7 | 330 | 173 | 0.49 |
| P900-220-5 | 2.6 | - | 200 | 0.51 |
| P900-240-5 | 2.5 | - | 204 | 0.48 |
| P900 | 2.3 | 48 | 0.83 |
| Feedstock | Scan Rate [Vs−1] | Voltage Range [∆V] | Charge Measured Q [As] | Capacity C [Fg−1] |
|---|---|---|---|---|
| P600-200-2 | 0.3 | 1.1 | 0.01034 | 2.16 |
| 0.1 | 1.1 | 0.00748 | 4.68 | |
| 0.05 | 1.1 | 0.00569 | 7.11 | |
| 0.01 | 1.1 | 0.00430 | 26.86 | |
| P600-220-2 | 0.3 | 0.7 | 0.00264 | 0.92 |
| 0.1 | 0.7 | 0.00152 | 1.58 | |
| 0.05 | 0.7 | 0.00037 | 0.77 | |
| 0.01 | 0.7 | 0.00001 | 0.15 | |
| P600-200-5 | 0.3 | 0.75 | 0.00309 | 1.84 |
| 0.1 | 0.75 | 0.00203 | 3.62 | |
| 0.05 | 0.75 | 0.00082 | 2.93 | |
| 0.01 | 0.75 | 0.00130 | 23.18 | |
| P600-220-5 | 0.3 | 0.2 | 0.00077 | 4.70 |
| 0.1 | 0.2 | 0.00075 | 13.76 | |
| 0.05 | 0.2 | 0.00076 | 27.85 | |
| 0.01 | 0.2 | 0.00073 | 134.15 | |
| P900-200-2 | 0.3 | 0.2 | 0.00104 | 1.28 |
| 0.1 | 0.2 | 0.00086 | 3.19 | |
| 0.05 | 0.2 | 0.00084 | 6.22 | |
| 0.01 | 0.2 | 0.00085 | 31.49 | |
| P900-220-2 | 0.3 | 0.2 | 0.00119 | 1.46 |
| 0.1 | 0.2 | 0.00127 | 4.67 | |
| 0.05 | 0.2 | 0.00134 | 9.84 | |
| 0.01 | 0.2 | 0.00129 | 47.28 | |
| P900-200-5 | 0.3 | 0.9 | 0.00593 | 1.46 |
| 0.1 | 0.9 | 0.00458 | 4.67 | |
| 0.05 | 0.9 | 0.00497 | 9.84 | |
| 0.01 | 0.9 | 0.00414 | 34.25 | |
| P900-220-5 | 0.3 | 0.2 | 0.00328 | 4.15 |
| 0.1 | 0.2 | 0.00315 | 11.94 | |
| 0.05 | 0.2 | 0.00308 | 23.40 | |
| 0.01 | 0.2 | 0.00313 | 118.70 |
| Electrochemical Properties | Examples | |||||
|---|---|---|---|---|---|---|
| Electric Conductivity | Specific Capacity | |||||
| physico-chemical properties | microscopic | high content of carbon-oxygen complexes | acidic surface oxides (carboxylic, lactonic, phenolic), formation between 200 and 700 °C, low stability | (−) O-containing surface functionalities increase the barrier for electron transfer leading to higher equivalent series resistance (ESR) | (−) higher ESR leads to decreased capacity(+) high content of O-functionalities lead to pseudcapacitive effects, better wettability = improved pore access and surface utilization | −P-600-220-5: high O-content on carbon surface → low EC, high specific capacity due to pseudocapacitive effects −P-900-220-5: lower O-content on surface due to higher temperature treatment, O incorporated in C-lattice and graphitized structure → higher EC values, higher SSA → specific capacity value due to double-layer capacitance |
| basic surface oxides, chemisorption during high temperature treatment, higher stability | ||||||
| neutral surface-oxides, chemisorption during high temperature treatment higher stability | ||||||
| high specific surface area | microporous | (+/−) influence of SSA on EC is overshadowed by other factors such as intrinsic EC of carbon particles and particle contact | ||||
| macroporous | ||||||
| macroscopic | electrode production | binder type and content, other additives, type of current collector etc. | (−) if high binder content (+) if low binder content and conductive additive | (+/−) depending on interfacial electrical resistance between carbon and collector, binder content etc. | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, V.; Jung, D.; Alhnidi, M.J.; Mackle, L.; Kruse, A. Bio-Based Carbon Materials from Potato Waste as Electrode Materials in Supercapacitors. Energies 2020, 13, 2406. https://doi.org/10.3390/en13092406
Hoffmann V, Jung D, Alhnidi MJ, Mackle L, Kruse A. Bio-Based Carbon Materials from Potato Waste as Electrode Materials in Supercapacitors. Energies. 2020; 13(9):2406. https://doi.org/10.3390/en13092406
Chicago/Turabian StyleHoffmann, Viola, Dennis Jung, Muhammad Jamal Alhnidi, Lukas Mackle, and Andrea Kruse. 2020. "Bio-Based Carbon Materials from Potato Waste as Electrode Materials in Supercapacitors" Energies 13, no. 9: 2406. https://doi.org/10.3390/en13092406