Neuroblastoma Interaction with the Tumour Microenvironment and Its Implications for Treatment and Disease Progression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to NB and the Tumour Microenvironment
2. The TME in NB and Its Link to NB Biology and Clinical Aspects
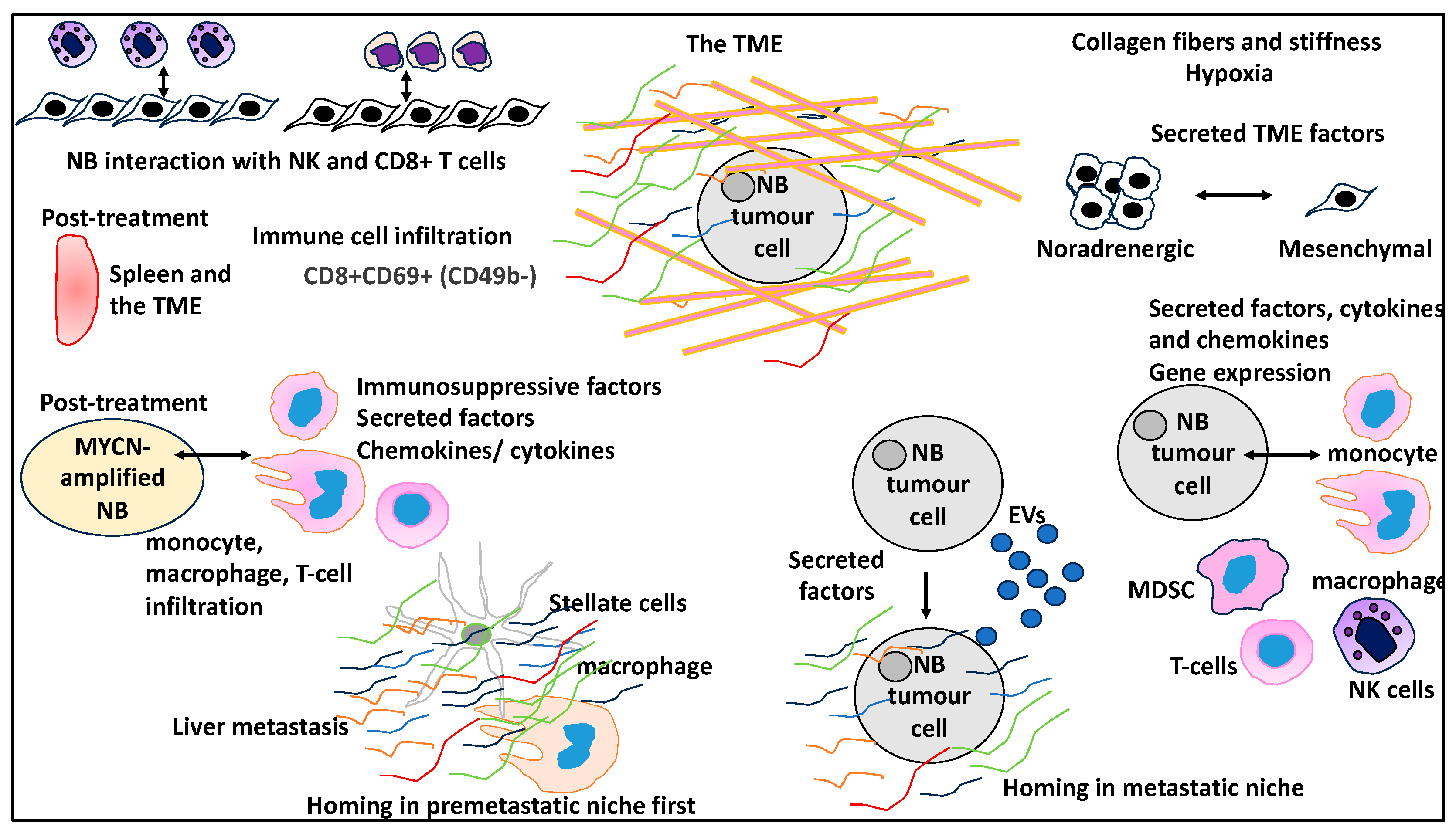
2.1. Immune and Stromal Landscapes of NB TME
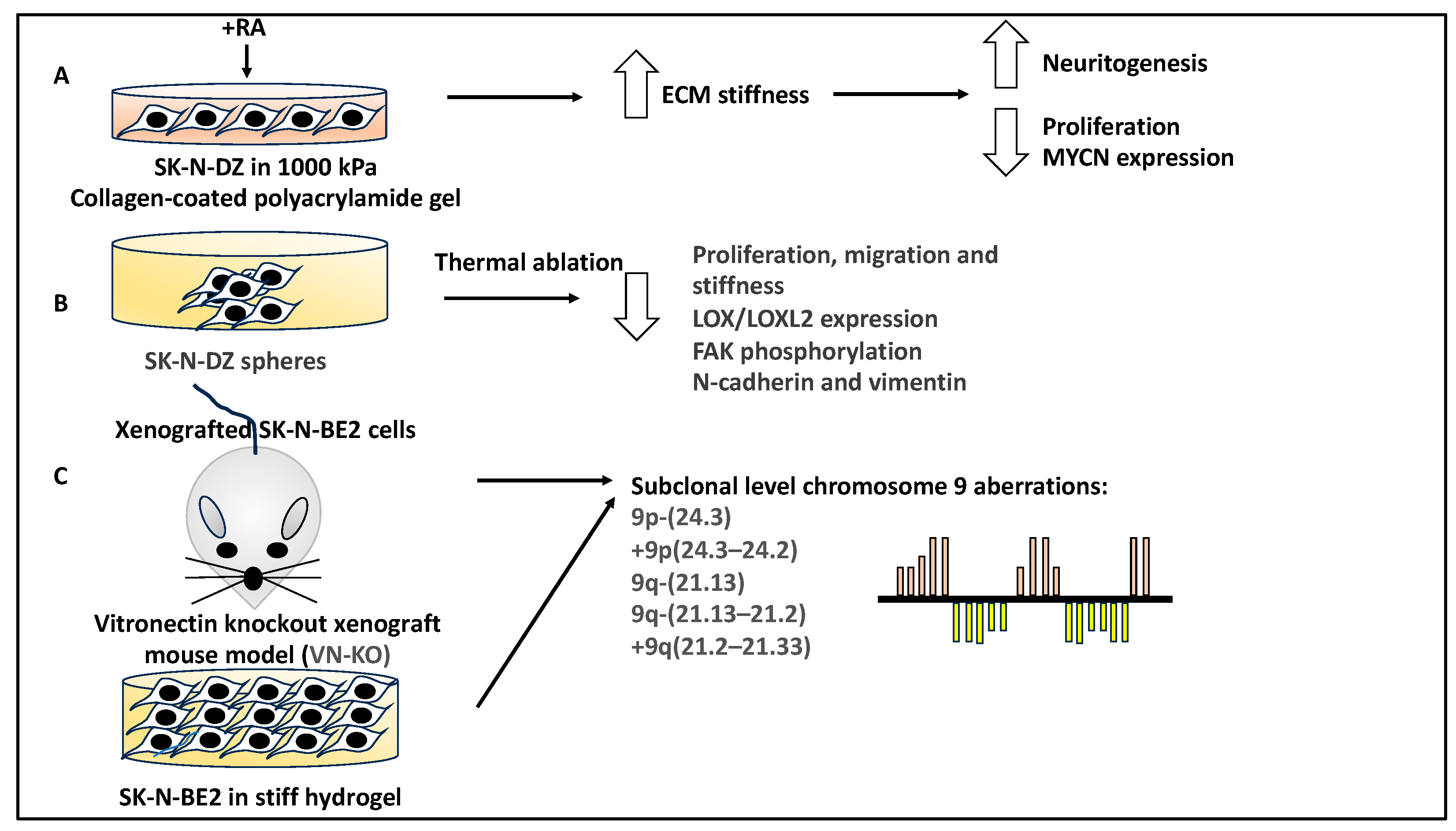
2.2. The Physical Properties of the ECM within the TME That Are Linked to NB Biology
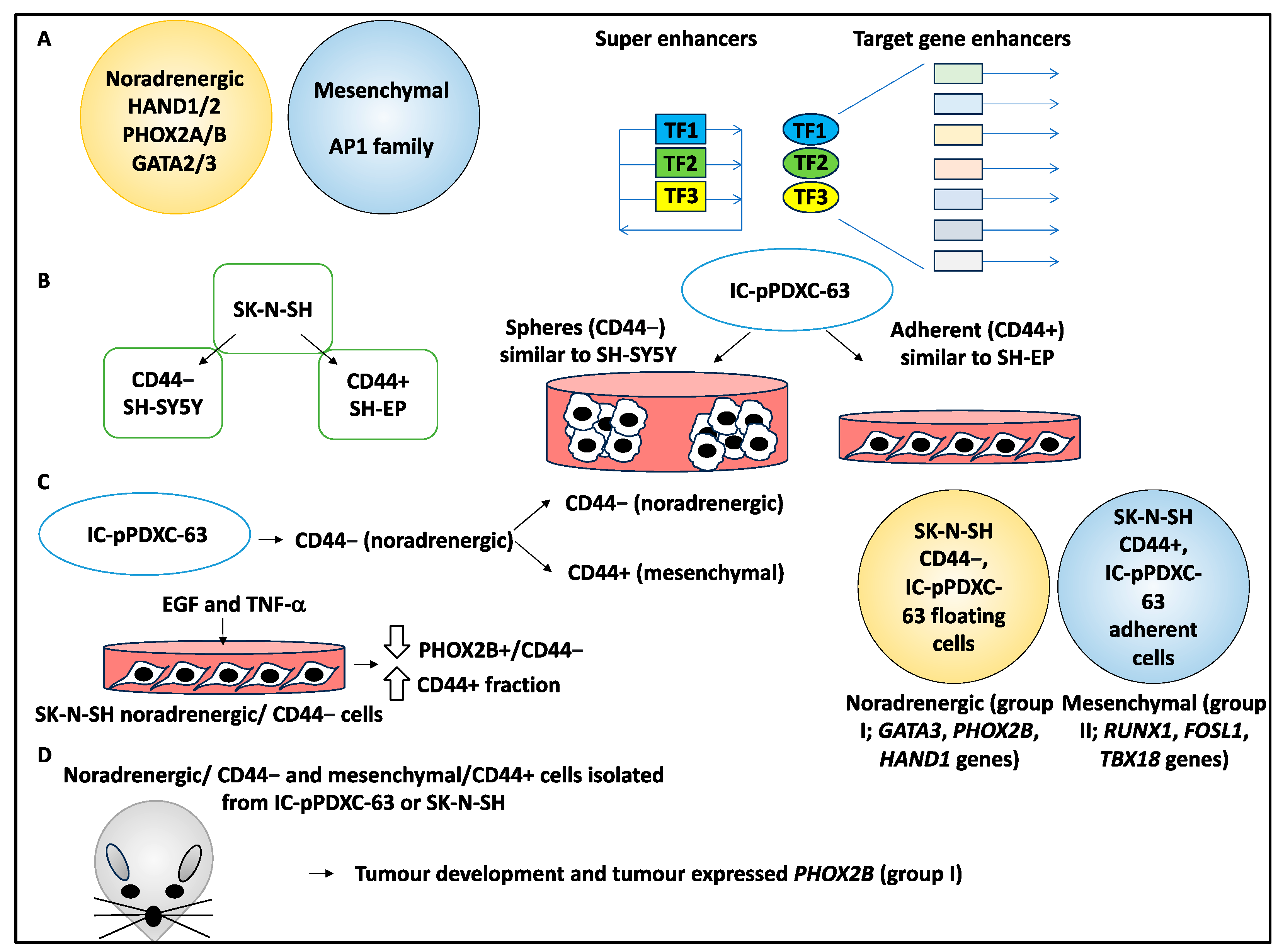
2.3. NB Mesenchymal to Noradrenergic Identity Transition Is Influenced by the TME
2.4. The Role of Differential Expression and Various Proteins in Influencing the TME in NB through Cytokines and Chemokines
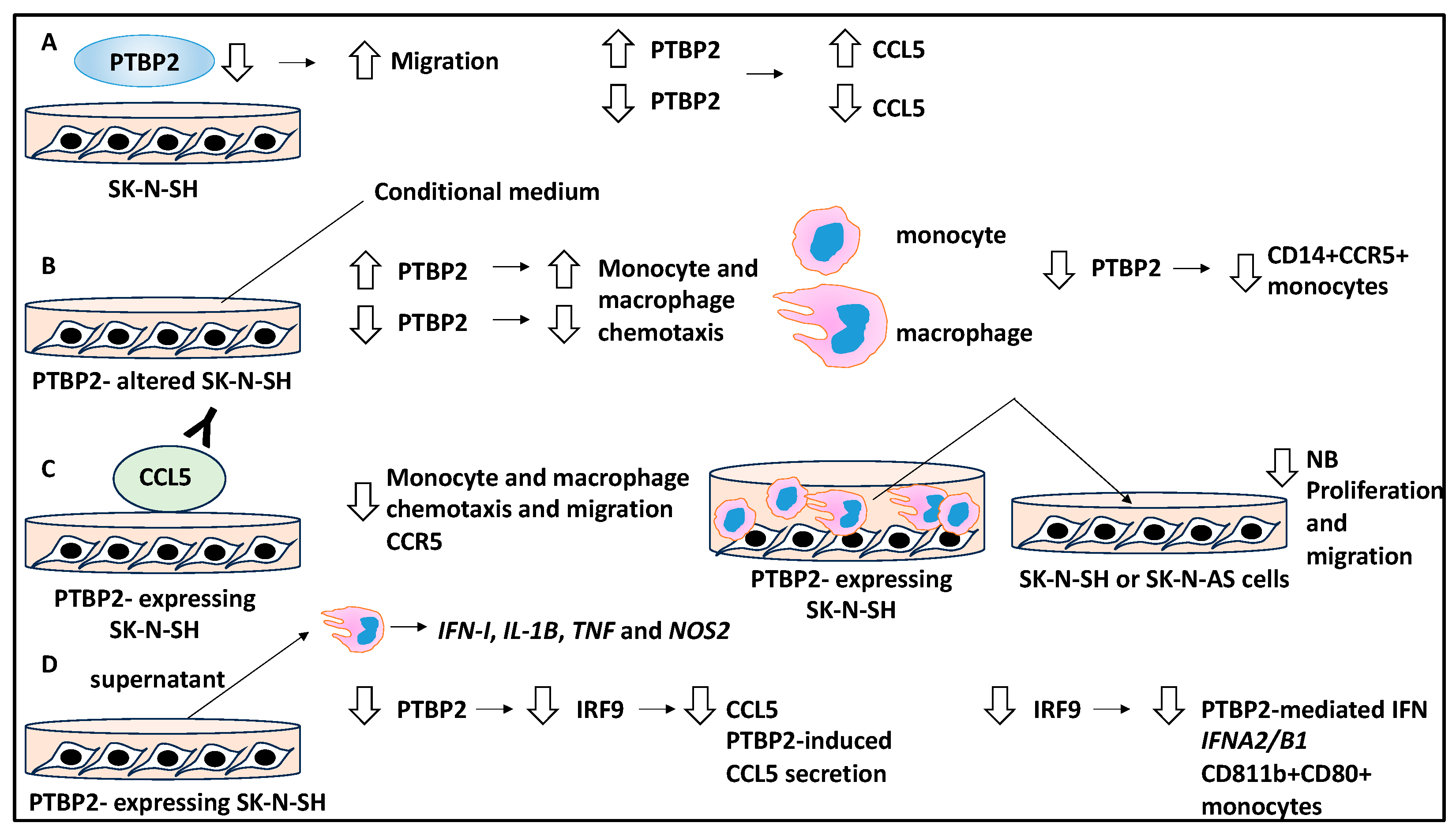
2.5. The Role of RNA-Binding Proteins in the TME That Influence NB Metastasis
2.6. The Contribution of EVs to NB PMN and Metastasis
2.7. Immune Cell Infiltration to the TME May Contribute to NB Relapse
2.8. Infiltration of Immune Cells to Spleens and TME Post-Treatment Linked to NB Treatment
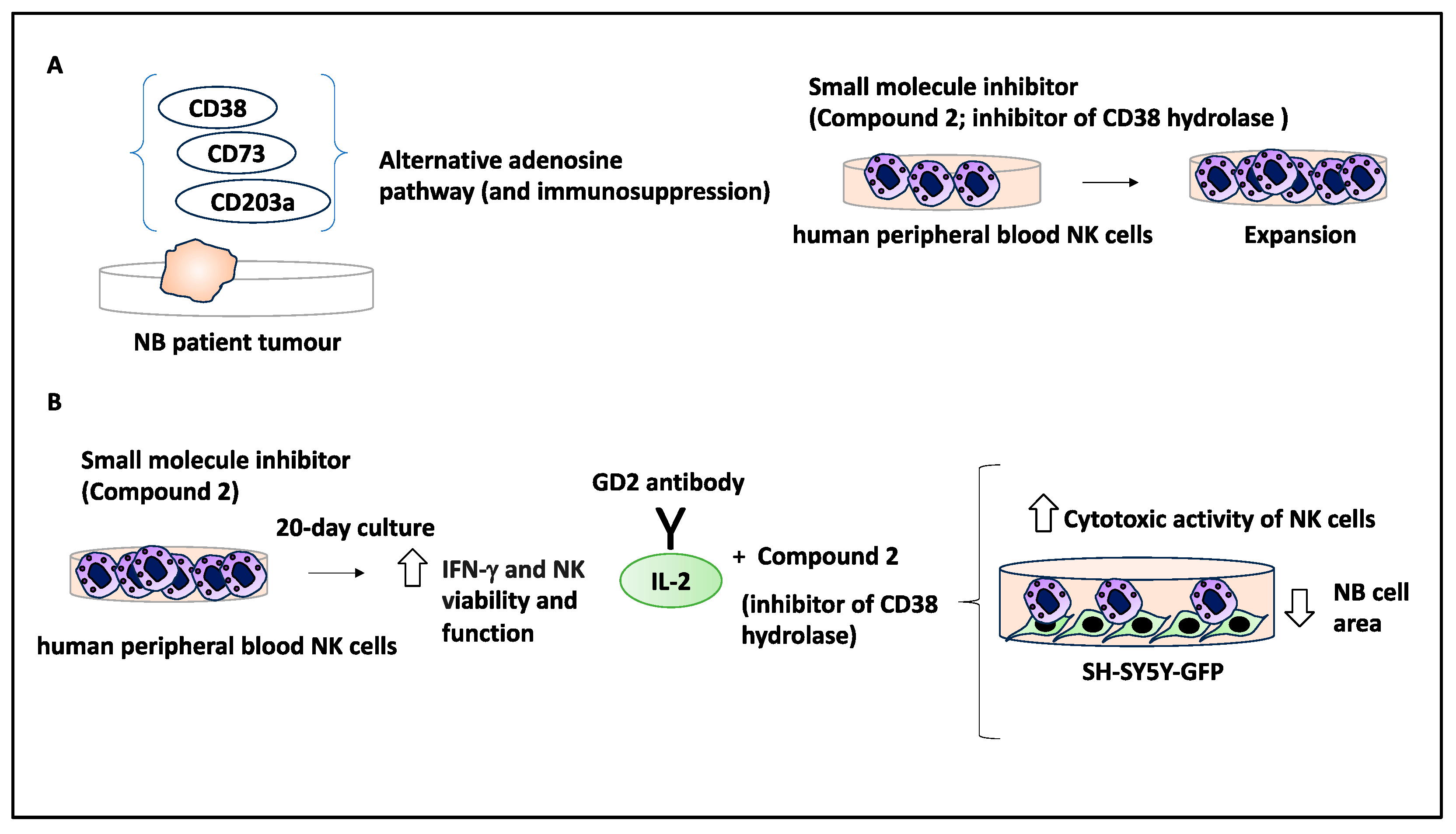
2.9. Small Molecule Inhibitors Could Stimulate an Immune Response in the TME
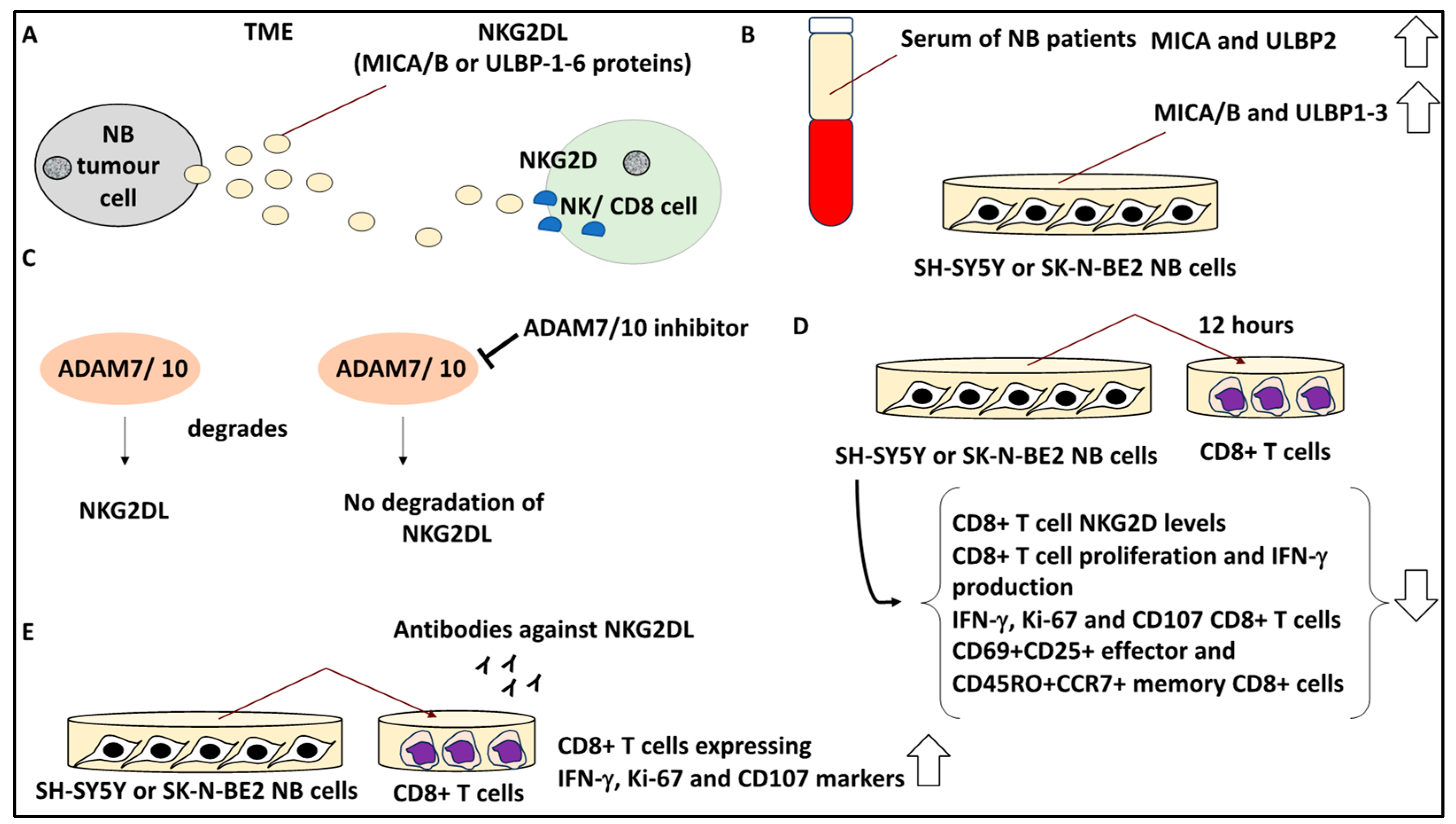
2.10. Targeting Soluble Ligands in the TME to Overcome Immunosuppression
2.11. Other Aspects of NB Interaction with the TME (CAR T Cells and Hypoxia)
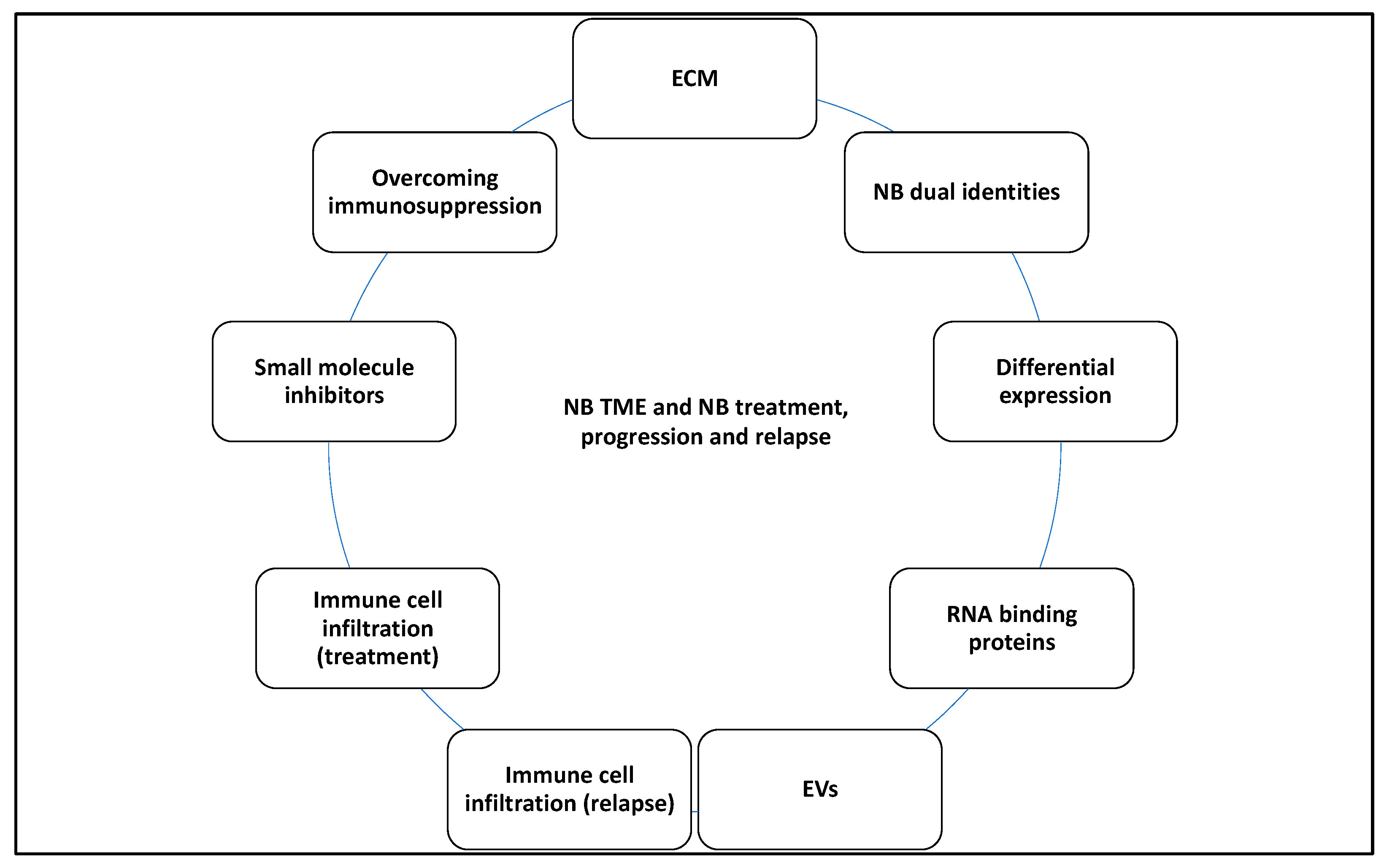
3. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADCC: | Antibody-dependent cell-mediated cytotoxicity |
| α-SMA: | α smooth muscle actin |
| B3GALT4: | Beta-1,3-galactosyltransferase |
| BM-MSCs: | Bone marrow-derived mesenchymal stem cells |
| CAFs: | Cancer-associated fibroblasts |
| CAR: | Chimeric antigen receptor |
| CRCs: | Core regulatory circuitries |
| ECM: | Extracellular matrix |
| EVs: | Extracellular vesicles |
| Fab-CHP: | FAM conjugated to collagen hybridising peptides |
| FAK: | Focal adhesion kinase |
| IDRFs: | Image-defined risk factors |
| IHC: | Immunohistochemistry |
| INRG: | International Neuroblastoma Risk Group |
| INRGSS: | International Neuroblastoma Risk Group Staging System |
| INSS: | International Neuroblastoma Staging System |
| IRF9: | Interferon regulatory factor 9 |
| NK: | Natural killer |
| NMT: | Noradrenergic-to-mesenchymal transition |
| MDSCs: | Myeloid-derived regulatory cells |
| MS: | Metastatic special |
| PBMC: | Peripheral blood mononuclear cells |
| PD-1: | Programmed cell death protein 1 |
| PDX: | Patient-derived xenograft |
| PMN: | Premetastatic niche |
| PTBP2: | Polypyrimidine tract binding protein 2 |
| TAMs: | Tumour-associated macrophages |
| TEVs: | Tumour extracellular vesicles |
| TFs: | Transcription factors |
| TME: | Tumour microenvironment |
| Treg: | Regulatory T cells |
| TrkA: | Neurotrophin receptor tropomyosin-related kinase |
| RA: | Retinoic acid |
| SCPs: | Schwann cell precursors |
| SEMA3A: | Semaphorin 3A |
| SHMT2: | Serine hydroxymethyltransferase 2 |
References
- Park, J.R.; Eggert, A.; Caron, H. Neuroblastoma: Biology, Prognosis, and Treatment. Hematol. Oncol. Clin. N. Am. 2010, 24, 65–86. [Google Scholar] [CrossRef]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.A.; Abdessalam, S.; Aldrink, J.H.; Austin, M.; Heaton, T.E.; Bruny, J.; Ehrlich, P.; Dasgupta, R.; Baertschiger, R.M.; Lautz, T.B.; et al. Update on neuroblastoma. J. Pediatr. Surg. 2019, 54, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef]
- Tomolonis, J.A.; Agarwal, S.; Shohet, J.M. Neuroblastoma pathogenesis: Deregulation of embryonic neural crest development. Cell Tissue Res. 2018, 372, 245–262. [Google Scholar] [CrossRef]
- Kameneva, P.; Artemov, A.V.; Kastriti, M.E.; Faure, L.; Olsen, T.K.; Otte, J.; Erickson, A.; Semsch, B.; Andersson, E.R.; Ratz, M.; et al. Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat. Genet. 2021, 53, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Bruno, G.; Nastasi, N.; Subbiani, A.; Boaretto, A.; Ciullini Mannurita, S.; Mattei, G.; Nardini, P.; Della Bella, C.; Magi, A.; Pini, A.; et al. β3-adrenergic receptor on tumor-infiltrating lymphocytes sustains IFN-γ-dependent PD-L1 expression and impairs anti-tumor immunity in neuroblastoma. Cancer Gene Ther. 2023, 30, 890–904. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Pritchard, J.; Berthold, F.; Carlsen, N.L.; Castel, V.; Castelberry, R.P.; De Bernardi, B.; Evans, A.E.; Favrot, M.; Hedborg, F. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 1993, 11, 1466–1477. [Google Scholar] [CrossRef]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.S.; Naranjo, A.; Zhang, F.F.; Cohn, S.L.; London, W.B.; Gastier-Foster, J.M.; Ramirez, N.C.; Pfau, R.; Reshmi, S.; Wagner, E.; et al. Revised Neuroblastoma Risk Classification System: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2021, 39, 3229–3241. [Google Scholar] [CrossRef]
- Peinemann, F.; Kahangire, D.A.; van Dalen, E.C.; Berthold, F. Rapid COJEC versus standard induction therapies for high-risk neuroblastoma. Cochrane Database Syst. Rev. 2015, 5, CD010774. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Prim. 2016, 2, 16078. [Google Scholar] [CrossRef]
- Bogen, D.; Brunner, C.; Walder, D.; Ziegler, A.; Abbasi, R.; Ladenstein, R.L.; Noguera, R.; Martinsson, T.; Amann, G.; Schilling, F.H.; et al. The genetic tumor background is an important determinant for heterogeneous MYCN-amplified neuroblastoma. Int. J. Cancer 2016, 139, 153–163. [Google Scholar] [CrossRef]
- Zafar, A.; Wang, W.; Liu, G.; Wang, X.; Xian, W.; McKeon, F.; Foster, J.; Zhou, J.; Zhang, R. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med. Res. Rev. 2021, 41, 961–1021. [Google Scholar] [CrossRef]
- Jahangiri, L. Profiling of the Prognostic Role of Extracellular Matrix-Related Genes in Neuroblastoma Using Databases and Integrated Bioinformatics. Onco 2022, 2, 85–112. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Noguera, R.; Nieto, O.A.; Tadeo, I.; Fariñas, F.; Alvaro, T. Extracellular matrix, biotensegrity and tumor microenvironment. An update and overview. Histol. Histopathol. 2012, 27, 693–705. [Google Scholar]
- Thirant, C.; Peltier, A.; Durand, S.; Kramdi, A.; Louis-Brennetot, C.; Pierre-Eugène, C.; Gautier, M.; Costa, A.; Grelier, A.; Zaïdi, S.; et al. Reversible transitions between noradrenergic and mesenchymal tumor identities define cell plasticity in neuroblastoma. Nat. Commun. 2023, 14, 2575. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; He, J.; Guo, H.; Lin, H.; Li, M.; Yang, T.; Wang, H.-Y.; Li, D.; Liu, J.; Li, L.; et al. PTBP2-Mediated Alternative Splicing of IRF9 Controls Tumor-Associated Monocyte/Macrophage Chemotaxis and Repolarization in Neuroblastoma Progression. Research 2023, 6, 33. [Google Scholar] [CrossRef] [PubMed]
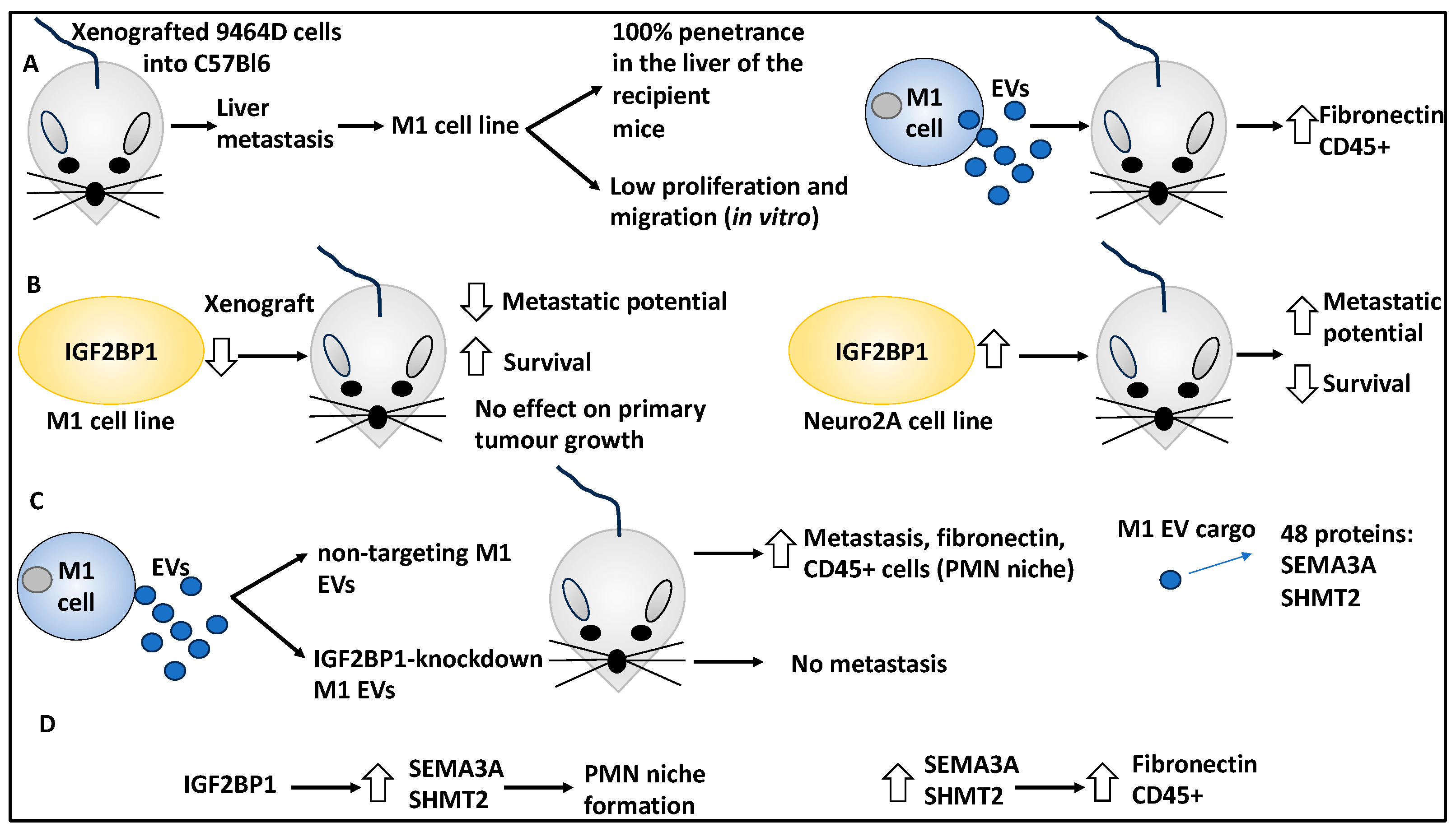
- Dhamdhere, M.R.; Gowda, C.P.; Singh, V.; Liu, Z.; Carruthers, N.; Grant, C.N.; Sharma, A.; Dovat, S.; Sundstrom, J.M.; Wang, H.-G.; et al. IGF2BP1 regulates the cargo of extracellular vesicles and promotes neuroblastoma metastasis. Oncogene 2023, 42, 1558–1571. [Google Scholar] [CrossRef]
- Verhoeven, B.M.; Mei, S.; Olsen, T.K.; Gustafsson, K.; Valind, A.; Lindström, A.; Gisselsson, D.; Fard, S.S.; Hagerling, C.; Kharchenko, P.V.; et al. The immune cell atlas of human neuroblastoma. Cell Rep. Med. 2022, 3, 100657. [Google Scholar] [CrossRef]
- Yuan, X.; Seneviratne, J.A.; Du, S.; Xu, Y.; Chen, Y.; Jin, Q.; Jin, X.; Balachandran, A.; Huang, S.; Xu, Y.; et al. Single-cell profiling of peripheral neuroblastic tumors identifies an aggressive transitional state that bridges an adrenergic-mesenchymal trajectory. Cell Rep. 2022, 41, 111455. [Google Scholar] [CrossRef]
- Jahangiri, L.; Ishola, T. Exosomes in Neuroblastoma Biology, Diagnosis, and Treatment. Life 2022, 12, 1714. [Google Scholar] [CrossRef]
- Marimpietri, D.; Petretto, A.; Raffaghello, L.; Pezzolo, A.; Gagliani, C.; Tacchetti, C.; Mauri, P.; Melioli, G.; Pistoia, V. Proteome profiling of neuroblastoma-derived exosomes reveal the expression of proteins potentially involved in tumor progression. PLoS ONE 2013, 8, e75054. [Google Scholar] [CrossRef]
- Fonseka, P.; Liem, M.; Ozcitti, C.; Adda, C.G.; Ang, C.-S.; Mathivanan, S. Exosomes from N-Myc amplified neuroblastoma cells induce migration and confer chemoresistance to non-N-Myc amplified cells: Implications of intra-tumour heterogeneity. J. Extracell. Vesicles 2019, 8, 1597614. [Google Scholar] [CrossRef]
- Sauvage, D.; Bosseler, M.; Viry, E.; Kanli, G.; Oudin, A.; Berchem, G.; Keunen, O.; Janji, B. The BET Protein Inhibitor JQ1 Decreases Hypoxia and Improves the Therapeutic Benefit of Anti-PD-1 in a High-Risk Neuroblastoma Mouse Model. Cells 2022, 11, 2783. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, F.; Dong, K. Soluble NKG2D ligands impair CD8(+) T cell antitumor function dependent of NKG2D downregulation in neuroblastoma. Oncol. Lett. 2023, 26, 297. [Google Scholar] [CrossRef]
- Hashimoto, O.; Yoshida, M.; Koma, Y.-I.; Yanai, T.; Hasegawa, D.; Kosaka, Y.; Nishimura, N.; Yokozaki, H. Collaboration of cancer-associated fibroblasts and tumour-associated macrophages for neuroblastoma development. J. Pathol. 2016, 240, 211–223. [Google Scholar] [CrossRef]
- Costa, A.; Thirant, C.; Kramdi, A.; Pierre-Eugène, C.; Louis-Brennetot, C.; Blanchard, O.; Surdez, D.; Gruel, N.; Lapouble, E.; Pierron, G.; et al. Single-cell transcriptomics reveals shared immunosuppressive landscapes of mouse and human neuroblastoma. J. Immunother. Cancer 2022, 10, e004807. [Google Scholar] [CrossRef]
- Das Thakur, M.; Franz, C.J.; Brennan, L.; Brouwer-Visser, J.; Tam, R.; Korski, K.; Koeppen, H.; Ziai, J.; Babitzki, G.; Ranchere-Vince, D.; et al. Immune contexture of paediatric cancers. Eur. J. Cancer 2022, 170, 179–193. [Google Scholar] [CrossRef]
- Truong, D.Q.; Ho, B.T.; Chau, G.-C.; Truong, D.K.; Pham, T.T.T.; Nakagawara, A.; Bui, C.-B. Collagen XI Alpha 1 (COL11A1) Expression in the Tumor Microenvironment Drives Neuroblastoma Dissemination. Pediatr. Dev. Pathol. 2022, 25, 91–98. [Google Scholar] [CrossRef]
- Lam, W.A.; Cao, L.; Umesh, V.; Keung, A.J.; Sen, S.; Kumar, S. Extracellular matrix rigidity modulates neuroblastoma cell differentiation and N-myc expression. Mol. Cancer 2010, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Bui, C.-B.; To, K.D.; Vu, D.M.; Nguyen, Q.-G.; Nguyen, H.T.; Nguyen, S.-B. Denatured collagen inhibits neuroblastoma tumor-sphere migration and growth via the LOX/LOXL2—FAK signaling pathway. J. Therm. Biol. 2023, 115, 103624. [Google Scholar] [CrossRef] [PubMed]
- López-Carrasco, A.; Martín-Vañó, S.; Burgos-Panadero, R.; Monferrer, E.; Berbegall, A.P.; Fernández-Blanco, B.; Navarro, S.; Noguera, R. Impact of extracellular matrix stiffness on genomic heterogeneity in MYCN-amplified neuroblastoma cell line. J. Exp. Clin. Cancer Res. 2020, 39, 226. [Google Scholar] [CrossRef]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- van Groningen, T.; Akogul, N.; Westerhout, E.M.; Chan, A.; Hasselt, N.E.; Zwijnenburg, D.A.; Broekmans, M.; Stroeken, P.; Haneveld, F.; Hooijer, G.K.J.; et al. A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat. Commun. 2019, 10, 1530. [Google Scholar] [CrossRef]
- Jahangiri, L.; Tsaprouni, L.; Trigg, R.M.; Williams, J.A.; Gkoutos, G.V.; Turner, S.D.; Pereira, J. Core regulatory circuitries in defining cancer cell identity across the malignant spectrum. Open Biol. 2020, 10, 200121. [Google Scholar] [CrossRef]
- Jahangiri, L.; Pucci, P.; Ishola, T.; Pereira, J.; Cavanagh, M.L.; Turner, S.D. Deep analysis of neuroblastoma core regulatory circuitries using online databases and integrated bioinformatics shows their pan-cancer roles as prognostic predictors. Discov. Oncol. 2021, 12, 56. [Google Scholar] [CrossRef]
- Sha, Y.-L.; Liu, Y.; Yang, J.-X.; Wang, Y.-Y.; Gong, B.-C.; Jin, Y.; Qu, T.-Y.; Xia, F.-T.; Han, L.; Zhao, Q. B3GALT4 remodels the tumor microenvironment through GD2-mediated lipid raft formation and the c-met/AKT/mTOR/IRF-1 axis in neuroblastoma. J. Exp. Clin. Cancer Res. 2022, 41, 314. [Google Scholar] [CrossRef]
- Jahangiri, L. Metastasis in Neuroblastoma and Its Link to Autophagy. Life 2023, 13, 818. [Google Scholar] [CrossRef]
- Jahangiri, L.; Ishola, T. Dormancy in Breast Cancer, the Role of Autophagy, lncRNAs, miRNAs and Exosomes. Int. J. Mol. Sci. 2022, 23, 5271. [Google Scholar] [CrossRef] [PubMed]
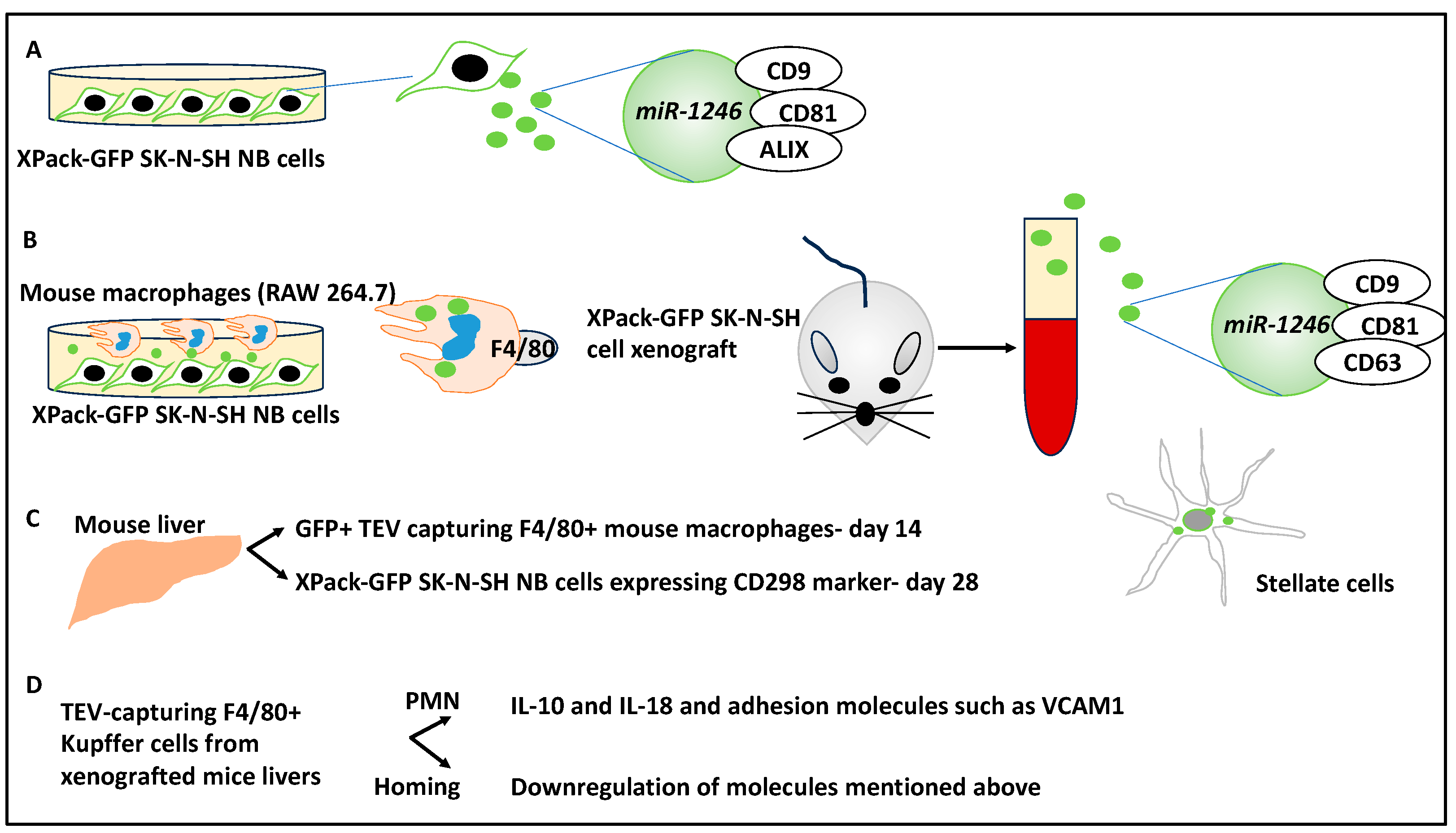
- Blavier, L.; Nakata, R.; Neviani, P.; Sharma, K.; Shimada, H.; Benedicto, A.; Matei, I.; Lyden, D.; DeClerck, Y.A. The capture of extracellular vesicles endogenously released by xenotransplanted tumours induces an inflammatory reaction in the premetastatic niche. J. Extracell. Vesicles 2023, 12, e12326. [Google Scholar] [CrossRef] [PubMed]
- Proestler, E.; Donzelli, J.; Nevermann, S.; Breitwieser, K.; Koch, L.F.; Best, T.; Fauth, M.; Wickström, M.; Harter, P.N.; Kogner, P.; et al. The multiple functions of miR-574-5p in the neuroblastoma tumor microenvironment. Front. Pharmacol. 2023, 14, 1183720. [Google Scholar] [CrossRef]
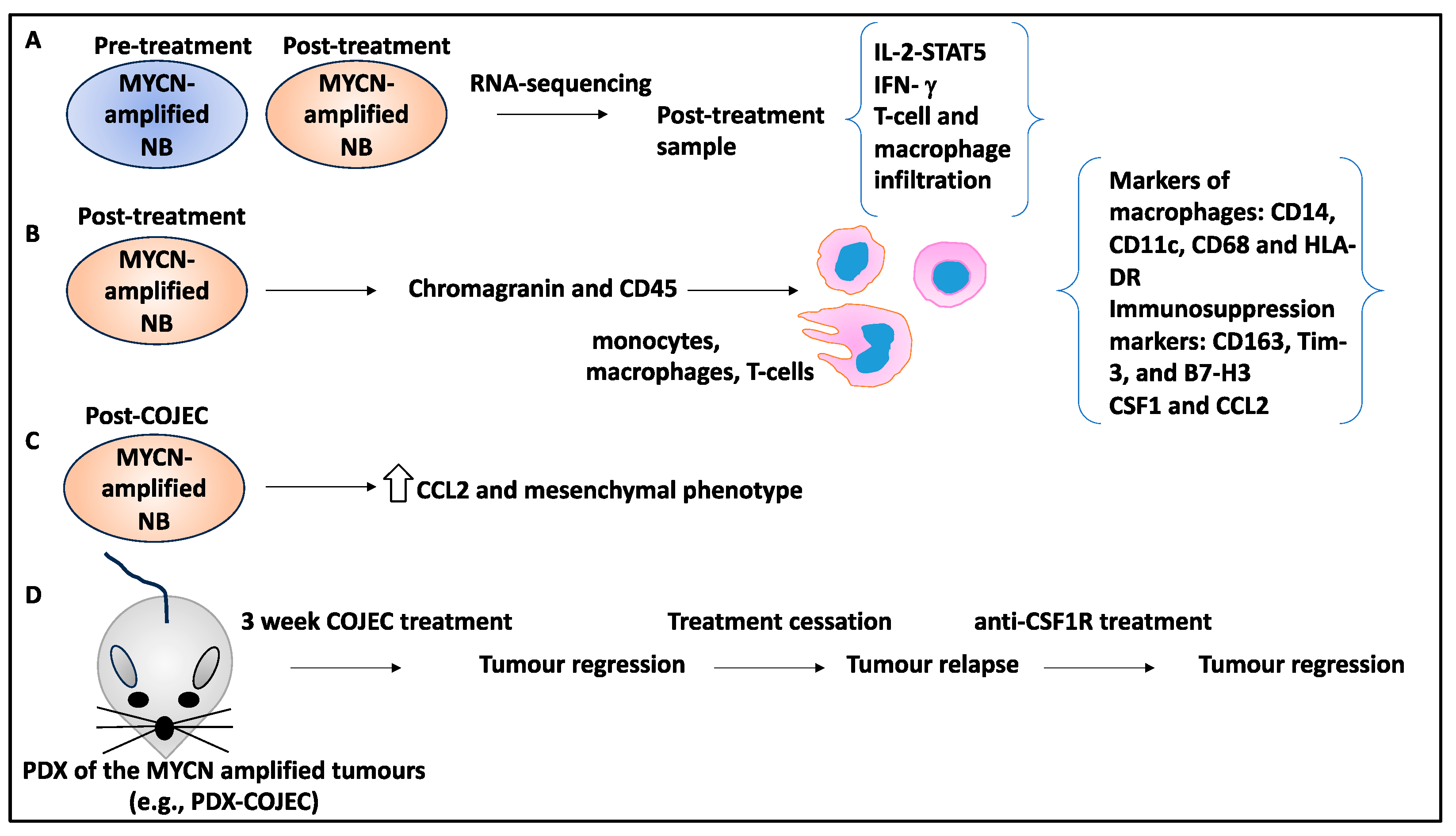
- Valind, A.; Verhoeven, B.M.; Enoksson, J.; Karlsson, J.; Christensson, G.; Mañas, A.; Aaltonen, K.; Jansson, C.; Bexell, D.; Baryawno, N.; et al. Macrophage infiltration promotes regrowth in MYCN-amplified neuroblastoma after chemotherapy. Oncoimmunology 2023, 12, 2184130. [Google Scholar] [CrossRef]
- Pierce, J.H.; Di Marco, E.; Cox, G.W.; Lombardi, D.; Ruggiero, M.; Varesio, L.; Wang, L.M.; Choudhury, G.G.; Sakaguchi, A.Y.; Di Fiore, P.P. Macrophage-colony-stimulating factor (CSF-1) induces proliferation, chemotaxis, and reversible monocytic differentiation in myeloid progenitor cells transfected with the human c-fms/CSF-1 receptor cDNA. Proc. Natl. Acad. Sci. USA 1990, 87, 5613–5617. [Google Scholar] [CrossRef]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef] [PubMed]
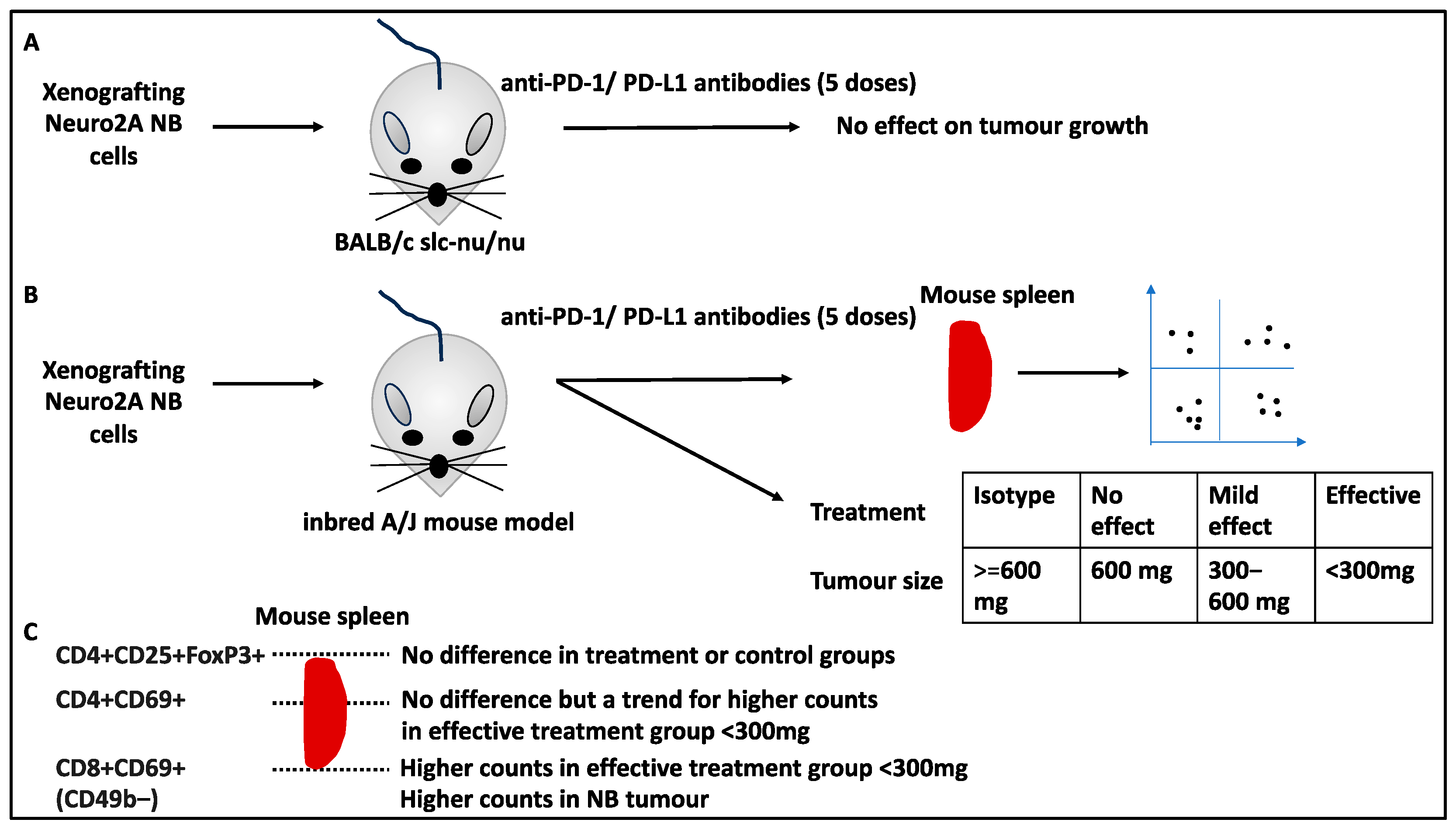
- Inoue, S.; Takeuchi, Y.; Horiuchi, Y.; Murakami, T.; Odaka, A. CD69 on Tumor-Infiltrating Cells Correlates with Neuroblastoma Suppression by Simultaneous PD-1 and PD-L1 Blockade. J. Surg. Res. 2023, 289, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Gur, H.; Ozen, F.; Can Saylan, C.; Atasever-Arslan, B. Dinutuximab in the Treatment of High-Risk Neuroblastoma in Children. Clin. Med. Insights Ther. 2017, 9, 1179559X17719106. [Google Scholar] [CrossRef]
- Mills, C.M.; Benton, T.Z.; Piña, I.; Francis, M.J.; Reyes, L.; Dolloff, N.G.; Peterson, Y.K.; Woster, P.M. Stimulation of natural killer cells with small molecule inhibitors of CD38 for the treatment of neuroblastoma. Chem. Sci. 2023, 14, 2168–2182. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Domaica, C.I.; Zwirner, N.W. Leveraging NKG2D Ligands in Immuno-Oncology. Front. Immunol. 2021, 12, 713158. [Google Scholar] [CrossRef]
- Nguyen, R.; Zhang, X.; Sun, M.; Abbas, S.; Seibert, C.; Kelly, M.C.; Shern, J.F.; Thiele, C.J. Anti-GD2 Antibodies Conjugated to IL15 and IL21 Mediate Potent Antitumor Cytotoxicity against Neuroblastoma. Clin. Cancer Res. 2022, 28, 3785–3796. [Google Scholar] [CrossRef]
- Sun, M.; Cao, Y.; Okada, R.; Reyes-González, J.M.; Stack, H.G.; Qin, H.; Li, N.; Seibert, C.; Kelly, M.C.; Ruppin, E.; et al. Preclinical optimization of a GPC2-targeting CAR T-cell therapy for neuroblastoma. J. Immunother. Cancer 2023, 11, e005881. [Google Scholar] [CrossRef]
- Correia, A.S.; Marques, L.; Vale, N. The Involvement of Hypoxia in the Response of Neuroblastoma Cells to the Exposure of Atorvastatin. Curr. Issues Mol. Biol. 2023, 45, 3333–3346. [Google Scholar] [CrossRef]
- Discher, D.E.; Janmey, P.; Wang, Y.-L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.J.; Renner, C.M.; Hale, I.; Yang, X.; Ponik, S.M.; Weisman, P.S.; Masters, K.S.; Kreeger, P.K. Scaffold stiffness influences breast cancer cell invasion via EGFR-linked Mena upregulation and matrix remodeling. Matrix Biol. 2020, 85–86, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.; Murphy, C.; O’Brien, F.J.; Piskareva, O. Three-dimensional In Vitro Biomimetic Model of Neuroblastoma using Collagen-based Scaffolds. J. Vis. Exp. 2021, 173, e62627. [Google Scholar]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugène, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, A.; Farina, A.R.; Cappabianca, L.; Desantis, G.; Tessitore, A.; Vetuschi, A.; Sferra, R.; Rucci, N.; Argenti, B.; Screpanti, I.; et al. TrkA alternative splicing: A regulated tumor-promoting switch in human neuroblastoma. Cancer Cell 2004, 6, 347–360. [Google Scholar] [CrossRef]
- Bao, R.; Spranger, S.; Hernandez, K.; Zha, Y.; Pytel, P.; Luke, J.J.; Gajewski, T.F.; Volchenboum, S.L.; Cohn, S.L.; Desai, A.V. Immunogenomic determinants of tumor microenvironment correlate with superior survival in high-risk neuroblastoma. J. Immunother. Cancer 2021, 9, e002417. [Google Scholar] [CrossRef]
- Liu, X.; Wills, C.A.; Chen, L.; Zhang, J.; Zhao, Y.; Zhou, M.; Sundstrom, J.M.; Schell, T.; Spiegelman, V.S.; Young, M.M.; et al. Small extracellular vesicles induce resistance to anti-GD2 immunotherapy unveiling tipifarnib as an adjunct to neuroblastoma immunotherapy. J. Immunother. Cancer 2022, 10, e004399. [Google Scholar] [CrossRef] [PubMed]
- Rohila, D.; Park, I.H.; Pham, T.V.; Jones, R.; Tapia, E.; Liu, K.X.; Tamayo, P.; Yu, A.; Sharabi, A.B.; Joshi, S. Targeting macrophage Syk enhances responses to immune checkpoint blockade and radiotherapy in high-risk neuroblastoma. Front. Immunol. 2023, 14, 1148317. [Google Scholar] [CrossRef] [PubMed]
- Vanichapol, T.; Chiangjong, W.; Panachan, J.; Anurathapan, U.; Chutipongtanate, S.; Hongeng, S. Secretory High-Mobility Group Box 1 Protein Affects Regulatory T Cell Differentiation in Neuroblastoma Microenvironment In Vitro. J. Oncol. 2018, 2018, 7946021. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahangiri, L. Neuroblastoma Interaction with the Tumour Microenvironment and Its Implications for Treatment and Disease Progression. Curr. Oncol. 2023, 30, 9116-9140. https://doi.org/10.3390/curroncol30100659
Jahangiri L. Neuroblastoma Interaction with the Tumour Microenvironment and Its Implications for Treatment and Disease Progression. Current Oncology. 2023; 30(10):9116-9140. https://doi.org/10.3390/curroncol30100659
Chicago/Turabian StyleJahangiri, Leila. 2023. "Neuroblastoma Interaction with the Tumour Microenvironment and Its Implications for Treatment and Disease Progression" Current Oncology 30, no. 10: 9116-9140. https://doi.org/10.3390/curroncol30100659