
Integrated Genomic and Bioinformatics Approaches to Identify Molecular Links between Endocrine Disruptors and Adverse Outcomes

 , and
, and
Abstract
:1. Introduction
2. Endocrine Disruptors
2.1. Pesticides and Biocides
2.2. Plastics and Microplastics
2.3. Perfluoroalkyl Substances (PFASs)
2.4. Polychlorinated Biphenyls (PCBs)
2.5. Synthetic Estrogens

{kind=link}
| Chemical | Class | Presence in Humans |
|---|---|---|
| BPA | Plasticizer | 2.68–4.74 ng/mL urine, 1.56–1.71 ng/mL serum, 0.74 ng/mL breast milk, 0.53–12.7 ng/g placenta [90] 3.78 ng/g adipose tissue, 1.48 ng/g liver, 0.91 ng/g brain [91]. |
| DEHP | Plasticizer | 34.05 ± 26.00 ng/mL breast milk [92] 0.3–1.0 ppm adipose tissue [93]. |
| DBP | Plasticizer | 26.61 ± 18.03 ng/mL breast milk [92]. |
| PFOA | PFASs | 44.4 ng/mL serum [94]. |
| PFOS | PFASs | 3.9 ng/mL serum [94], 41.9 ng/g liver [95], 1.24 ng/g placenta [96]. |
| DDT | Pesticide | 162.95 ± 156.44 ng/g lipid (breast milk) [92], 0.969–3.710 mg/kg adipose tissue [97]. |
| PCBs | PCBs | 175.05 ± 85.22 ng/g lipid (breast milk) [92], 1.51–3.65 ng/g adipose tissue [97], 18.9–816 ng/kg adipose tissue [98]. |
3. Environmental and Human Health Implications of EDC Exposure
3.1. Impacts on Aquatic Species
3.1.1. Fertility
3.1.2. Cancer
3.1.3. Metabolic Disease
3.1.4. Neurological Effects
3.2. Health Impacts in Animals
3.2.1. Fertility
3.2.2. Cancer
3.2.3. Metabolic Disease
3.2.4. Neurological Effects
3.2.5. Asthma
3.3. Health Effects in Humans
3.3.1. Fertility
3.3.2. Cancer
3.3.3. Metabolic Disease
3.3.4. Neurological Effects
3.3.5. Asthma
4. Genomics and Bioinformatics Approaches in Endocrine Disruptor Research
4.1. RNA Sequencing
4.2. RNAseq for the Study of Endocrine Disruption
5. Risk Assessment Based on Genomic Technologies
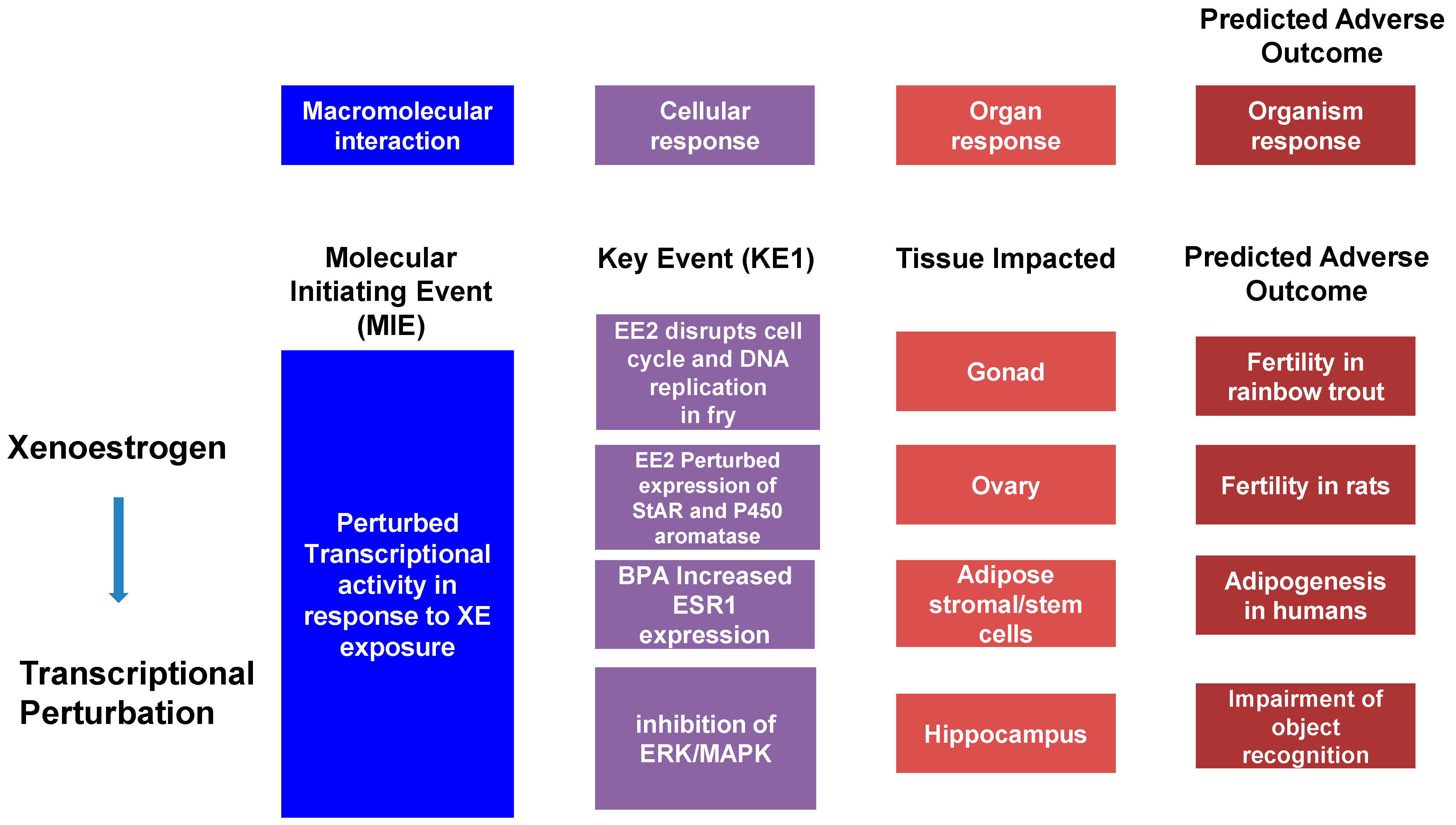
6. The Adverse Outcomes Framework
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, G.; White, P.A. The Mutagenic Hazards of Aquatic Sediments: A Review. Mutat. Res. 2004, 567, 151–225. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.E.; Ruggeri, B.; Sprague, L.J.; Eckhardt-Ludka, C.; Lapira, J.; Wick, I.; Soverchia, L.; Ubaldi, M.; Polzonetti-Magni, A.M.; Vidal-Dorsch, D.; et al. Analysis of Endocrine Disruption in Southern California Coastal Fish Using an Aquatic Multispecies Microarray. Environ. Health Perspect. 2009, 117, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Grün, F.; Watanabe, H.; Zamanian, Z.; Maeda, L.; Arima, K.; Cubacha, R.; Gardiner, D.M.; Kanno, J.; Iguchi, T.; Blumberg, B. Endocrine-Disrupting Organotin Compounds Are Potent Inducers of Adipogenesis in Vertebrates. Mol. Endocrinol. 2006, 20, 2141–2155. [Google Scholar] [CrossRef]
- Lam, S.H.; Mathavan, S.; Tong, Y.; Li, H.; Karuturi, R.K.M.; Wu, Y.; Vega, V.B.; Liu, E.T.; Gong, Z. Zebrafish Whole-Adult-Organism Chemogenomics for Large-Scale Predictive and Discovery Chemical Biology. PLoS Genet. 2008, 4, e1000121. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Paull, G.C.; Coe, T.S.; Katsu, Y.; Urushitani, H.; Iguchi, T.; Tyler, C.R. Sexual Reprogramming and Estrogenic Sensitization in Wild Fish Exposed to Ethinylestradiol. Environ. Sci. Technol. 2009, 43, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Soto, A.M.; Vandenberg, L.N.; Maffini, M.V.; Sonnenschein, C. Does Breast Cancer Start in the Womb? Basic Clin. Pharmacol. Toxicol. 2008, 102, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Vandenberg, L.N.; Maffini, M.V.; Sonnenschein, C.; Rubin, B.S.; Soto, A.M. Bisphenol-A and the Great Divide: A Review of Controversies in the Field of Endocrine Disruption. Endocr. Rev. 2009, 30, 75–95. [Google Scholar] [CrossRef]
- La Merrill, M.A.; Vandenberg, L.N.; Smith, M.T.; Goodson, W.; Browne, P.; Patisaul, H.B.; Guyton, K.Z.; Kortenkamp, A.; Cogliano, V.J.; Woodruff, T.J.; et al. Consensus on the Key Characteristics of Endocrine-Disrupting Chemicals as a Basis for Hazard Identification. Nat. Rev. Endocrinol. 2020, 16, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Vajda, A.M.; Barber, L.B.; Gray, J.L.; Lopez, E.M.; Woodling, J.D.; Norris, D.O. Reproductive Disruption in Fish Downstream from an Estrogenic Wastewater Effluent. Environ. Sci. Technol. 2008, 42, 3407–3414. [Google Scholar] [CrossRef]
- Toporova, L.; Balaguer, P. Nuclear Receptors Are the Major Targets of Endocrine Disrupting Chemicals. Mol. Cell. Endocrinol. 2020, 502, 110665. [Google Scholar] [CrossRef]
- Litwa, E.; Rzemieniec, J.; Wnuk, A.; Lason, W.; Krzeptowski, W.; Kajta, M. RXRalpha, PXR and CAR Xenobiotic Receptors Mediate the Apoptotic and Neurotoxic Actions of Nonylphenol in Mouse Hippocampal Cells. J. Steroid Biochem. Mol. Biol. 2016, 156, 43–52. [Google Scholar] [CrossRef]
- Huang, W.; Wu, Q.; Xu, F.; Li, L.; Li, J.; Que, H.; Zhang, G. Functional Characterization of Retinoid X Receptor with an Emphasis on the Mediation of Organotin Poisoning in the Pacific Oyster (Crassostrea gigas). Gene 2020, 753, 144780. [Google Scholar] [CrossRef] [PubMed]
- Inderbinen, S.G.; Engeli, R.T.; Rohrer, S.R.; Di Renzo, E.; Aengenheister, L.; Buerki-Thurnherr, T.; Odermatt, A. Tributyltin and Triphenyltin Induce 11β-Hydroxysteroid Dehydrogenase 2 Expression and Activity through Activation of Retinoid X Receptor α. Toxicol. Lett. 2020, 322, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Milton, F.A.; Lacerda, M.G.; Sinoti, S.B.P.; Mesquita, P.G.; Prakasan, D.; Coelho, M.S.; de Lima, C.L.; Martini, A.G.; Pazzine, G.T.; Borin, M.d.F.; et al. Dibutyltin Compounds Effects on PPARγ/RXRα Activity, Adipogenesis, and Inflammation in Mammalians Cells. Front. Pharm. 2017, 8, 507. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.; Madureira, T.V.; Ferreira, N.; Pinheiro, I.; Castro, L.F.; Rocha, E. Peroxisome Proliferator-Activated Receptor Gamma (PPARgamma) in Brown Trout: Interference of Estrogenic and Androgenic Inputs in Primary Hepatocytes. Environ. Toxicol. Pharm. 2016, 46, 328–336. [Google Scholar] [CrossRef]
- Fang, H.; Fang, W.; Cao, H.; Luo, S.; Cong, J.; Liu, S.; Pan, F.; Jia, X. Di-(2-Ethylhexyl)-Phthalate Induces Apoptosis via the PPARγ/PTEN/AKT Pathway in Differentiated Human Embryonic Stem Cells. Food Chem. Toxicol. 2019, 131, 110552. [Google Scholar] [CrossRef] [PubMed]
- Shoaito, H.; Petit, J.; Chissey, A.; Auzeil, N.; Guibourdenche, J.; Gil, S.; Laprévote, O.; Fournier, T.; Degrelle, S.A. The Role of Peroxisome Proliferator–Activated Receptor Gamma (PPARγ) in Mono(2-Ethylhexyl) Phthalate (MEHP)-Mediated Cytotrophoblast Differentiation. Environ. Health Perspect. 2019, 127, 27003. [Google Scholar] [CrossRef]
- Parillo, F.; Maranesi, M.; Brecchia, G.; Gobbetti, A.; Boiti, C.; Zerani, M. In Vivo Chronic and in Vitro Acute Effects of Di(2-Ethylhexyl) Phthalate on Pseudopregnant Rabbit Corpora Lutea: Possible Involvement of Peroxisome Proliferator-Activated Receptor Gamma. Biol. Reprod. 2014, 90, 41. [Google Scholar] [CrossRef]
- Kim, Y.S.; Hwang, K.A.; Hyun, S.H.; Nam, K.H.; Lee, C.K.; Choi, K.C. Bisphenol A and Nonylphenol Have the Potential to Stimulate the Migration of Ovarian Cancer Cells by Inducing Epithelial-Mesenchymal Transition via an Estrogen Receptor Dependent Pathway. Chem. Res. Toxicol. 2015, 28, 662–671. [Google Scholar] [CrossRef]
- Kitraki, E.; Nalvarte, I.; Alavian-Ghavanini, A.; Ruegg, J. Developmental Exposure to Bisphenol A Alters Expression and DNA Methylation of Fkbp5, an Important Regulator of the Stress Response. Mol. Cell. Endocrinol. 2015, 417, 191–199. [Google Scholar] [CrossRef]
- Jiang, J.; Ma, L.; Yuan, L.; Wang, X.; Zhang, W. Study on Developmental Abnormalities in Hypospadiac Male Rats Induced by Maternal Exposure to Di-n-Butyl Phthalate (DBP). Toxicology 2007, 232, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lehmann, K.P.; Sar, M.; Young, S.S.; Gaido, K.W. Gene Expression Profiling Following in Utero Exposure to Phthalate Esters Reveals New Gene Targets in the Etiology of Testicular Dysgenesis. Biol. Reprod. 2005, 73, 180–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fudvoye, J.; Lopez-Rodriguez, D.; Franssen, D.; Parent, A.-S. Endocrine Disrupters and Possible Contribution to Pubertal Changes. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101300. [Google Scholar] [CrossRef]
- Saaristo, M.; Wong, B.B.; Mincarelli, L.; Craig, A.; Johnstone, C.P.; Allinson, M.; Lindstrom, K.; Craft, J.A. Characterisation of the Transcriptome of Male and Female Wild-Type Guppy Brains with RNA-Seq and Consequences of Exposure to the Pharmaceutical Pollutant, 17alpha-Ethinyl Estradiol. Aquat. Toxicol. 2017, 186, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Komada, M.; Gendai, Y.; Kagawa, N.; Nagao, T. Prenatal Exposure to Di(2-Ethylhexyl) Phthalate Impairs Development of the Mouse Neocortex. Toxicol. Lett. 2016, 259, 69–79. [Google Scholar] [CrossRef]
- Braun, J.M.; Kalkbrenner, A.E.; Calafat, A.M.; Yolton, K.; Ye, X.; Dietrich, K.N.; Lanphear, B.P. Impact of Early-Life Bisphenol A Exposure on Behavior and Executive Function in Children. Pediatrics 2011, 128, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, Z.; Han, H.; Luo, G.; Zhou, B.; Wang, S.; Wang, J. Impairment of Object Recognition Memory by Maternal Bisphenol A Exposure Is Associated with Inhibition of Akt and ERK/CREB/BDNF Pathway in the Male Offspring Hippocampus. Toxicology 2016, 341–343, 56–64. [Google Scholar] [CrossRef]
- Chen, F.; Zhou, L.; Bai, Y.; Zhou, R.; Chen, L. Hypothalamic-Pituitary-Adrenal Axis Hyperactivity Accounts for Anxiety- and Depression-like Behaviors in Rats Perinatally Exposed to Bisphenol A. J. Biomed. Res. 2015, 29, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.J.; Du, J.C.; Chiou, H.C.; Yang, S.H.; Liao, K.W.; Yang, W.; Chung, M.Y.; Chien, L.C.; Hwang, B.; Chen, M.L. Attention Deficit/Hyperactivity Disorder and Urinary Nonylphenol Levels: A Case-Control Study in Taiwanese Children. PLoS ONE 2016, 11, e0149558. [Google Scholar] [CrossRef]
- Morii, K.; Nishisaka, M.; Nakamura, S.; Oda, T.; Aoyama, Y.; Yamamoto, T.; Kishida, H.; Okushin, H.; Uesaka, K. A Case of Synthetic Oestrogen-Induced Autoimmune Hepatitis with Microvesicular Steatosis. J. Clin. Pharm. 2014, 39, 573–576. [Google Scholar] [CrossRef]
- Tao, J.; Sun, L.X.; Le, X.C. Study of the Effects of Bisphenol A Using Human Fetal Lung Fibroblasts. J. Environ. Sci. 2016, 48, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Koike, E.; Yasuda, Y.; Shiota, M.; Shimaoka, M.; Tsuritani, M.; Konishi, H.; Yamasaki, H.; Okumoto, K.; Hoshiai, H. Exposure to Ethinyl Estradiol Prenatally and/or after Sexual Maturity Induces Endometriotic and Precancerous Lesions in Uteri and Ovaries of Mice. Congenit. Anom. 2013, 53, 9–17. [Google Scholar] [CrossRef]
- Hu, W.Y.; Shi, G.B.; Hu, D.P.; Nelles, J.L.; Prins, G.S. Actions of Estrogens and Endocrine Disrupting Chemicals on Human Prostate Stem/Progenitor Cells and Prostate Cancer Risk. Mol. Cell. Endocrinol. 2012, 354, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Oral, D.; Erkekoglu, P.; Kocer-Gumusel, B.; Chao, M.W. Epithelial-Mesenchymal Transition: A Special Focus on Phthalates and Bisphenol A. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Huang, C.; Ma, X.; Wu, R.; Zhu, W.; Li, X.; Liang, Z.; Deng, F.; Wu, J.; Geng, S.; et al. Phthalates Promote Prostate Cancer Cell Proliferation through Activation of ERK5 and P38. Environ. Toxicol. Pharm. 2018, 63, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-A.; Hwang, K.-A.; Lee, H.-R.; Yi, B.-R.; Jeung, E.-B.; Choi, K.-C. Cell Growth of BG-1 Ovarian Cancer Cells Is Promoted by Di-n-Butyl Phthalate and Hexabromocyclododecane via Upregulation of the Cyclin D and Cyclin-Dependent Kinase-4 Genes. Mol. Med. Rep. 2012, 5, 761–766. [Google Scholar] [CrossRef]
- Grun, F.; Blumberg, B. Perturbed Nuclear Receptor Signaling by Environmental Obesogens as Emerging Factors in the Obesity Crisis. Rev. Endocr. Metab. Disord. 2007, 8, 161–171. [Google Scholar] [CrossRef]
- Marmugi, A.; Lasserre, F.; Beuzelin, D.; Ducheix, S.; Huc, L.; Polizzi, A.; Chetivaux, M.; Pineau, T.; Martin, P.; Guillou, H.; et al. Adverse Effects of Long-Term Exposure to Bisphenol A during Adulthood Leading to Hyperglycaemia and Hypercholesterolemia in Mice. Toxicology 2014, 325, 133–143. [Google Scholar] [CrossRef]
- Li, J.; Lai, H.; Chen, S.; Zhu, H.; Lai, S. Gender Differences in the Associations between Urinary Bisphenol A and Body Composition among American Children: The National Health and Nutrition Examination Survey, 2003–2006. J. Epidemiol. 2017, 27, 228–234. [Google Scholar] [CrossRef]
- Luzio, A.; Monteiro, S.M.; Rocha, E.; Fontainhas-Fernandes, A.A.; Coimbra, A.M. Development and Recovery of Histopathological Alterations in the Gonads of Zebrafish (Danio rerio) after Single and Combined Exposure to Endocrine Disruptors (17alpha-Ethinylestradiol and Fadrozole). Aquat. Toxicol. 2016, 175, 90–105. [Google Scholar] [CrossRef]
- Young, B.J.; Lopez, G.C.; Cristos, D.S.; Crespo, D.C.; Somoza, G.M.; Carriquiriborde, P. Intersex and Liver Alterations Induced by Long-Term Sublethal Exposure to 17alpha-Ethinylestradiol in Adult Male Cnesterodon decemmaculatus (Pisces: Poeciliidae). Environ. Toxicol. Chem. 2016, 36, 1738–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernabo, I.; Biasone, P.; Macirella, R.; Tripepi, S.; Brunelli, E. Liver Histology and Ultrastructure of the Italian Newt (Lissotriton Italicus): Normal Structure and Modifications after Acute Exposure to Nonylphenol Ethoxylates. Exp. Toxicol. Pathol. 2014, 66, 455–468. [Google Scholar] [CrossRef]
- Zhang, W.; Shen, X.Y.; Zhang, W.W.; Chen, H.; Xu, W.P.; Wei, W. The Effects of Di 2-Ethyl Hexyl Phthalate (DEHP) on Cellular Lipid Accumulation in HepG2 Cells and Its Potential Mechanisms in the Molecular Level. Toxicol. Mech. Methods 2017, 27, 245–252. [Google Scholar] [CrossRef]
- Huff, M.; da Silveira, W.A.; Carnevali, O.; Renaud, L.; Hardiman, G. Systems Analysis of the Liver Transcriptome in Adult Male Zebrafish Exposed to the Plasticizer (2-Ethylhexyl) Phthalate (DEHP). Sci. Rep. 2018, 8, 2118. [Google Scholar] [CrossRef] [Green Version]
- Radke, E.G.; Galizia, A.; Thayer, K.A.; Cooper, G.S. Phthalate Exposure and Metabolic Effects: A Systematic Review of the Human Epidemiological Evidence. Environ. Int. 2019, 132, 104768. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Cornelis, M.C.; Townsend, M.K.; Tobias, D.K.; Eliassen, A.H.; Franke, A.A.; Hauser, R.; Hu, F.B. Association of Urinary Concentrations of Bisphenol A and Phthalate Metabolites with Risk of Type 2 Diabetes: A Prospective Investigation in the Nurses’ Health Study (NHS) and NHSII Cohorts. Environ. Health Perspect. 2014, 122, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Badamasi, I.; Odong, R.; Masembe, C. Threats Posed by Xenoestrogenic Chemicals to the Aquatic Ecosystem, Fish Reproduction and Humans: A Review. Afr. J. Aquat. Sci. 2020, 45, 243–258. [Google Scholar] [CrossRef]
- Hutchinson, T.H.; Lyons, B.P.; Thain, J.E.; Law, R.J. Evaluating Legacy Contaminants and Emerging Chemicals in Marine Environments Using Adverse Outcome Pathways and Biological Effects-Directed Analysis. Mar. Pollut. Bull. 2013, 74, 517–525. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Hunt, P.A.; Gore, A.C. Endocrine Disruptors and the Future of Toxicology Testing—Lessons from CLARITY–BPA. Nat. Rev. Endocrinol. 2019, 15, 366–374. [Google Scholar] [CrossRef]
- Schneider, M.; Pons, J.-L.; Labesse, G.; Bourguet, W. In Silico Predictions of Endocrine Disruptors Properties. Endocrinology 2019, 160, 2709–2716. [Google Scholar] [CrossRef]
- Kar, S.; Sangem, P.; Anusha, N.; Senthilkumaran, B. Endocrine Disruptors in Teleosts: Evaluating Environmental Risks and Biomarkers. Aquac. Fish. 2021, 6, 1–26. [Google Scholar] [CrossRef]
- Carson, R. Silent Spring; Houghton Mifflin: New York, NY, USA, 1962. [Google Scholar]
- Eskenazi, B.; Chevrier, J.; Rosas, L.G.; Anderson, H.A.; Bornman, M.S.; Bouwman, H.; Chen, A.; Cohn, B.A.; de Jager, C.; Henshel, D.S.; et al. The Pine River Statement: Human Health Consequences of DDT Use. Environ. Health Perspect. 2009, 117, 1359–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, C.; Taylor, A.R.; Kenney, W.F.; Schlenk, D.; Gan, J. Historical Record and Fluxes of DDTs at the Palos Verdes Shelf Superfund Site, California. Sci. Total Environ. 2017, 581, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.W.; Reish, D.J.; Spies, R.B.; Brady, M.E.; Segelhorst, E.W. (Eds.) Human Impacts. In Ecology of the Southern California Bight; University of California Press: Berkeley, CA, USA, 2020; pp. 682–766. [Google Scholar]
- Siriwong, W.; Thirakhupt, K.; Sitticharoenchai, D.; Rohitrattana, J.; Thongkongowm, P.; Borjan, M.; Robson, M. DDT and Derivatives in Indicator Species of the Aquatic Food Web of Rangsit Agricultural Area, Central Thailand. Ecol. Indic. 2009, 9, 878–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, P.A.; Rasmussen, J.B.; Blaise, C. Genotoxic Substances in the St. Lawrence System I: Industrial Genotoxins Sorbed to Particulate Matter in the St. Lawrence, St. Maurice, and Saguenay Rivers, Canada. Environ. Toxicol. Chem. 1998, 17, 286–303. [Google Scholar] [CrossRef]
- White, P.A.; Rasmussen, J.B.; Blaise, C. Sorption of Organic Genotoxins to Particulate Matter in Industrial Effluents. Environ. Mol. Mutagenes. 1996, 27, 140–151. [Google Scholar] [CrossRef]
- Safe, S. Molecular Biology of the Ah Receptor and Its Role in Carcinogenesis. Toxicol. Lett. 2001, 120, 1–7. [Google Scholar] [CrossRef]
- Safe, S.; Wormke, M. Inhibitory Aryl Hydrocarbon Receptor-Estrogen Receptor Alpha Cross-Talk and Mechanisms of Action. Chem. Res. Toxicol. 2003, 16, 807–816. [Google Scholar] [CrossRef]
- Matthews, J.; Gustafsson, J.-A. Estrogen Receptor and Aryl Hydrocarbon Receptor Signaling Pathways. Nucl. Recept. Signal. 2006, 4, e016. [Google Scholar] [CrossRef]
- Biamis, C.; Driscoll, K.O.; Hardiman, G. Microplastic Toxicity: A Review of the Role of Marine Sentinel Species in Assessing the Environmental and Public Health Impacts. Case Stud. Chem. Environ. Eng. 2021, 3, 100073. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Kang, S.; Wang, Z.; Wu, C. Microplastics in Freshwater Sediment: A Review on Methods, Occurrence, and Sources. Sci. Total Environ. 2021, 754, 141948. [Google Scholar] [CrossRef] [PubMed]
- Coyle, R.; Hardiman, G.; Driscoll, K.O. Microplastics in the Marine Environment: A Review of Their Sources, Distribution Processes, Uptake and Exchange in Ecosystems. Case Stud. Chem. Environ. Eng. 2020, 2, 100010. [Google Scholar] [CrossRef]
- Letcher, R.J.; Bustnes, J.O.; Dietz, R.; Jenssen, B.M.; Jørgensen, E.H.; Sonne, C.; Verreault, J.; Vijayan, M.M.; Gabrielsen, G.W. Exposure and Effects Assessment of Persistent Organohalogen Contaminants in Arctic Wildlife and Fish. Sci. Total Environ. 2010, 408, 2995–3043. [Google Scholar] [CrossRef]
- Luoma, S.N. Bioavailability of Trace Metals to Aquatic Organisms—A Review. Sci. Total Environ. 1983, 28, 1–22. [Google Scholar] [CrossRef]
- Burton, G., Jr. Allen Sediment Quality Criteria in Use around the World. Limnology 2002, 3, 65–76. [Google Scholar] [CrossRef]
- Barboza, L.G.A.; Gimenez, B.C.G. Microplastics in the Marine Environment: Current Trends and Future Perspectives. Mar. Pollut. Bull. 2015, 97, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Arancio, A.L.; Cole, K.D.; Dominguez, A.R.; Cohenour, E.R.; Kadie, J.; Maloney, W.C.; Cilliers, C.; Schuh, S.M. Bisphenol A, Bisphenol AF, Di-n-Butyl Phthalate, and 17β-Estradiol Have Shared and Unique Dose-Dependent Effects on Early Embryo Cleavage Divisions and Development in Xenopus Laevis. Reprod. Toxicol. 2019, 84, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Vom Saal, F.S.; Vandenberg, L.N. Update on the Health Effects of Bisphenol A: Overwhelming Evidence of Harm. Endocrinology 2021, 162, bqaa171. [Google Scholar] [CrossRef]
- Harbinder, S.; Lazzara, C.A.; Klar, A.J. Implication of the Strand-Specific Imprinting and Segregation Model: Integrating in Utero Hormone Exposure, Stem Cell and Lateral Asymmetry Hypotheses in Breast Cancer Aetiology. Hered. Genet 2013, 2013, 005. [Google Scholar] [CrossRef] [Green Version]
- Henley, D.V.; Korach, K.S. Endocrine-Disrupting Chemicals Use Distinct Mechanisms of Action to Modulate Endocrine System Function. Endocrinology 2006, 147, S25–S32. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.J.; Samandar, E.; Reidy, J.A.; Hauser, R.; Needham, L.L.; Calafat, A.M. Metabolite Profiles of Di-n-Butyl Phthalate in Humans and Rats. Environ. Sci. Technol. 2007, 41, 7576–7580. [Google Scholar] [CrossRef]
- Bao, A.M.; Man, X.M.; Guo, X.J.; Dong, H.B.; Wang, F.Q.; Sun, H.; Wang, Y.B.; Zhou, Z.M.; Sha, J.H. Effects of Di-n-Butyl Phthalate on Male Rat Reproduction Following Pubertal Exposure. Asian J. 2011, 13, 702–709. [Google Scholar] [CrossRef]
- PFAS—Fidra. Available online: https://www.fidra.org.uk/project/pfas/ (accessed on 4 November 2021).
- Russell, M.H.; Berti, W.R.; Szostek, B.; Buck, R.C. Investigation of the Biodegradation Potential of a Fluoroacrylate Polymer Product in Aerobic Soils. Environ. Sci. Technol. 2008, 42, 800–807. [Google Scholar] [CrossRef]
- Washington, J.W.; Ellington, J.J.; Jenkins, T.M.; Evans, J.J.; Yoo, H.; Hafner, S.C. Degradability of an Acrylate-Linked, Fluorotelomer Polymer in Soil. Environ. Sci. Technol. 2009, 43, 6617–6623. [Google Scholar] [CrossRef]
- Lau, C.; Anitole, K.; Hodes, C.; Lai, D.; Pfahles-Hutchens, A.; Seed, J. Perfluoroalkyl Acids: A Review of Monitoring and Toxicological Findings. Toxicol. Sci. 2007, 99, 366–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenland, K.; Fletcher, T.; Savitz, D.A. Epidemiologic Evidence on the Health Effects of Perfluorooctanoic Acid (PFOA). Environ. Health Perspect. 2010, 118, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Gao, P.; Xiang, P.; Zhang, X.; Cui, X.; Ma, L.Q. Molecular Mechanisms of PFOA-Induced Toxicity in Animals and Humans: Implications for Health Risks. Environ. Int. 2017, 99, 43–54. [Google Scholar] [CrossRef]
- Kimbrough, R.D. Polychlorinated Biphenyls (PCBs) and Human Health: An Update. Crit. Rev. Toxicol. 1995, 25, 133–163. [Google Scholar] [CrossRef]
- La Rocca, C.; Mantovani, A. From Environment to Food: The Case of PCB. Ann.-Ist. Super. Sanita 2006, 42, 410–416. [Google Scholar] [PubMed]
- Serdar, B.; LeBlanc, W.G.; Norris, J.M.; Dickinson, L.M. Potential Effects of Polychlorinated Biphenyls (PCBs) and Selected Organochlorine Pesticides (OCPs) on Immune Cells and Blood Biochemistry Measures: A Cross-Sectional Assessment of the NHANES 2003-2004 Data. Environ. Health 2014, 13, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simhadri, J.J.; Loffredo, C.A.; Trnovec, T.; Murinova, L.P.; Nunlee-Bland, G.; Koppe, J.G.; Schoeters, G.; Jana, S.S.; Ghosh, S. Biomarkers of Metabolic Disorders and Neurobehavioral Diseases in a PCB-Exposed Population: What We Learned and the Implications for Future Research. Environ. Res. 2020, 191, 110211. [Google Scholar] [CrossRef]
- Buha Djordjevic, A.; Antonijevic, E.; Curcic, M.; Milovanovic, V.; Antonijevic, B. Endocrine-Disrupting Mechanisms of Polychlorinated Biphenyls. Curr. Opin. Toxicol. 2020, 19, 42–49. [Google Scholar] [CrossRef]
- Tournaire, M.; Devouche, E.; Epelboin, S.; Cabau, A.; Dunbavand, A.; Levadou, A. Birth Defects in Children of Men Exposed in Utero to Diethylstilbestrol (DES). Therapies 2018, 73, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xie, X.; Liu, H. Effects of Diethylstilbestrol on Zebrafish Gonad Development and Endocrine Disruption Mechanism. Biomolecules 2021, 11, 941. [Google Scholar] [CrossRef]
- Imamichi, Y.; Sekiguchi, T.; Kitano, T.; Kajitani, T.; Okada, R.; Inaoka, Y.; Miyamoto, K.; Uwada, J.; Takahashi, S.; Nemoto, T.; et al. Diethylstilbestrol Administration Inhibits Theca Cell Androgen and Granulosa Cell Estrogen Production in Immature Rat Ovary. Sci. Rep. 2017, 7, 8374. [Google Scholar] [CrossRef] [Green Version]
- Hunter, D.; Heng, K.; Mann, N.; Anand-Ivell, R.; Ivell, R. Maternal Exposure to Dibutyl Phthalate (DBP) or Diethylstilbestrol (DES) Leads to Long-Term Changes in Hypothalamic Gene Expression and Sexual Behavior. Int. J. Mol. Sci. 2021, 22, 4163. [Google Scholar] [CrossRef]
- Lee, J.; Choi, K.; Park, J.; Moon, H.-B.; Choi, G.; Lee, J.J.; Suh, E.; Kim, H.-J.; Eun, S.-H.; Kim, G.-H.; et al. Bisphenol A Distribution in Serum, Urine, Placenta, Breast Milk, and Umbilical Cord Serum in a Birth Panel of Mother-Neonate Pairs. Sci. Total Environ. 2018, 626, 1494–1501. [Google Scholar] [CrossRef] [PubMed]
- Geens, T.; Neels, H.; Covaci, A. Distribution of Bisphenol-A, Triclosan and n-Nonylphenol in Human Adipose Tissue, Liver and Brain. Chemosphere 2012, 87, 796–802. [Google Scholar] [CrossRef]
- Schlumpf, M.; Kypke, K.; Wittassek, M.; Angerer, J.; Mascher, H.; Mascher, D.; Vökt, C.; Birchler, M.; Lichtensteiger, W. Exposure Patterns of UV Filters, Fragrances, Parabens, Phthalates, Organochlor Pesticides, PBDEs, and PCBs in Human Milk: Correlation of UV Filters with Use of Cosmetics. Chemosphere 2010, 81, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Overturf, M.L.; Druilhet, R.E.; Liehr, J.G.; Kirkendall, W.M.; Caprioli, R.M. Phthalate Esters in Normal and Pathological Human Kidneys. Bull. Environ. Contam. Toxicol. 1979, 22, 536–542. [Google Scholar] [CrossRef]
- Pitter, G.; Da Re, F.; Canova, C.; Barbieri, G.; Zare Jeddi, M.; Daprà, F.; Manea, F.; Zolin, R.; Bettega, A.M.; Stopazzolo, G.; et al. Serum Levels of Perfluoroalkyl Substances (PFAS) in Adolescents and Young Adults Exposed to Contaminated Drinking Water in the Veneto Region, Italy: A Cross-Sectional Study Based on a Health Surveillance Program. Environ. Health Perspect. 2020, 128, 27007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, F.; Nadal, M.; Navarro-Ortega, A.; Fàbrega, F.; Domingo, J.L.; Barceló, D.; Farré, M. Accumulation of Perfluoroalkyl Substances in Human Tissues. Environ. Int. 2013, 59, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Mamsen, L.S.; Björvang, R.D.; Mucs, D.; Vinnars, M.-T.; Papadogiannakis, N.; Lindh, C.H.; Andersen, C.Y.; Damdimopoulou, P. Concentrations of Perfluoroalkyl Substances (PFASs) in Human Embryonic and Fetal Organs from First, Second, and Third Trimester Pregnancies. Environ. Int. 2019, 124, 482–492. [Google Scholar] [CrossRef]
- Wang, N.; Kong, D.; Shan, Z.; Shi, L.; Cai, D.; Cao, Y.; Liu, Y.; Pang, G. Simultaneous Determination of Pesticides, Polycyclic Aromatic Hydrocarbons, Polychlorinated Biphenyls and Phthalate Esters in Human Adipose Tissue by Gas Chromatography-Tandem Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 898, 38–52. [Google Scholar] [CrossRef]
- Johnson-Restrepo, B.; Kannan, K.; Rapaport, D.P.; Rodan, B.D. Polybrominated Diphenyl Ethers and Polychlorinated Biphenyls in Human Adipose Tissue from New York. Environ. Sci. Technol. 2005, 39, 5177–5182. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen Signaling Multiple Pathways to Impact Gene Transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Depiereux, S.; Le Gac, F.; De Meulder, B.; Pierre, M.; Helaers, R.; Guiguen, Y.; Kestemont, P.; Depiereux, E. Meta-Analysis of Microarray Data of Rainbow Trout Fry Gonad Differentiation Modulated by Ethynylestradiol. PLoS ONE 2015, 10, e0135799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardinali, M.; Maradonna, F.; Olivotto, I.; Bortoluzzi, G.; Mosconi, G.; Polzonetti-Magni, A.M.; Carnevali, O. Temporary Impairment of Reproduction in Freshwater Teleost Exposed to Nonylphenol. Reprod. Toxicol. 2004, 18, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Golshan, M.; Hatef, A.; Socha, M.; Milla, S.; Butts, I.A.; Carnevali, O.; Rodina, M.; Sokolowska-Mikolajczyk, M.; Fontaine, P.; Linhart, O.; et al. Di-(2-Ethylhexyl)-Phthalate Disrupts Pituitary and Testicular Hormonal Functions to Reduce Sperm Quality in Mature Goldfish. Aquat. Toxicol. 2015, 163, 16–26. [Google Scholar] [CrossRef]
- Kong, H.J.; Lee, I.K.; Kim, J.; Kim, W.J.; Kim, H.S.; Cho, W.S.; Kim, D.W.; Park, J.Y.; An, C.M. RNA-Seq-Based Transcriptome Analysis of Korean Rose Bitterling (Rhodeus uyekii) Exposed to Synthetic Estrogen 17-Alpha-Ethinylestradiol (EE2). Mar. Genom. 2015, 24 Pt 3, 233–236. [Google Scholar] [CrossRef]
- Cocci, P.; Mosconi, G.; Palermo, F.A. Effects of 4-Nonylphenol on Hepatic Gene Expression of Peroxisome Proliferator-Activated Receptors and Cytochrome P450 Isoforms (CYP1A1 and CYP3A4) in Juvenile Sole (Solea solea). Chemosphere 2013, 93, 1176–1181. [Google Scholar] [CrossRef]
- Derouiche, L.; Keller, M.; Martini, M.; Duittoz, A.H.; Pillon, D. Developmental Exposure to Ethinylestradiol Affects Reproductive Physiology, the GnRH Neuroendocrine Network and Behaviors in Female Mouse. Front. Neurosci. 2015, 9, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.G.; Kim, J.Y.; Chung, J.Y.; Kim, Y.J.; Park, J.E.; Oh, S.; Yoon, Y.D.; Yoo, K.S.; Yoo, Y.H.; Kim, J.M. Bisphenol A Exposure during Adulthood Causes Augmentation of Follicular Atresia and Luteal Regression by Decreasing 17beta-Estradiol Synthesis via Downregulation of Aromatase in Rat Ovary. Environ. Health Perspect. 2013, 121, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, B.L.; Kassim, N.M.; Mohd, M.A. Assessment of Pubertal Development in Juvenile Male Rats after Sub-Acute Exposure to Bisphenol A and Nonylphenol. Toxicol. Lett. 2003, 143, 261–270. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, E.; Kim, T.H.; Choi, J.S.; Lee, J.; Jung, K.K.; Kwack, S.J.; Kim, K.B.; Kang, T.S.; Han, S.Y.; et al. Effects of Di(2-Ethylhexyl) Phthalate on Regulation of Steroidogenesis or Spermatogenesis in Testes of Sprague-Dawley Rats. J. Health Sci. 2009, 55, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Stenz, L.; Escoffier, J.; Rahban, R.; Nef, S.; Paoloni-Giacobino, A. Testicular Dysgenesis Syndrome and Long-Lasting Epigenetic Silencing of Mouse Sperm Genes Involved in the Reproductive System after Prenatal Exposure to DEHP. PLoS ONE 2017, 12, e0170441. [Google Scholar] [CrossRef] [Green Version]
- Giribabu, N.; Sainath, S.B.; Sreenivasula Reddy, P. Prenatal Di-n-Butyl Phthalate Exposure Alters Reproductive Functions at Adulthood in Male Rats. Environ. Toxicol. 2014, 29, 534–544. [Google Scholar] [CrossRef]
- Nelli, G.; Pamanji, S.R. Di-n-Butyl Phthalate Prompts Interruption of Spermatogenesis, Steroidogenesis, and Fertility Associated with Increased Testicular Oxidative Stress in Adult Male Rats. Environ. Sci. Pollut. Res. Int. 2017, 24, 18563–18574. [Google Scholar] [CrossRef]
- Singh, S.; Li, S.S. Epigenetic Effects of Environmental Chemicals Bisphenol A and Phthalates. Int. J. Mol. Sci. 2012, 13, 10143–10153. [Google Scholar] [CrossRef]
- Hilakivi-Clarke, L.; Warri, A.; Bouker, K.B.; Zhang, X.; Cook, K.L.; Jin, L.; Zwart, A.; Nguyen, N.; Hu, R.; Cruz, M.I.; et al. Effects of In Utero Exposure to Ethinyl Estradiol on Tamoxifen Resistance and Breast Cancer Recurrence in a Preclinical Model. J. Natl. Cancer Inst. 2017, 109, djw188. [Google Scholar] [CrossRef]
- Dong, Y.; Araki, M.; Hirane, M.; Tanabe, E.; Fukushima, N.; Tsujiuchi, T. Effects of Bisphenol A and 4-Nonylphenol on Cellular Responses through the Different Induction of LPA Receptors in Liver Epithelial WB-F344 Cells. J. Recept. Signal Transduct. 2014, 34, 201–204. [Google Scholar] [CrossRef]
- Yu, J.; Luo, Y.; Yang, X.F.; Yang, M.X.; Yang, J.; Yang, X.S.; Zhou, J.; Gao, F.; He, L.T.; Xu, J. Effects of Perinatal Exposure to Nonylphenol on Delivery Outcomes of Pregnant Rats and Inflammatory Hepatic Injury in Newborn Rats. Braz. J. Med. Biol. Res. 2016, 49, e5647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zhang, W.; Rui, B.B.; Yang, S.M.; Xu, W.P.; Wei, W. Di(2-Ethylhexyl) Phthalate Exacerbates Non-Alcoholic Fatty Liver in Rats and Its Potential Mechanisms. Environ. Toxicol. Pharm. 2016, 42, 38–44. [Google Scholar] [CrossRef]
- Chen, M.; Zhao, S.; Guo, W.-H.; Zhu, Y.-P.; Pan, L.; Xie, Z.-W.; Sun, W.-L.; Jiang, J.-T. Maternal Exposure to Di-n-Butyl Phthalate (DBP) Aggravate Gestational Diabetes Mellitus via FoxM1 Suppression by PSTAT1 Signalling. Ecotoxicol. Environ. Saf. 2020, 205, 111154. [Google Scholar] [CrossRef] [PubMed]
- Pillon, D.; Cadiou, V.; Angulo, L.; Duittoz, A.H. Maternal Exposure to 17-Alpha-Ethinylestradiol Alters Embryonic Development of GnRH-1 Neurons in Mouse. Brain Res. 2012, 1433, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Zheng, Y.L.; Zhang, Y.Q.; Han, B.P.; Chen, L.T.; Li, J.; Li, F.; Shan, Q. Chronic Application of Nonylphenol-Induced Apoptosis via Suppression of Bcl-2 Transcription and up-Regulation of Active Caspase-3 in Mouse Brain. Neurosci. Lett. 2008, 439, 147–152. [Google Scholar] [CrossRef]
- Win-Shwe, T.T.; Yanagisawa, R.; Koike, E.; Nitta, H.; Takano, H. Expression Levels of Neuroimmune Biomarkers in Hypothalamus of Allergic Mice after Phthalate Exposure. J. Appl. Toxicol. 2013, 33, 1070–1078. [Google Scholar] [CrossRef]
- Aung, K.H.; Win-Shwe, T.-T.; Kanaya, M.; Takano, H.; Tsukahara, S. Involvement of Hemeoxygenase-1 in Di(2-Ethylhexyl) Phthalate (DEHP)-Induced Apoptosis of Neuro-2a Cells. J. Toxicol. Sci. 2014, 39, 217–229. [Google Scholar] [CrossRef] [Green Version]
- Luu, B.E.; Green, S.R.; Childers, C.L.; Holahan, M.R.; Storey, K.B. The Roles of Hippocampal MicroRNAs in Response to Acute Postnatal Exposure to Di(2-Ethylhexyl) Phthalate in Female and Male Rats. Neurotoxicology 2017, 59, 98–104. [Google Scholar] [CrossRef]
- He, M.; Ichinose, T.; Yoshida, S.; Takano, H.; Nishikawa, M.; Shibamoto, T.; Sun, G. Exposure to Bisphenol A Enhanced Lung Eosinophilia in Adult Male Mice. Allergy Asthma Clin. Immunol. 2016, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Suen, J.L.; Hsu, S.H.; Hung, C.H.; Chao, Y.S.; Lee, C.L.; Lin, C.Y.; Weng, T.H.; Yu, H.S.; Huang, S.K. A Common Environmental Pollutant, 4-Nonylphenol, Promotes Allergic Lung Inflammation in a Murine Model of Asthma. Allergy 2013, 68, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.S.; Shaw, B.R.; Wu, A.H. Plasticizers, Antioxidants, and Other Contaminants Found in Air Delivered by PVC Tubing Used in Respiratory Therapy. Biomed. Chromatogr. 2003, 17, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Han, M.; Ren, Y.; Yang, X.; Duan, L.; Zeng, Y.; Li, J. Dibutyl Phthalate Aggravated Asthma-like Symptoms through Oxidative Stress and Increasing Calcitonin Gene-Related Peptide Release. Ecotoxicol. Environ. Saf. 2020, 199, 110740. [Google Scholar] [CrossRef] [PubMed]
- Santangeli, S.; Maradonna, F.; Olivotto, I.; Piccinetti, C.C.; Gioacchini, G.; Carnevali, O. Effects of BPA on Female Reproductive Function: The Involvement of Epigenetic Mechanism. Gen. Comp. Endocrinol. 2016, 245, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Vitku, J.; Heracek, J.; Sosvorova, L.; Hampl, R.; Chlupacova, T.; Hill, M.; Sobotka, V.; Bicikova, M.; Starka, L. Associations of Bisphenol A and Polychlorinated Biphenyls with Spermatogenesis and Steroidogenesis in Two Biological Fluids from Men Attending an Infertility Clinic. Environ. Int. 2016, 89–90, 166–173. [Google Scholar] [CrossRef]
- Noorimotlagh, Z.; Haghighi, N.J.; Ahmadimoghadam, M.; Rahim, F. An Updated Systematic Review on the Possible Effect of Nonylphenol on Male Fertility. Environ. Sci. Pollut. Res. Int. 2016, 24, 3298–3314. [Google Scholar] [CrossRef]
- Chen, M.; Tang, R.; Fu, G.; Xu, B.; Zhu, P.; Qiao, S.; Chen, X.; Xu, B.; Qin, Y.; Lu, C.; et al. Association of Exposure to Phenols and Idiopathic Male Infertility. J. Hazard. Mater. 2013, 250–251, 115–121. [Google Scholar] [CrossRef]
- Forte, M.; Di Lorenzo, M.; Carrizzo, A.; Valiante, S.; Vecchione, C.; Laforgia, V.; De Falco, M. Nonylphenol Effects on Human Prostate Non Tumorigenic Cells. Toxicology 2016, 357–358, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.P.; Chien, M.H.; Chern, I.Y. Impact of Low Concentrations of Phthalates on the Effects of 17beta-Estradiol in MCF-7 Breast Cancer Cells. Taiwan J. Obs. Gynecol. 2016, 55, 826–834. [Google Scholar] [CrossRef]
- Miao, Y.; Wang, R.; Lu, C.; Zhao, J.; Deng, Q. Lifetime Cancer Risk Assessment for Inhalation Exposure to Di(2-Ethylhexyl) Phthalate (DEHP). Environ. Sci. Pollut. Res. Int. 2017, 24, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.-P.; Chien, M.-H.; Chen, H.-Y.; Ng, Y.-T. Effects of Phthalates on Normal Human Breast Cells Co-Cultured with Different Fibroblasts. PLoS ONE 2018, 13, e0199596. [Google Scholar] [CrossRef] [PubMed]
- Ohlstein, J.F.; Strong, A.L.; McLachlan, J.A.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Bisphenol A Enhances Adipogenic Differentiation of Human Adipose Stromal/Stem Cells. J. Mol. Endocrinol. 2014, 53, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Papaconstantinou, A.D. Effects of Beta-Estradiol and Bisphenol A on Heat Shock Protein Levels and Localization in the Mouse Uterus Are Antagonized by the Antiestrogen ICI 182,780. Toxicol. Sci. 2001, 63, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Dyson, J.; Day, C. Treatment of Non-Alcoholic Fatty Liver Disease. Dig. Dis. 2014, 32, 597–604. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Non-Alcoholic Fatty Liver Disease. BMC Med. 2017, 15, 45. [Google Scholar] [CrossRef] [Green Version]
- Lallukka, S.; Yki-Jarvinen, H. Non-Alcoholic Fatty Liver Disease and Risk of Type 2 Diabetes. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yki-Jarvinen, H. Non-Alcoholic Fatty Liver Disease as a Cause and a Consequence of Metabolic Syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Yki-Jarvinen, H. Fat in the Liver and Insulin Resistance. Ann. Med. 2005, 37, 347–356. [Google Scholar] [CrossRef]
- Zhou, A.; Chang, H.; Huo, W.; Zhang, B.; Hu, J.; Xia, W.; Chen, Z.; Xiong, C.; Zhang, Y.; Wang, Y.; et al. Prenatal Exposure to Bisphenol A and Risk of Allergic Diseases in Early Life. Pediatr. Res. 2017, 81, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, B.G.; Maarsingh, H.; Meurs, H.; Gosens, R. Airway Structural Components Drive Airway Smooth Muscle Remodeling in Asthma. Proc. Am. Thorac. Soc. 2009, 6, 683–692. [Google Scholar] [CrossRef]
- Kuo, P.L.; Hsu, Y.L.; Tsai, M.J.; Lien, C.T.; Huang, M.S.; Ko, Y.C. Nonylphenol Induces Bronchial Epithelial Apoptosis via Fas-Mediated Pathway and Stimulates Bronchial Epithelium to Secrete IL-6 and IL-8, Causing Bronchial Smooth Muscle Proliferation and Migration. Basic Clin. Pharmacol. Toxicol. 2012, 110, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, L.; Druilhe, A.; Dombret, M.C.; Aubier, M.; Pretolani, M. Airway Structural Alterations Selectively Associated with Severe Asthma. Am. J. Respir. Crit. Care Med. 2003, 167, 1360–1368. [Google Scholar] [CrossRef]
- Franken, C.; Lambrechts, N.; Govarts, E.; Koppen, G.; Den Hond, E.; Ooms, D.; Voorspoels, S.; Bruckers, L.; Loots, I.; Nelen, V.; et al. Phthalate-Induced Oxidative Stress and Association with Asthma-Related Airway Inflammation in Adolescents. Int. J. Hyg. Environ. Health 2017, 220, 468–477. [Google Scholar] [CrossRef]
- Wang, I.J.; Karmaus, W.J.; Chen, S.L.; Holloway, J.W.; Ewart, S. Effects of Phthalate Exposure on Asthma May Be Mediated through Alterations in DNA Methylation. Clin. Epigenet. 2015, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-M.; Kim, J.; Cheong, H.-K.; Jeon, B.-H.; Ahn, K. Exposure to Phthalates Aggravates Pulmonary Function and Airway Inflammation in Asthmatic Children. PLoS ONE 2018, 13, e0208553. [Google Scholar] [CrossRef]
- Bhasker, C.R.; Hardiman, G. Advances in Pharmacogenomics Technologies. Pharmacogenomics 2010, 11, 481–485. [Google Scholar] [CrossRef]
- Miracle, A.L.; Ankley, G.T. Ecotoxicogenomics: Linkages between Exposure and Effects in Assessing Risks of Aquatic Contaminants to Fish. Reprod. Toxicol. 2005, 19, 321–326. [Google Scholar] [CrossRef]
- Martín-Díaz, M.L.; DelValls, T.A.; Riba, I.; Blasco, J. Integrative Sediment Quality Assessment Using a Biomarker Approach: Review of 3 Years of Field Research. Cell. Biol. Toxicol. 2008, 24, 513–526. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Bourguignon, J.-P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Grün, F.; Blumberg, B. Endocrine Disrupters as Obesogens. Mol. Cell. Endocrinol. 2009, 304, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M.E.; Sprague, L.J.; Ribecco, C.; Ruggeri, B.; Lekmine, N.; Ludka, C.; Wick, I.; Soverchia, L.; Ubaldi, M.; Šášik, R.; et al. Application of a Targeted Endocrine Q-PCR Panel to Monitor the Effects of Pollution in Southern California Flatfish. Endocr. Disruptors 2014, 2, e969598. [Google Scholar] [CrossRef]
- Baker, M.E.; Vidal-Dorsch, D.E.; Ribecco, C.; Sprague, L.J.; Angert, M.; Lekmine, N.; Ludka, C.; Martella, A.; Ricciardelli, E.; Bay, S.M.; et al. Molecular Analysis of Endocrine Disruption in Hornyhead Turbot at Wastewater Outfalls in Southern California Using a Second Generation Multi-Species Microarray. PLoS ONE 2013, 8, e75553. [Google Scholar] [CrossRef] [Green Version]
- Hardiman, G. Microarray Technology—Advances, Applications, Future Prospects. Pharmacogenomics 2007, 8, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Rao, M.S.; Van Vleet, T.R.; Ciurlionis, R.; Buck, W.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Liguori, M.J. Comparison of RNA-Seq and Microarray Gene Expression Platforms for the Toxicogenomic Evaluation of Liver From Short-Term Rat Toxicity Studies. Front. Genet. 2019, 9, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casneuf, T.; Van de Peer, Y.; Huber, W. In Situ Analysis of Cross-Hybridisation on Microarrays and the Inference of Expression Correlation. BMC Bioinform. 2007, 8, 461. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Grennan, K.; Badner, J.; Zhang, D.; Gershon, E.; Jin, L.; Liu, C. Removing Batch Effects in Analysis of Expression Microarray Data: An Evaluation of Six Batch Adjustment Methods. PLoS ONE 2011, 6, e17238. [Google Scholar] [CrossRef] [Green Version]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef] [Green Version]
- Microarray Innovations: Technology and Experimentation. Available online: https://www.routledge.com/Microarray-Innovations-Technology-and-Experimentation/Hardiman/p/book/9780367385811 (accessed on 20 July 2021).
- Rouse, R.J.D.; Field, K.; Lapira, J.; Lee, A.; Wick, I.; Eckhardt, C.; Bhasker, C.R.; Soverchia, L.; Hardiman, G. Development and Application of a Microarray Meter Tool to Optimize Microarray Experiments. BMC Res. Notes 2008, 1, 45. [Google Scholar] [CrossRef] [Green Version]
- Wick, I.; Hardiman, G. Biochip Platforms as Functional Genomics Tools for Drug Discovery. Curr. Opin. Drug Discov. Dev. 2005, 8, 347–354. [Google Scholar]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and Microarray in Transcriptome Profiling of Activated T Cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N.; Forrest, A.R.; Kolle, G.; Gardiner, B.B.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem Cell Transcriptome Profiling via Massive-Scale MRNA Sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The Transcriptional Landscape of the Yeast Genome Defined by RNA Sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [Green Version]
- da Silveira, W.A.; Hazard, E.S.; Chung, D.; Hardiman, G. Molecular Profiling of RNA Tumors Using High-Throughput RNA Sequencing: From Raw Data to Systems Level Analyses. Methods Mol. Biol. 2019, 1908, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Davis-Turak, J.; Courtney, S.M.; Hazard, E.S.; Glen, W.B.; da Silveira, W.A.; Wesselman, T.; Harbin, L.P.; Wolf, B.J.; Chung, D.; Hardiman, G. Genomics Pipelines and Data Integration: Challenges and Opportunities in the Research Setting. Expert Rev. Mol. Diagn. 2017, 17, 225–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigden, D.J.; Fernández, X.M. The 2021 Nucleic Acids Research Database Issue and the Online Molecular Biology Database Collection. Nucleic Acids Res. 2021, 49, D1–D9. [Google Scholar] [CrossRef]
- Lawler, M.; Siu, L.L.; Rehm, H.L.; Chanock, S.J.; Alterovitz, G.; Burn, J.; Calvo, F.; Lacombe, D.; Teh, B.T.; North, K.N.; et al. All the World’s a Stage: Facilitating Discovery Science and Improved Cancer Care through the Global Alliance for Genomics and Health. Cancer Discov. 2015, 5, 1133–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leet, J.K.; Richter, C.A.; Cornman, R.S.; Berninger, J.P.; Bhandari, R.K.; Nicks, D.K.; Zajicek, J.L.; Blazer, V.S.; Tillitt, D.E. Effects of Early Life Stage Exposure of Largemouth Bass to Atrazine or a Model Estrogen (17α-Ethinylestradiol). PeerJ 2020, 8, e9614. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.; Yang, Q.; Wang, W.; Liu, Q.; Liu, W.; Liu, S. Effects of 17 α-Methyltestosterone on the Transcriptome, Gonadal Histology and Sex Steroid Hormones in Pseudorasbora Parva. Theriogenology 2020, 155, 88–97. [Google Scholar] [CrossRef]
- Renaud, L.; Agarwal, N.; Richards, D.J.; Falcinelli, S.; Hazard, E.S.; Carnevali, O.; Hyde, J.; Hardiman, G. Transcriptomic Analysis of Short-Term 17α-Ethynylestradiol Exposure in Two Californian Sentinel Fish Species Sardine (Sardinops sagax) and Mackerel (Scomber japonicus). Environ. Pollut. 2019, 244, 926–937. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, A.; Pierron, F.; Gourves, P.-Y.; Klopp, C.; Lagarde, G.; Pereto, C.; Dufour, V.; Gonzalez, P.; Coynel, A.; Budzinski, H.; et al. Whole-Transcriptome Response to Wastewater Treatment Plant and Stormwater Effluents in the Asian Clam, Corbicula fluminea. Ecotoxicol. Environ. Saf. 2018, 165, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Legrand, E.; Forget-Leray, J.; Duflot, A.; Olivier, S.; Thomé, J.-P.; Danger, J.-M.; Boulangé-Lecomte, C. Transcriptome Analysis of the Copepod Eurytemora Affinis upon Exposure to Endocrine Disruptor Pesticides: Focus on Reproduction and Development. Aquat. Toxicol. 2016, 176, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Mo, J.; Qi, Q.; Peng, J.; Qi, G.; Kanerva, M.; Iwata, H.; Li, Q. Prediction of Adverse Effects of Effluents Containing Phenolic Compounds in the Ba River on the Ovary of Fish (Hemiculter leucisculus) Using Transcriptomic and Metabolomic Analyses. Sci. Total Environ. 2021, 801, 149554. [Google Scholar] [CrossRef]
- National Primary Drinking Water Regulations|US EPA. Available online: https://www.epa.gov/ground-water-and-drinking-water/national-primary-drinking-water-regulations (accessed on 20 July 2021).
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the Pharmaceutical Industry: New Estimates of R&D Costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, T. A Tough Road: Cost to Develop One New Drug Is $2.6 Billion; Approval Rate for Drugs Entering Clinical Development Is Less than 12%—Policy & Medicine. Available online: https://www.policymed.com/2014/12/a-tough-road-cost-to-develop-one-new-drug-is-26-billion-approval-rate-for-drugs-entering-clinical-de.html (accessed on 20 July 2021).
- Boverhof, D.R.; Zacharewski, T.R. Toxicogenomics in Risk Assessment: Applications and Needs. Toxicol. Sci. 2006, 89, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, Y.; Nakatsu, N.; Yamashita, T.; Ono, A.; Ohno, Y.; Urushidani, T.; Yamada, H. Open TG-GATEs: A Large-Scale Toxicogenomics Database. Nucleic Acids Res. 2015, 43, D921–D927. [Google Scholar] [CrossRef]
- Kiyosawa, N.; Manabe, S.; Sanbuissho, A.; Yamoto, T. Gene Set-Level Network Analysis Using a Toxicogenomics Database. Genomics 2010, 96, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costs of Animal and Non-Animal Testing—Humane Society International. Available online: https://www.hsi.org/news-media/time_and_cost/ (accessed on 20 July 2021).
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [Green Version]
- Emerging Contaminants. Available online: https://www.wqa.org/whats-in-your-water/emerging-contaminants (accessed on 20 July 2021).
- Glossary: Uncertainty Factor. Available online: https://www.greenfacts.org/glossary/tuv/uncertainty-factor-safety-factor.htm (accessed on 20 July 2021).
- Baker, M.; Šášik, R.; Gerwick, L.; Hardiman, G. The Praeger Handbook of Environmental Health; Greenwood Publishing Group Inc.: Westport, CT, USA, 2012. [Google Scholar]
- Liu, Z.; Huang, R.; Roberts, R.; Tong, W. Toxicogenomics: A 2020 Vision. Trends Pharm. Sci. 2019, 40, 92–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libbrecht, M.W.; Noble, W.S. Machine Learning Applications in Genetics and Genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Ganter, B.; Tugendreich, S.; Pearson, C.I.; Ayanoglu, E.; Baumhueter, S.; Bostian, K.A.; Brady, L.; Browne, L.J.; Calvin, J.T.; Day, G.-J.; et al. Development of a Large-Scale Chemogenomics Database to Improve Drug Candidate Selection and to Understand Mechanisms of Chemical Toxicity and Action. J. Biotechnol. 2005, 119, 219–244. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. In Statistical Genomics; Mathé, E., Davis, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1418, pp. 93–110. ISBN 978-1-4939-3576-5. [Google Scholar]
- Papatheodorou, I.; Fonseca, N.A.; Keays, M.; Tang, Y.A.; Barrera, E.; Bazant, W.; Burke, M.; Füllgrabe, A.; Fuentes, A.M.-P.; George, N.; et al. Expression Atlas: Gene and Protein Expression across Multiple Studies and Organisms. Nucleic Acids Res. 2018, 46, D246–D251. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef]
- Keenan, A.B.; Jenkins, S.L.; Jagodnik, K.M.; Koplev, S.; He, E.; Torre, D.; Wang, Z.; Dohlman, A.B.; Silverstein, M.C.; Lachmann, A.; et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018, 6, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Mav, D.; Shah, R.R.; Howard, B.E.; Auerbach, S.S.; Bushel, P.R.; Collins, J.B.; Gerhold, D.L.; Judson, R.S.; Karmaus, A.L.; Maull, E.A.; et al. A Hybrid Gene Selection Approach to Create the S1500+ Targeted Gene Sets for Use in High-Throughput Transcriptomics. PLoS ONE 2018, 13, e0191105. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Reyero, N. Are Adverse Outcome Pathways Here to Stay? Environ. Sci. Technol. 2015, 49, 3–9. [Google Scholar] [CrossRef]
- Perkins, E.J.; Antczak, P.; Burgoon, L.; Falciani, F.; Garcia-Reyero, N.; Gutsell, S.; Hodges, G.; Kienzler, A.; Knapen, D.; McBride, M.; et al. Adverse Outcome Pathways for Regulatory Applications: Examination of Four Case Studies with Different Degrees of Completeness and Scientific Confidence. Toxicol. Sci. 2015, 148, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Villeneuve, D.L.; Crump, D.; Garcia-Reyero, N.; Hecker, M.; Hutchinson, T.H.; LaLone, C.A.; Landesmann, B.; Lettieri, T.; Munn, S.; Nepelska, M.; et al. Adverse Outcome Pathway (AOP) Development I: Strategies and Principles. Toxicol. Sci. 2014, 142, 312–320. [Google Scholar] [CrossRef] [Green Version]
- What EDCs Are|Endocrine Society. Available online: https://www.endocrine.org/topics/edc/what-edcs-are (accessed on 20 July 2021).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verga, J.U.; Huff, M.; Owens, D.; Wolf, B.J.; Hardiman, G. Integrated Genomic and Bioinformatics Approaches to Identify Molecular Links between Endocrine Disruptors and Adverse Outcomes. Int. J. Environ. Res. Public Health 2022, 19, 574. https://doi.org/10.3390/ijerph19010574
Verga JU, Huff M, Owens D, Wolf BJ, Hardiman G. Integrated Genomic and Bioinformatics Approaches to Identify Molecular Links between Endocrine Disruptors and Adverse Outcomes. International Journal of Environmental Research and Public Health. 2022; 19(1):574. https://doi.org/10.3390/ijerph19010574
Chicago/Turabian StyleVerga, Jacopo Umberto, Matthew Huff, Diarmuid Owens, Bethany J. Wolf, and Gary Hardiman. 2022. "Integrated Genomic and Bioinformatics Approaches to Identify Molecular Links between Endocrine Disruptors and Adverse Outcomes" International Journal of Environmental Research and Public Health 19, no. 1: 574. https://doi.org/10.3390/ijerph19010574