
Critical Functions of Histone Deacetylases (HDACs) in Modulating Inflammation Associated with Cardiovascular Diseases

 , ,
, , {kind=link}
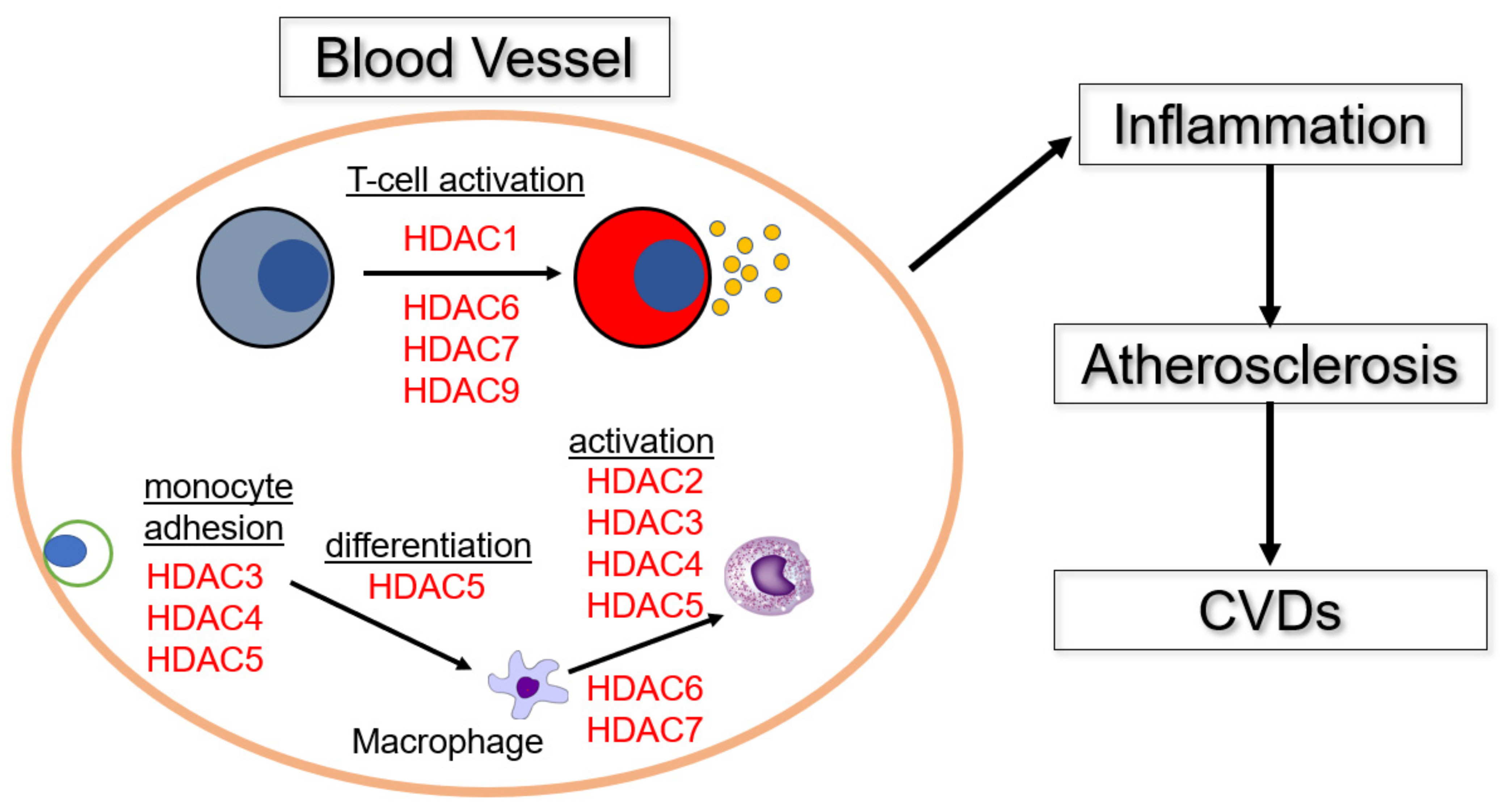{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Classes of HDACs
3. HDACs and Inflammations
3.1. Class I HDACs
3.2. Class IIa HDACs
3.3. Class IIb HDACs
3.4. Class IV HDAC
3.5. Class III HDACs
4. Pathophysiology of Inflammation in Cardiovascular Disease
5. The Function of HDACs in Regulation of Atherosclerosis and Cardiovascular Diseases
5.1. Cardiac Hypertrophy
5.2. Atherosclerosis
5.3. Arrhythmia
5.4. Ischemic Heart Diseases
5.5. Cardiac Fibrosis
6. HDACs as a Therapeutic Target for CVD
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peterson, C.L. HDAC’s at work: Everyone doing their part. Mol. Cell 2002, 9, 921–922. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, S.; Seth, S. A translational perspective on histone acetylation modulators in psychiatric disorders. Psychopharmacology 2018, 235, 1867–1873. [Google Scholar] [CrossRef]
- Shankar, S.; Srivastava, R.K. Histone deacetylase inhibitors: Mechanisms and clinical significance in cancer: HDAC inhibitor-induced apoptosis. Adv. Exp. Med. Biol. 2008, 615, 261–298. [Google Scholar] [CrossRef] [PubMed]
- Loeuillet, C.; Touquet, B.; Guichou, J.F.; Labesse, G.; Sereno, D. A tiny change makes a big difference in the anti-parasitic activities of an HDAC inhibitor. Int. J. Mol. Sci. 2019, 20, 2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.N.; Choijookhuu, N.; Takagi, H.; Srisowanna, N.; Huynh, M.N.N.; Yamaguchi, Y.; Oo, P.S.; Kyaw, M.T.H.; Sato, K.; Yamaguchi, R.; et al. The HDAC inhibitor, SAHA, prevents colonic inflammation by suppressing pro-inflammatory cytokines and chemokines in DSS-induced colitis. Acta Histochem. Cytochem. 2018, 51, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Tang, R.; Wang, S.; Xiong, Y.; Wang, W.; Chen, G.; Zhang, K.; Li, P.; Tang, Y.D. Isoform-selective HDAC inhibitor mocetinostat (MGCD0103) alleviates myocardial ischemia/reperfusion injury via mitochondrial protection through the HDACs/CREB/PGC-1α signaling pathway. J. Cardiovasc. Pharmacol. 2021, 79, 217–228. [Google Scholar] [CrossRef]
- Mishra, N.; Reilly, C.M.; Brown, D.R.; Ruiz, P.; Gilkeson, G.S. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Investig. 2003, 111, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hong, Y.; Huang, H. Triptolide Attenuates Inflammatory Response in Membranous Glomerulo-Nephritis Rat via Downregulation of NF-κB Signaling Pathway. Kidney Blood Press. Res. 2016, 41, 901–910. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Ridker, P.M.; Hansson, G.K.; Leducq Transatlantic Network on Atherothrombosis. Inflammation in atherosclerosis: From pathophysiology to practice. J. Am. Coll. Cardiol. 2009, 54, 2129–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aday, A.W.; Ridker, P.M. Targeting residual inflammatory risk: A shifting paradigm for atherosclerotic disease. Front. Cardiovasc. Med. 2019, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC inhibitor: From cancer to cardiovascular diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [Green Version]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenberg, A. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370(Pt. 3), 737–749. [Google Scholar] [CrossRef]
- Li, J.; Lin, Q.; Wang, W.; Wade, P.; Wong, J. Specific targeting and constitutive association of histone deacetylase complexes during transcriptional repression. Genes Dev. 2002, 16, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef] [Green Version]
- McKinsey, T.A. The biology and therapeutic implications of HDACs in the heart. Handb. Exp. Pharmacol. 2011, 206, 57–78. [Google Scholar] [CrossRef]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [Green Version]
- Borradaile, N.M.; Pickering, J.G. NAD+, sirtuins, and cardiovascular disease. Curr. Pharm. Des. 2009, 15, 110–117. [Google Scholar] [CrossRef]
- Drogaris, P.; Villeneuve, V.; Pomiès, C.; Lee, E.H.; Bourdeau, V.; Bonneil, E.; Ferbeyre, G.; Verreault, A.; Thibault, P. Histone deacetylase inhibitors globally enhance h3/h4 tail acetylation without without affecting h3 lysine 56 acetylation. Sci. Rep. 2012, 2, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone de acetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, M.D.; Haynes, D.R. Epigenetic regulation of inflammation: Progressing from broad acting histone deacetylase (HDAC) inhibitors to targeting specific HDACs. Inflammopharmacology 2013, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Göschl, L.; Preglej, T.; Boucheron, N.; Saferding, V.; Müller, L.; Platzer, A.; Hirahara, K.; Shih, H.Y.; Backlund, J.; Matthias, P.; et al. Histone deacetylase 1 (HDAC1): A key player of T cell-mediated arthritis. J. Autoimmun. 2020, 108, 102379. [Google Scholar] [CrossRef]
- Barnes, P.J. Role of HDAC2 in the pathophysiology of COPD. Annu. Rev. Physiol. 2009, 71, 451–464. [Google Scholar] [CrossRef]
- Ito, K.; Caramori, G.; Lim, S.; Oates, T.; Chung, K.F.; Barnes, P.J.; Adcock, I.M. Expression and activity of histone deacetylases in human asthmatic airways. Am. J. Respir. Crit. Care Med. 2002, 166, 392–396. [Google Scholar] [CrossRef]
- Liao, W.; Lim, A.Y.H.; Tan, W.S.D.; Abisheganaden, J.; Wong, W.S.F. Restoration of HDAC2 and Nrf2 by andrographolide overcomes corticosteroid resistance in chronic obstructive pulmonary disease. Br. J. Pharmacol. 2020, 177, 3662–3673. [Google Scholar] [CrossRef]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kim, K.; Park, D.; Lee, E.; Lee, H.; Lee, Y.S.; Choe, J.; Jeoung, D. Histone deacetylase 3 mediates allergic skin inflammation by regulating expression of MCP1 protein. J. Biol. Chem. 2012, 287, 25844–25859. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Kobayashi, M.; Yano, K.; Miura, M.; Izumi, A.; Mataki, C.; Doi, T.; Hamakubo, T.; Reid, P.C.; Hume, D.A.; et al. Histone deacetylase inhibitor reduces monocyte adhesion to endothelium through the suppression of vascular cell adhesion molecule-1 expression. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2652–2659. [Google Scholar] [CrossRef] [Green Version]
- Winkler, A.R.; Nocka, K.N.; Williams, C.M. Smoke exposure of human macrophages reduces HDAC3 activity, resulting in enhanced inflammatory cytokine production. Pulm. Pharmacol. Ther. 2012, 25, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Mullican, S.E.; Gaddis, C.A.; Alenghat, T.; Nair, M.G.; Giacomin, P.R.; Everett, L.J.; Feng, D.; Steger, D.J.; Schug, J.; Artis, D.; et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011, 25, 2480–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.M.; Hogg, J.C.; et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Han, S.; Lu, J.; Zhang, Y.; Cheng, C.; Han, L.; Wang, X.; Li, L.; Liu, C.; Huang, B. Recruitment of histone deacetylase 4 by transcription factors represses interleukin-5 transcription. Biochem. J. 2006, 400, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Usui, T.; Okada, M.; Mizuno, W.; Oda, M.; Ide, N.; Morita, T.; Hara, Y.; Yamawaki, H. HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1894–H1904. [Google Scholar] [CrossRef] [Green Version]
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.; Carducci, M.A.; Atadja, P.; Pili, R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006, 66, 8814–8821. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wu, D.; Xu, J.; Liu, L.; Jiao, W.; Yu, J.; Chen, G. Suppression of NLRP3 Inflammasome by Dihydroarteannuin via the HIF-1α and JAK3/STAT3 Signaling Pathway Contributes to Attenuation of Collagen-Induced Arthritis in Mice. Front. Pharmacol. 2022, 13, 884881. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, G.; Yang, X. HDAC5 promotes Mycoplasma pneumoniae-induced inflammation in macrophages through NF-κB activation. Life Sci. 2019, 221, 13–19. [Google Scholar] [CrossRef]
- Wang, W.; Ha, C.H.; Jhun, B.S.; Wong, C.; Jain, M.K.; Jin, Z.G. Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood 2010, 115, 2971–2979. [Google Scholar] [CrossRef] [Green Version]
- Rivadeneira, F.; Styrkarsdottir, U.; Estrada, K.; Halldorsson, B.V.; Hsu, Y.H.; Richards, J.B.; Zillikens, M.C.; Kavvoura, F.K.; Amin, N.; Aulchenko, Y.S.; et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat. Genet. 2009, 41, 1199–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, M.D.; Fairlie, D.P.; Bartold, P.M.; Rainsford, K.D.; Le, G.T.; Lucke, A.J.; Holding, C.A.; Haynes, D.R. Inhibitors of histone deacetylases in class I and class II suppress human osteoclasts in vitro. J. Cell. Physiol. 2011, 226, 3233–3241. [Google Scholar] [CrossRef]
- Crotti, T.N.; Smith, M.D.; Weedon, H.; Ahern, M.J.; Findlay, D.M.; Kraan, M.; Tak, P.P.; Haynes, D.R. Receptor activator NF-κB ligand (RANKL) expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathy, osteoarthritis, and from normal patients: Semiquantitative and quantitative analysis. Ann. Rheum. Dis. 2002, 61, 1047–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotti, T.; Smith, M.D.; Hirsch, R.; Soukoulis, S.; Weedon, H.; Capone, M.; Ahern, M.J.; Haynes, D. Receptor activator NF κB ligand (RANKL) and osteoprotegerin (OPG) protein expression in periodontitis. J. Periodontal Res. 2003, 38, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Kasler, H.G.; Young, B.D.; Mottet, D.; Lim, H.W.; Collins, A.M.; Olson, E.N.; Verdin, E. Histone deacetylase 7 regulates cell survival and TCR signaling in CD4/CD8 double-positive thymocytes. J. Immunol. 2011, 186, 4782–4793. [Google Scholar] [CrossRef]
- Kato, H.; Tamamizu-Kato, S.; Shibasaki, F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J. Biol. Chem. 2004, 279, 41966–41974. [Google Scholar] [CrossRef] [Green Version]
- Pham, L.; Kaiser, B.; Romsa, A.; Schwarz, T.; Gopalakrishnan, R.; Jensen, E.D.; Mansky, K.C. HDAC3 and HDAC7 have opposite effects on osteoclast differentiation. J. Biol. Chem. 2011, 286, 12056–12065. [Google Scholar] [CrossRef] [Green Version]
- Fischle, W.; Dequiedt, F.; Fillion, M.; Hendzel, M.J.; Voelter, W.; Verdin, E. Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J. Biol. Chem. 2001, 276, 35826–35835. [Google Scholar] [CrossRef] [Green Version]
- Isselbacher, E.M.; Lino Cardenas, C.L.; Lindsay, M.E. Hereditary influence in thoracic aortic aneurysm and dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.; Cao, Q.; Reilly, C.M.; Young, N.L.; Garcia, B.A.; Mishra, N. Histone deacetylase 9 deficiency protects against effector T cell-mediated systemic autoimmunity. J. Biol. Chem. 2011, 286, 28833–28843. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Li, H.; Li, K.; Fan, X.D. HDAC9 promotes brain ischemic injury by provoking IκBα/NF-κB and MAPKs signaling pathways. Biochem. Biophys. Res. Commun. 2018, 503, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E. HDAC6 Regulates LPS-Tolerance in Astrocytes. PLoS ONE 2011, 6, e25804. [Google Scholar] [CrossRef] [PubMed]
- de Zoeten, E.F.; Wang, L.; Butler, K.; Beier, U.H.; Akimova, T.; Sai, H.; Bradner, J.E.; Mazitschek, R.; Kozikowski, A.P.; Matthias, P.; et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3+ T-regulatory cells. Mol. Cell. Biol. 2011, 31, 2066–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halili, M.A.; Andrews, M.R.; Labzin, L.I.; Schroder, K.; Matthias, G.; Cao, C.; Lovelace, E.; Reid, R.C.; Le, G.T.; Hume, D.A.; et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J. Leukoc. Biol. 2010, 87, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Sun, J.; Liu, W.; Li, W.; Jia, J.; Ou, F.; Su, K.; Zheng, Y.; Zhang, Z.; Sun, Y. HDAC10 upregulation contributes to interleukin 1β-mediated inflammatory activation of synovium-derived mesenchymal stem cells in temporomandibular joint. J. Cell. Physiol. 2019, 234, 12646–12662. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation andinsulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef]
- Yanginlar, C.; Logie, C. HDAC11 is a regulator of diverseimmune functions. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 54–59. [Google Scholar] [CrossRef]
- Wang, X.; Wu, Y.; Jiao, J.; Huang, Q. Mycobacterium tuberculosisinfection induces IL-10 gene expression by disturbing histonedeacetylase 6 and histonedeacetylase 11 equilibrium in macrophages. Tuberculosis 2018, 108, 118–123. [Google Scholar] [CrossRef]
- Larsson, L.; Thorbert-Mros, S.; Rymo, L.; Berglundh, T. Influence of epigenetic modifications of the interleukin-10 promoter on IL10 gene expression. Eur. J. Oral Sci. 2012, 120, 14–20. [Google Scholar] [CrossRef]
- Zhu, Q.; Tang, T.; Liu, H.; Sun, Y.; Wang, X.; Liu, Q.; Yang, L.; Lei, Z.; Huang, Z.; Chen, Z.; et al. Pterostilbene Attenuates Cocultured BV-2 Microglial Inflammation-Mediated SH-SY5Y Neuronal Oxidative Injury via SIRT-1 Signalling. Oxid. Med. Cell. Longev. 2020, 2020, 3986348. [Google Scholar] [CrossRef]
- Zhao, H.; Mei, X.; Yang, D.; Tu, G. Resveratrol inhibits inflammation after spinal cord injury via SIRT-1/NF-κB signaling pathway. Neurosci. Lett. 2021, 762, 136151. [Google Scholar] [CrossRef]
- Kumar, J.; Haldar, C.; Verma, R. Melatonin Ameliorates LPS-Induced Testicular Nitro-oxidative Stress (iNOS/TNFα) and Inflammation (NF-kB/COX-2) via Modulation of SIRT-1. Reprod. Sci. 2021, 28, 3417–3430. [Google Scholar] [CrossRef]
- Grabiec, A.M.; Krausz, S.; de Jager, W.; Burakowski, T.; Groot, D.; Sanders, M.E.; Prakken, B.J.; Maslinski, W.; Eldering, E.; Tak, P.P.; et al. Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J. Immunol. 2010, 184, 2718–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiboub, M.; Zhao, J.; Li Yim, A.Y.F.; Schilderink, R.; Verseijden, C.; van Hamersveld, P.H.P.; Duarte, J.M.; Hakvoort, T.B.M.; Admiraal, I.; Harker, N.R.; et al. HDAC3 Mediates the Inflammatory Response and LPS Tolerance in Human Monocytes and Macrophages. Front. Immunol. 2020, 11, 550769. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Morita, T.; Okada, M.; Yamawaki, H. Histone deacetylase 4 controls neointimal hyperplasia via stimulating proliferation and migration of vascular smooth muscle cells. Hypertension 2014, 63, 397–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, Y.S.; Haas, S.; Hackstein, H.; Bein, G.; Hernandez-Santana, M.; Lehrach, H.; Sauer, S.; Seitz, H. Identification of novel transcriptional regulators involved in macrophage differentiation and activation in U937 cells. BMC Immunol. 2009, 10, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Reduced histone deacetylase in COPD: Clinical implications. Chest 2006, 129, 151–155. [Google Scholar] [CrossRef]
- Aung, H.T.; Schroder, K.; Himes, S.R.; Brion, K.; van Zuylen, W.; Trieu, A.; Suzuki, H.; Hayashizaki, Y.; Hume, D.A.; Sweet, M.J.; et al. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J. 2006, 20, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Grausenburger, R.; Bilic, I.; Boucheron, N.; Zupkovitz, G.; El-Housseiny, L.; Tschismarov, R.; Zhang, Y.; Rembold, M.; Gaisberger, M.; Hartl, A.; et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J. Immunol. 2010, 185, 3489–3497. [Google Scholar] [CrossRef]
- Zhao, T.C.; Wang, Z.; Zhao, T.Y. The important role of histone deacetylases in modulating vascular physiology and arteriosclerosis. Atherosclerosis 2020, 303, 36–42. [Google Scholar] [CrossRef]
- Daugherty, A.; Webb, N.R.; Rateri, D.L.; King, V.L. Thematic review series: The immune system and atherogenesis. Cytokine regulation of macrophage functions in atherogenesis. J. Lipid Res. 2005, 46, 1812–1822. [Google Scholar] [CrossRef] [Green Version]
- Deckert-Schlüter, M.; Schlüter, D.; Theisen, F.; Wiestler, O.D.; Hof, H. Activation of the innate immune system in murine congenital Toxoplasma encephalitis. J. Neuroimmunol. 1994, 53, 47–51. [Google Scholar] [CrossRef]
- Dong, N.; Wang, Y. MiR-30a Regulates S100A12-induced Retinal Microglial Activation and Inflammation by Targeting NLRP3. Curr. Eye Res. 2019, 44, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jung, J.W.; Park, S.B.; Roh, K.; Lee, S.Y.; Kim, J.H.; Kang, S.K.; Kang, K.S. Histone deacetylase regulates high mobility group A2-targeting microRNAs in human cord blood-derived multipotent stem cell aging. Cell. Mol. Life Sci. 2011, 68, 325–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Qiu, Y.; Zheng, Z.; Yu, L.; Shi, S.; Wu, X. NOD-like receptor protein 3 and high mobility group box-1 are associated with prognosis of patients with congenital heart disease. J. Int. Med. Res. 2020, 48, 300060519884500. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Molla, M.D.; Ayelign, B.; Dessie, G.; Geto, Z.; Admasu, T.D. Caspase-1 as a regulatory molecule of lipid metabolism. Lipids Health Dis. 2020, 19, 34. [Google Scholar] [CrossRef] [Green Version]
- Libby, P. Interleukin-1 beta as a target for atherosclerosis therapy: Biological basis of CANTOS and beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Zakynthinos, E.; Pappa, N. Inflammatory biomarkers in coronary artery disease. J. Cardiol. 2009, 53, 317–333. [Google Scholar] [CrossRef] [Green Version]
- Avina-Zubieta, J.A.; Thomas, J.; Sadatsafavi, M.; Lehman, A.J.; Lacaille, D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: A meta-analysis of observational studies. Ann. Rheum. Dis. 2012, 71, 1524–1529. [Google Scholar] [CrossRef]
- Esdaile, J.M.; Abrahamowicz, M.; Grodzicky, T.; Li, Y.; Panaritis, C.; du Berger, R.; Côte, R.; Grover, S.A.; Fortin, P.R.; Clarke, A.E.; et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001, 44, 2331–2337. [Google Scholar] [CrossRef]
- Khalid, U.; Ahlehoff, O.; Gislason, G.H.; Kristensen, S.L.; Skov, L.; Torp-Pedersen, C.; Hansen, P.R. Psoriasis and risk of heart failure: A nationwide cohort study. Eur. J. Heart Fail. 2014, 16, 743–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaptoge, S.; Seshasai, S.R.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; Rumley, A.; Lowe, G.D.; et al. Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanfield, B.A.; Purves, T.; Palmer, S.; Sullenger, B.; Welty-Wolf, K.; Haines, K.; Agarwal, S.; Kasotakis, G. IL-10 and class 1 histone deacetylases act synergistically and independently on the secretion of proinflammatory mediators in alveolar macrophages. PLoS ONE 2021, 16, e0245169. [Google Scholar] [CrossRef] [PubMed]
- Eom, G.H.; Cho, Y.K.; Ko, J.H.; Shin, S.; Choe, N.; Kim, Y.; Joung, H.; Kim, H.S.; Nam, K.I.; Kee, H.J.; et al. Casein kinase-2α1 induces hypertrophic response by phosphorylation of histone deacetylase 2 S394 and its activation in the heart. Circulation 2011, 123, 2392–2403. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, C.M.; Luo, Y.; Yin, Z.; Zhang, M.; Zhu, W.; Wang, T.; Floss, T.; Goettlicher, M.; Noppinger, P.R.; Wurst, W.; et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat. Med. 2007, 13, 324–331. [Google Scholar] [CrossRef]
- Kee, H.J.; Eom, G.H.; Joung, H.; Shin, S.; Kim, J.R.; Cho, Y.K.; Choe, N.; Sim, B.W.; Jo, D.; Jeong, M.H.; et al. Activation of histone deacetylase 2 by inducible heat shock protein 70 in cardiac hypertrophy. Circ. Res. 2008, 103, 1259–1269. [Google Scholar] [CrossRef]
- Trivedi, C.M.; Lu, M.M.; Wang, Q.; Epstein, J.A. Transgenic overexpression of Hdac3 in the heart produces increased postnatal cardiac myocyte proliferation but does not induce hypertrophy. J. Biol. Chem. 2008, 283, 26484–26489. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Mukherjee, B.; Mukhopadhyay, R.; Naskar, K.; Sundar, S.; Dujardin, J.C.; Roy, S. Imipramine exploits histone deacetylase 11 to increase the IL-12/IL-10 ratio in macrophages infected with antimony-resistant Leishmania donovani and clears organ parasites in experimental infection. J. Immunol. 2014, 193, 4083–4094. [Google Scholar] [CrossRef] [Green Version]
- Cornwell, J.D.; McDermott, J.C. MEF2 in cardiac hypertrophy in response to hypertension. Trends Cardiovasc. Med. 2022, in press. [Google Scholar] [CrossRef]
- Bush, E.W.; McKinsey, T.A. Targeting histone deacetylases for heart failure. Expert Opin. Ther. Targets 2009, 13, 767–784. [Google Scholar] [CrossRef]
- He, T.; Huang, J.; Chen, L.; Han, G.; Stanmore, D.; Krebs-Haupenthal, J.; Avkiran, M.; Hagenmüller, M.; Backs, J. Cyclic AMP represses pathological MEF2 activation by myocyte-specific hypo-phosphorylation of HDAC5. J. Mol. Cell. Cardiol. 2020, 145, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Calalb, M.B.; McKinsey, T.A.; Newkirk, S.; Huynh, K.; Sucharov, C.C.; Bristow, M.R. Increased phosphorylation-dependent nuclear export of class II histone deacetylases in failing human heart. Clin. Transl. Sci. 2009, 2, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Yang, X.; Zimmerman, R.J.; Wang, Q.; Ross, M.A.; Granger, J.M.; Luczak, E.D.; Bedja, D.; Jiang, H.; Feng, N. CaMKII exacerbates heart failure progression by activating class I HDACs. J. Mol. Cell. Cardiol. 2020, 149, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Gallo, P.; Latronico, M.V.; Gallo, P.; Grimaldi, S.; Borgia, F.; Todaro, M.; Jones, P.; Gallinari, P.; De Francesco, R.; Ciliberto, G.; et al. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardiovasc. Res. 2008, 80, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Eom, G.H.; Nam, Y.S.; Oh, J.G.; Choe, N.; Min, H.K.; Yoo, E.K.; Kang, G.; Nguyen, V.H.; Min, J.J.; Kim, J.K.; et al. Regulation of acetylation of histone deacetylase 2 by p300/CBP-associated factor/histone deacetylase 5 in the development of cardiac hypertrophy. Circ. Res. 2014, 114, 1133–1143. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Wei, A.; Chen, F.; Chen, X.; Ding, W.; Ding, Z.; Wu, Z.; Du, R.; Cao, W. Enhancing PPARγ by HDAC inhibition reduces foam cell formation and atherosclerosis in ApoE deficient mice. Pharmacol. Res. 2020, 160, 105059. [Google Scholar] [CrossRef]
- Stuart, S.D.F.; De Jesus, N.M.; Lindsey, M.L.; Ripplinger, C.M. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J. Mol. Cell. Cardiol. 2016, 91, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Lambert, A.A.; Papst, P.; Pitts, K.R. Agonist-induced nuclear export of GFP-HDAC5 in isolated adult rat ventricular myocytes. J. Pharmacol. Toxicol. Methods 2009, 59, 135–140. Available online: https://pubmed.ncbi.nlm.nih.gov/19328241/#:~:text=May%2DJun%202009%3B59,j.vascn.2009.03.002 (accessed on 26 March 2009). [CrossRef]
- Kim, G.R.; Cho, S.N.; Kim, H.S.; Yu, S.Y.; Choi, S.Y.; Ryu, Y.; Lin, M.Q.; Jin, L.; Kee, H.J.; Jeong, M.H. Histone deacetylase and GATA-binding factor 6 regulate arterial remodeling in angiotensin II-induced hypertension. J. Hypertens. 2016, 34, 2206–2219. [Google Scholar] [CrossRef]
- Ismat, F.A.; Zhang, M.; Kook, H.; Huang, B.; Zhou, R.; Ferrari, V.A.; Epstein, J.A.; Patel, V.V. Homeobox protein Hop functions in the adult cardiac conduction system. Circ. Res. 2005, 96, 898–903. [Google Scholar] [CrossRef] [Green Version]
- Kook, H.; Lepore, J.J.; Gitler, A.D.; Lu, M.M.; Yung, W.W.-M.; Mackay, J.; Zhou, R.; Ferrari, V.; Gruber, P.; Epstein, J.A. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J. Clin. Investig. 2003, 112, 863–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weintraub, S.; Frishman, W.H. A Novel Calcium Channel Blocker: Etripamil: What Is the Future of Intranasal Drug Delivery in the Treatment of Cardiac Arrhythmias? Cardiol. Rev. 2021, 29, 253–258. [Google Scholar] [CrossRef]
- Zhao, T.C.; Cheng, G.; Zhang, L.X.; Tseng, Y.T.; Padbury, J.F. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc. Res. 2007, 76, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Sohn, I.S.; Nam, K.I.; Park, J.E.; Qian, Y.R.; Yin, Z.; Ahn, Y.; Jeong, M.H.; Bang, Y.-J.; Kim, N.; et al. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 2006, 113, 51–59. [Google Scholar] [CrossRef]
- Kong, Y.; Tannous, P.; Lu, G.; Berenji, K.; Rothermel, B.A.; Olson, E.N.; Hill, J.A. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 2006, 113, 2579–2588. [Google Scholar] [CrossRef]
- Guo, W.; Shan, B.; Klingsberg, R.C.; Qin, X.; Lasky, J.A. Abrogation of TGF-beta1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L864–L870. [Google Scholar] [CrossRef] [Green Version]
- Nural-Guvener, H.F.; Zakharova, L.; Nimlos, J.; Popovic, S.; Mastroeni, D.; Gaballa, M.A. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair 2014, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.J.; Ziaeian, B.; Fonarow, G.C. Heart Failure with Mid-Range (Borderline) Ejection Fraction: Clinical Implications and Future Directions. JACC Heart Fail. 2017, 5, 763–771. [Google Scholar] [CrossRef]
- Wallner, M.; Eaton, D.M.; Berretta, R.M.; Liesinger, L.; Schittmayer, M.; Gindlhuber, J.; Wu, J.; Jeong, M.Y.; Lin, Y.H.; Borghetti, G.; et al. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Sci. Transl. Med. 2020, 12, eaay7205. [Google Scholar] [CrossRef]
- Jeong, M.Y.; Lin, Y.H.; Wennersten, S.A.; Demos-Davies, K.M.; Cavasin, M.A.; Mahaffey, J.H.; Monzani, V.; Saripalli, C.; Mascagni, P.; Reece, T.B.; et al. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci. Transl. Med. 2018, 10, eaao0144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travers, J.G.; Wennersten, S.A.; Peña, B.; Bagchi, R.A.; Smith, H.E.; Hirsch, R.A.; Vanderlinden, L.A.; Lin, Y.H.; Dobrinskikh, E.; Demos-Davies, K.M.; et al. HDAC Inhibition Reverses Preexisting Diastolic Dysfunction and Blocks Covert Extracellular Matrix Remodeling. Circulation 2021, 143, 1874–1890. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhang, L.; Zhuang, S.; Qin, G.J.; Zhao, T.C. HDAC4 degradation mediates HDAC inhibition-induced protective effects against hypoxia/reoxygenation injury. J. Cell. Physiol. 2015, 230, 1321–1331. [Google Scholar] [CrossRef] [Green Version]
- Slegtenhorst, B.R.; Dor, F.J.; Rodriguez, H.; Voskuil, F.J.; Tullius, S.G. Ischemia/reperfusion Injury and its Consequences on Immunity and Inflammation. Curr. Transplant. Rep. 2014, 1, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.H.; Seok, Y.M.; Song, M.J.; Lee, H.A.; Kurz, T.; Kim, I. Histone deacetylase inhibition attenuates cardiac hypertrophy and fibrosis through acetylation of mineralocorticoid receptor in spontaneously hypertensive rats. Mol. Pharmacol. 2015, 87, 782–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.S.; Weng, S.C.; Tseng, P.H.; Lin, H.P.; Chen, C.S. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J. Biol. Chem. 2005, 280, 38879–38887. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Edelstein, L.C.; Micheva-Viteva, S.; Phelan, B.D.; Dougherty, J.P. Short communication: Activation of latent HIV type 1 gene expression by suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor approved for use to treat cutaneous T cell lymphoma. AIDS Res. Hum. Retrovir. 2009, 25, 883–887. Available online: https://pubmed.ncbi.nlm.nih.gov/19689202/#:~:text=2009%20Sep%3B25,10.1089/aid.2008.0294 (accessed on 26 March 2009). [CrossRef] [Green Version]
- Raedler, L.A. Farydak (Panobinostat): First HDAC inhibitor approved for patients with relapsed multiple myeloma. Am. Health Drug Benefits 2016, 9, 84–87. [Google Scholar]
- Chen, J.B.; Chern, T.R.; Wei, T.T.; Chen, C.C.; Lin, J.H.; Fang, J.M. Design and synthesis of dual-action inhibitors targeting histone deacetylases and 3-hydroxy-3-methylglutaryl coenzyme A reductase for cancer treatment. J. Med. Chem. 2013, 56, 3645–3655. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lin, J.H.; Chou, C.W.; Chang, Y.F.; Yeh, S.H.; Chen, C.C. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter associated HDAC1/2. Cancer Res. 2008, 68, 2375–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulthinee, S.; Yano, N.; Zhuang, S.; Wang, L.; Zhao, T.C. Critical Functions of Histone Deacetylases (HDACs) in Modulating Inflammation Associated with Cardiovascular Diseases. Pathophysiology 2022, 29, 471-485. https://doi.org/10.3390/pathophysiology29030038
Kulthinee S, Yano N, Zhuang S, Wang L, Zhao TC. Critical Functions of Histone Deacetylases (HDACs) in Modulating Inflammation Associated with Cardiovascular Diseases. Pathophysiology. 2022; 29(3):471-485. https://doi.org/10.3390/pathophysiology29030038
Chicago/Turabian StyleKulthinee, Supaporn, Naohiro Yano, Shougang Zhuang, Lijiang Wang, and Ting C. Zhao. 2022. "Critical Functions of Histone Deacetylases (HDACs) in Modulating Inflammation Associated with Cardiovascular Diseases" Pathophysiology 29, no. 3: 471-485. https://doi.org/10.3390/pathophysiology29030038