
The Genomic Landscape of a Restricted ALL Cohort from Patients Residing on the U.S./Mexico Border

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants and Sequencing
2.2. Bioinformatics
2.3. Gene Inclusion and Classification of Mutations
3. Results
3.1. Patient Demographics
3.2. The Genomic Landscape of ALL
3.2.1. ALL Borderland Patients Harbor Tier Mutations Detected in First-Level Cancer Biomarkers with Therapeutic Potential
3.2.2. ALL Borderland Patients Harbor Tier Mutations Detected in Second-Level Biomarkers with Diagnostic and Prognostic Value for Hematologic Malignancies
3.2.3. ALL Borderland Patients Harbor Tier Mutations Detected in Third-Level Biomarkers with Diagnostic Value for Leukemia
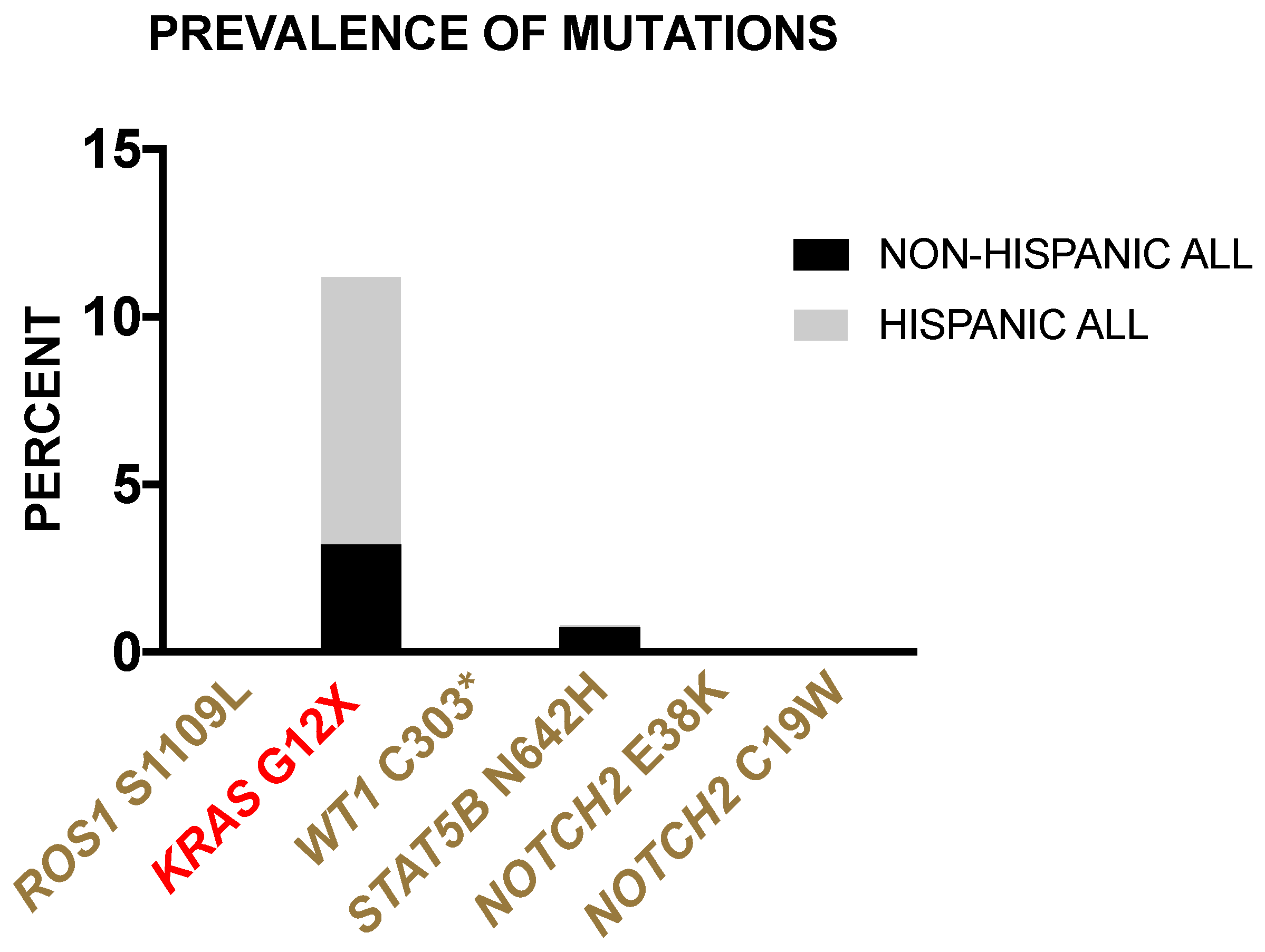
3.2.4. Prevalence of Borderland Tier Mutations in Hispanic and Non-Hispanic ALL Patients from the TARGET-ALL Phase II Database
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nguyen, K.; Devidas, M.; Cheng, S.C.; La, M.; Raetz, E.A.; Carroll, W.L.; Winick, N.J.; Hunger, S.P.; Gaynon, P.S.; Loh, M.L.; et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: A Children’s Oncology Group study. Leukemia 2008, 22, 2142–2150. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.D.; Goding Sauer, A.; Ortiz, A.P.; Fedewa, S.A.; Pinheiro, P.S.; Tortolero-Luna, G.; Martinez-Tyson, D.; Jemal, A.; Siegel, R.L. Cancer Statistics for Hispanics/Latinos, 2018. CA Cancer J. Clin. 2018, 68, 425–445. [Google Scholar] [CrossRef] [Green Version]
- Karol, S.E.; Pui, C.H. Personalized therapy in pediatric high-risk B-cell acute lymphoblastic leukemia. Ther. Adv. Hematol. 2020, 11, 2040620720927575. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Roberts, K.G.; Yang, Y.L.; Payne-Turner, D.; Lin, W.; Files, J.K.; Dickerson, K.; Gu, Z.; Taunton, J.; Janke, L.J.; Chen, T.; et al. Oncogenic role and therapeutic targeting of ABL-class and JAK-STAT activating kinase alterations in Ph-like ALL. Blood Adv. 2017, 1, 1657–1671. [Google Scholar] [CrossRef]
- Moorman, A.V. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica 2016, 101, 407–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, M.R.; Tallman, M.S.; Abboud, C.N.; Altman, J.K.; Appelbaum, F.R.; Arber, D.A.; Bhatt, V.; Bixby, D.; Blum, W.; Coutre, S.E.; et al. Acute Myeloid Leukemia, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 926–957. [Google Scholar] [CrossRef] [PubMed]
- Coccaro, N.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Next-Generation Sequencing in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2019, 20, 2929. [Google Scholar] [CrossRef] [Green Version]
- Loree, J.M.; Anand, S.; Dasari, A.; Unger, J.M.; Gothwal, A.; Ellis, L.M.; Varadhachary, G.; Kopetz, S.; Overman, M.J.; Raghav, K. Disparity of Race Reporting and Representation in Clinical Trials Leading to Cancer Drug Approvals From 2008 to 2018. JAMA Oncol. 2019, e191870. [Google Scholar] [CrossRef]
- Dickmann, L.J.; Schutzman, J.L. Racial and Ethnic Composition of Cancer Clinical Drug Trials: How Diverse Are We? Oncologist 2018, 23, 243–246. [Google Scholar] [CrossRef] [Green Version]
- Heredia, N.I.; Krasny, S.; Strong, L.L.; Von Hatten, L.; Nguyen, L.; Reininger, B.M.; McNeill, L.H.; Fernandez, M.E. Community Perceptions of Biobanking Participation: A Qualitative Study among Mexican-Americans in Three Texas Cities. Public Health Genom. 2017, 20, 46–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bencomo-Alvarez, A.E.; Gonzalez, M.A.; Rubio, A.J.; Olivas, I.M.; Lara, J.J.; Padilla, O.; Orazi, A.; Corral, J.; Philipovskiy, A.; Gaur, S.; et al. Ethnic and border differences on blood cancer presentation and outcomes: A Texas population-based study. Cancer 2020, 127, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Mittendorf, K.F.; Holt, M.; Lenoue-Newton, M.; Maurer, I.; Miller, C.; Stachowiak, M.; Botyrius, M.; Cole, J.; Micheel, C.; et al. The My Cancer Genome clinical trial data model and trial curation workflow. J. Am. Med. Inform. Assoc. 2020, 27, 1057–1066. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Lavretsky, P.; McInerney, N.R.; Mohl, J.E.; Brown, J.I.; James, H.F.; McCracken, K.G.; Fleischer, R.C. Assessing changes in genomic divergence following a century of human-mediated secondary contact among wild and captive-bred ducks. Mol. Ecol. 2020, 29, 578–595. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Leung, M.-Y.; Knapka, J.A.; Wagler, A.E.; Rodriguez, G.; Kirken, R.A. OncoMiner: A Pipeline for Bioinformatics Analysis of Exonic Sequence Variants in Cancer. In Big Data Analytics in Genomics; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Tabbò, F.; Pizzi, M.; Kyriakides, P.W.; Ruggeri, B.; Inghirami, G. Oncogenic kinase fusions: An evolving arena with innovative clinical opportunities. Oncotarget 2016, 7, 25064–25086. [Google Scholar] [CrossRef] [Green Version]
- Uguen, A.; De Braekeleer, M. ROS1 fusions in cancer: A review. Future Oncol 2016, 12, 1911–1928. [Google Scholar] [CrossRef] [PubMed]
- Gumy-Pause, F.; Wacker, P.; Sappino, A.P. ATM gene and lymphoid malignancies. Leukemia 2004, 18, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, J.; Matheson, E.; Minto, L.; Blair, H.; Case, M.; Halsey, C.; Swidenbank, I.; Ponthan, F.; Kirschner-Schwabe, R.; Groeneveld-Krentz, S.; et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 2014, 124, 3420–3430. [Google Scholar] [CrossRef]
- Oshima, K.; Khiabanian, H.; da Silva-Almeida, A.C.; Tzoneva, G.; Abate, F.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Carpenter, Z.; Penson, A.; Perez-Garcia, A.; et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, 11306–11311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, J.; Touzart, A.; Leprêtre, S.; Graux, C.; Bargetzi, M.; Lhermitte, L.; Hypolite, G.; Leguay, T.; Hicheri, Y.; Guillerm, G.; et al. DNMT3A mutation is associated with increased age and adverse outcome in adult T-cell acute lymphoblastic leukemia. Haematologica 2019, 104, 1617–1625. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.Q.; Chen, C.J.; Jing, Y.; Qin, J.Y.; Li, Y.; Chen, G.F.; Zhou, W.; Li, Y.H.; Wang, J.; Li, D.W.; et al. Characteristics and prognostic significance of genetic mutations in acute myeloid leukemia based on a targeted next-generation sequencing technique. Cancer Med. 2020, 9, 8457–8467. [Google Scholar] [CrossRef]
- Krauth, M.T.; Alpermann, T.; Bacher, U.; Eder, C.; Dicker, F.; Ulke, M.; Kuznia, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; et al. WT1 mutations are secondary events in AML, show varying frequencies and impact on prognosis between genetic subgroups. Leukemia 2015, 29, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Weng, W.W.; Chen, P.; Zhang, Y.; Ruan, J.F.; Ba, D.D.; Xu, W.Q.; Tang, Y.M. Low expression of TET2 gene in pediatric acute lymphoblastic leukemia is associated with poor clinical outcome. Int. J. Lab. Hematol. 2019, 41, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.Z.; Jiang, Q.; Xu, L.P.; Jiang, H.; Wang, Y.; Zhao, X.S.; Li, Z.R.; Lai, Y.Y.; Liu, Y.R.; Zhang, X.H.; et al. The prognostic significance of Wilms’ tumor gene 1 (WT1) expression at diagnosis in adults with Ph-negative B cell precursor acute lymphoblastic leukemia. Ann. Hematol. 2019, 98, 2551–2559. [Google Scholar] [CrossRef]
- Rampal, R.; Figueroa, M.E. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica 2016, 101, 672–679. [Google Scholar] [CrossRef] [Green Version]
- Bandapalli, O.R.; Schuessele, S.; Kunz, J.B.; Rausch, T.; Stutz, A.M.; Tal, N.; Geron, I.; Gershman, N.; Izraeli, S.; Eilers, J.; et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica 2014, 99, e188–e192. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Tsirigos, A.; Vlierberghe, P.V.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres-Marco, D.; da Ros, V.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.D.; Canté-Barrett, K.; Pieters, R.; Meijerink, J.P. The relevance of PTEN-AKT in relation to NOTCH1-directed treatment strategies in T-cell acute lymphoblastic leukemia. Haematologica 2016, 101, 1010–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.; Vosberg, S.; Schlee, C.; Heesch, S.; Schwartz, S.; Gökbuget, N.; Hoelzer, D.; Graf, A.; Krebs, S.; Bartram, I.; et al. Mutational spectrum of adult T-ALL. Oncotarget 2015, 6, 2754–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, H.A.; Lu, J.W.; Lin, T.Y.; Tsai, C.H.; Chou, W.C.; Lin, C.C.; Kuo, Y.Y.; Liu, C.Y.; Tseng, M.H.; Chiang, Y.C.; et al. Clinico-biological significance of suppressor of cytokine signaling 1 expression in acute myeloid leukemia. Blood Cancer J. 2017, 7, e588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takam Kamga, P.; Dal Collo, G.; Midolo, M.; Adamo, A.; Delfino, P.; Mercuri, A.; Cesaro, S.; Mimiola, E.; Bonifacio, M.; Andreini, A.; et al. Inhibition of Notch Signaling Enhances Chemosensitivity in B-cell Precursor Acute Lymphoblastic Leukemia. Cancer Res. 2019, 79, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, J.M.; Pedersen, M.B.; Hansen, M.C.; Plesner, T.L.; Frederiksen, H.; Møller, M.; Hamilton-Dutoit, S.; Noergaard, P.; Mortensen, B.K.; Fiore, D.; et al. Shared Genomic Alterations in Patients with Co-Existing Myeloproliferative Neoplasms and Angioimmunoblastic T-Cell Lymphoma. Blood 2019, 134, 2776. [Google Scholar] [CrossRef]
- Kiel, M.J.; Velusamy, T.; Betz, B.L.; Zhao, L.; Weigelin, H.G.; Chiang, M.Y.; Huebner-Chan, D.R.; Bailey, N.G.; Yang, D.T.; Bhagat, G.; et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J. Exp. Med. 2012, 209, 1553–1565. [Google Scholar] [CrossRef] [Green Version]
- Messina, M.; Chiaretti, S.; Wang, J.; Fedullo, A.L.; Peragine, N.; Gianfelici, V.; Piciocchi, A.; Brugnoletti, F.; Di Giacomo, F.; Pauselli, S.; et al. Prognostic and therapeutic role of targetable lesions in B-lineage acute lymphoblastic leukemia without recurrent fusion genes. Oncotarget 2016, 7, 13886–13901. [Google Scholar] [CrossRef]
- Jain, N.; Roberts, K.G.; Jabbour, E.; Patel, K.; Eterovic, A.K.; Chen, K.; Zweidler-McKay, P.; Lu, X.; Fawcett, G.; Wang, S.A.; et al. Ph-like acute lymphoblastic leukemia: A high-risk subtype in adults. Blood 2017, 129, 572–581. [Google Scholar] [CrossRef] [Green Version]
- Kunz, J.B.; Rausch, T.; Bandapalli, O.R.; Eilers, J.; Pechanska, P.; Schuessele, S.; Assenov, Y.; Stutz, A.M.; Kirschner-Schwabe, R.; Hof, J.; et al. Pediatric T-cell lymphoblastic leukemia evolves into relapse by clonal selection, acquisition of mutations and promoter hypomethylation. Haematologica 2015, 100, 1442–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Diagnosis | Subtype | Status | Ethnicity |
|---|---|---|---|---|
| P1 | ALL | T-ALL | Relapse | Hispanic |
| P2 | ALL | pre-T-ALL | Relapse | Hispanic |
| P3 | ALL | pre-T-ALL | New onset | Hispanic |
| P4 | ALL | pre-T-ALL | New onset | Hispanic |
| P5 | ALL | early pre-B-ALL | Relapse | Hispanic |
| P6 | ALL | early pre-B-ALL | Relapse | Hispanic |
| P7 | ALL | pre-T-ALL | Relapse | Hispanic |
| P8 | ALL | early pre-B-ALL | New onset | Hispanic |
| P9 | ALL | early pre-B-ALL | New onset | Hispanic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grant, A.H.; Ayala-Marin, Y.M.; Mohl, J.E.; Robles-Escajeda, E.; Rodriguez, G.; Dutil, J.; Kirken, R.A. The Genomic Landscape of a Restricted ALL Cohort from Patients Residing on the U.S./Mexico Border. Int. J. Environ. Res. Public Health 2021, 18, 7345. https://doi.org/10.3390/ijerph18147345
Grant AH, Ayala-Marin YM, Mohl JE, Robles-Escajeda E, Rodriguez G, Dutil J, Kirken RA. The Genomic Landscape of a Restricted ALL Cohort from Patients Residing on the U.S./Mexico Border. International Journal of Environmental Research and Public Health. 2021; 18(14):7345. https://doi.org/10.3390/ijerph18147345
Chicago/Turabian StyleGrant, Alice Hernandez, Yoshira Marie Ayala-Marin, Jonathon Edward Mohl, Elisa Robles-Escajeda, Georgialina Rodriguez, Julie Dutil, and Robert Arthur Kirken. 2021. "The Genomic Landscape of a Restricted ALL Cohort from Patients Residing on the U.S./Mexico Border" International Journal of Environmental Research and Public Health 18, no. 14: 7345. https://doi.org/10.3390/ijerph18147345