Valproic Acid Impacts the Growth of Growth Plate Chondrocytes



Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Reagents and Antibodies
2.3. Cell Culture
2.4. AED Treatment and MTT Assay
2.5. RNA Extraction and Real-Time Polymerase Chain Reaction (RT-PCR)
2.6. Protein Extraction and Western Blotting
2.7. Flow Cytometry
2.8. Statistical Analysis
3. Results
3.1. VPA Markedly Reduces the Number of Chondrocytes
3.2. VPA Has No Effect on Cartilage Matrix Gene Expression
3.3. VPA Induces Chondrocyte Apoptosis, Noncleaved and Cleaved Caspase 3 Expression
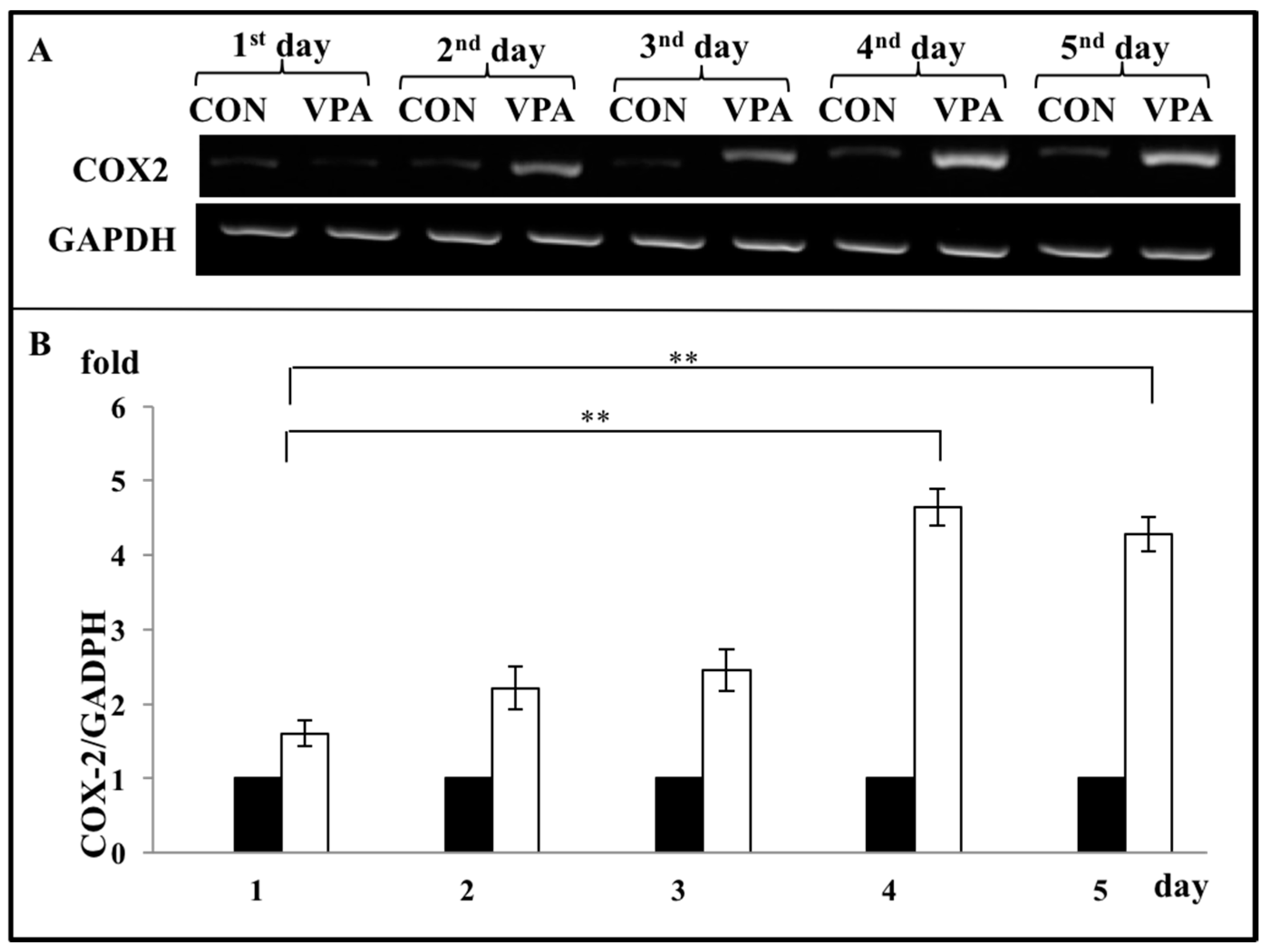
3.4. VPA Increases COX-2 Expression in Growth Plate Chondrocytes
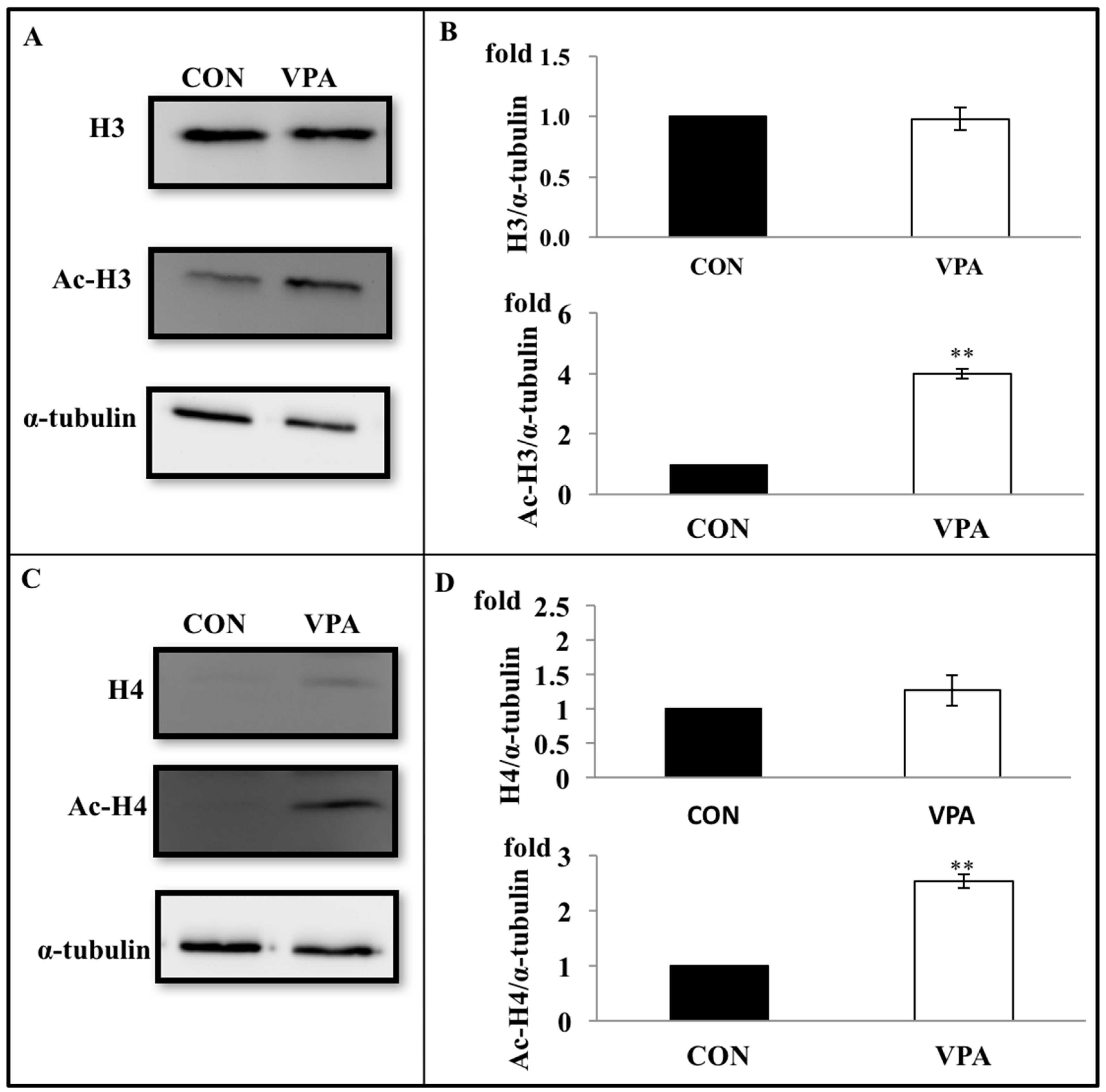
3.5. VPA Increases Histone Acetylation in Growth Plate Chondrocytes
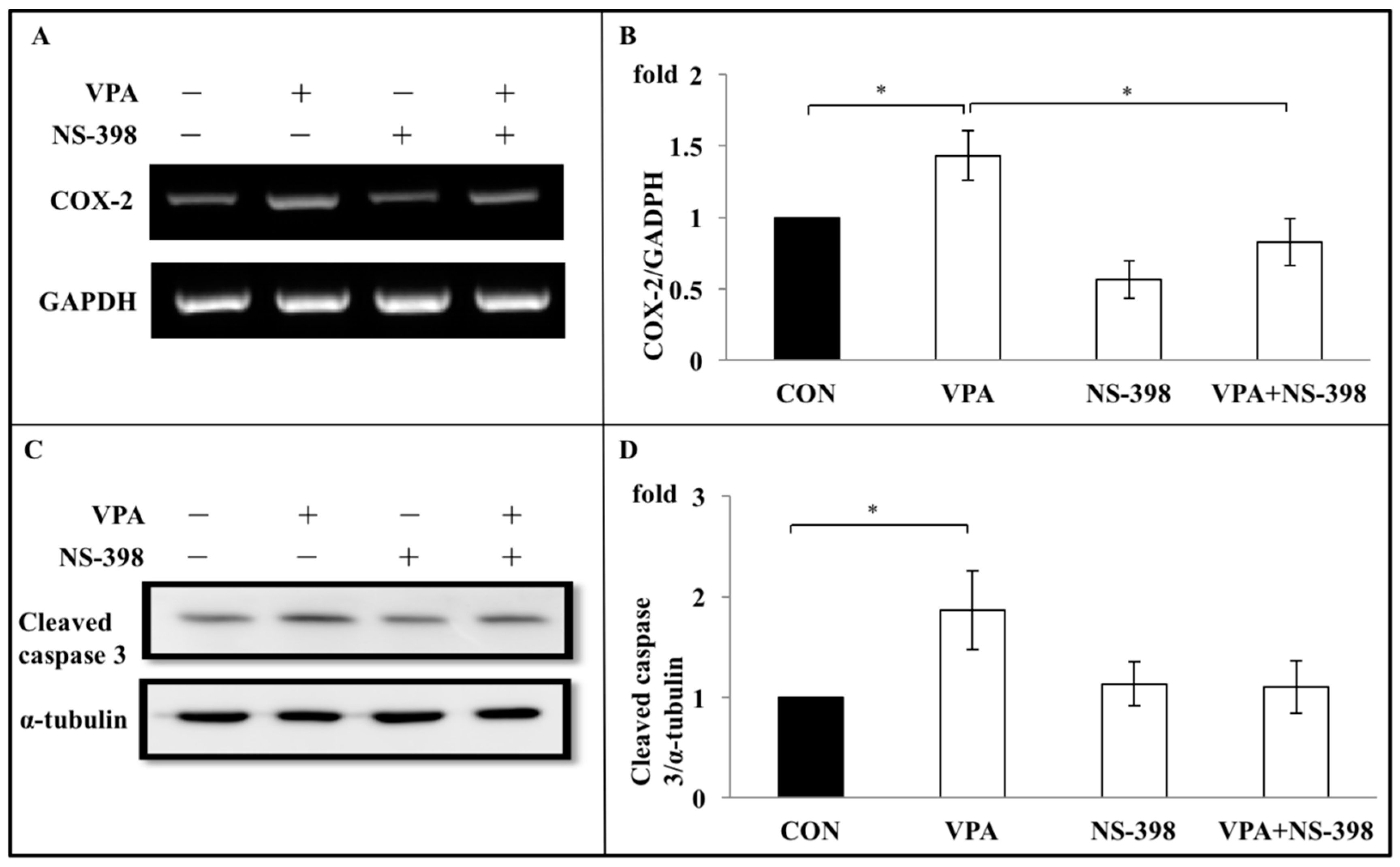
3.6. Apoptosis Induced by VPA on Rat Growth Plate Chondrocyte Are Through COX-2 Dependent Inflammation Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Savage, N. Epidemiology: The complexities of epilepsy. Nature 2014, 511, S2–S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, M.; Marson, A.G.; Ramaratnam, S. Epilepsy (generalised). BMJ Clin. Evid. 2012, 2012. [Google Scholar]
- Chen, C.C.; Chen, T.F.; Hwang, Y.C.; Wen, Y.R.; Chiu, Y.H.; Wu, C.Y.; Chen, R.C.; Chen, T.H.; Liou, H.H. Population-based survey on prevalence of adult patients with epilepsy in Taiwan (Keelung communitybased integrated screening no. 12). Epilepsy Res. 2006, 72, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Estimation of the burden of active and life-time epilepsy: A meta-analytic approach. Epilepsia 2010, 51, 883–890. [Google Scholar] [CrossRef]
- Kwan, P.; Brodie, M.J. Early identification of refractory epilepsy. N. Engl. J. Med. 2000, 342, 314–319. [Google Scholar] [CrossRef]
- McCorry, D.; Chadwick, D.; Marson, A. Current drug treatment of epilepsy in adults. Lancet. Neurol. 2004, 3, 729–735. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, H.W. Metabolic and hormonal disturbances in women with epilepsy on antiepileptic drug monotherapy. Epilepsia 2007, 48, 1366–1370. [Google Scholar] [CrossRef]
- Greenwood, R.S. Adverse effects of antiepileptic drugs. Epilepsia 2000, 41, S42–S52. [Google Scholar] [CrossRef]
- Robinson, P.B.; Harris, M.; Harvey, W. Abnormal skeletal and dental growth in epileptic children. Br. Dent. J. 1983, 154, 9–13. [Google Scholar] [CrossRef]
- Morijiri, Y.; Sato, T. Factors causing rickets in institutionalised handicapped children on anticonvulsant therapy. Arch. Dis. Child 1981, 56, 446–449. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.Y.; Ronen, G.M.; Atkinson, S.A. Long-term valproate and lamotrigine treatment may be a marker for reduced growth and bone mass in children with epilepsy. Epilepsia 2001, 42, 1141–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rim, Y.A.; Nam, Y.; Ju, J.H. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, M.T.; Kozlowski, B.W.; Blyler, E.M.; Trahms, C.M.; Taylor, M.L.; Hogan, M.P. Vitamin D, calcium, and bone status in children with developmental delay in relation to anticonvulsant use and ambulatory status. Am. J. Clin. Nutr. 1997, 65, 1042–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Onodera, K.; Shinoda, H.; Mayanagi, H. Phenytoin and its metabolite, 5-(4-hydroxyphenyl)- 5-phenylhydantoin, show bone resorption in cultured neonatal mouse calvaria. Jpn. J. Pharmacol. 2000, 82, 82–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, N.; Seino, Y.; Ishida, H.; Seino, Y.; Tanaka, H.; Tsutsumi, C.; Ogata, K.; Kiyohara, K.; Kato, H.; Nozawa, M.; et al. Increased circulating levels of gamma-carboxyglutamic acid-containing protein and decreased bone mass in children on anticonvulsant therapy. Calcif. Tissue Int. 1989, 44, 80–85. [Google Scholar] [CrossRef]
- Chung, S.; Ahn, C. Effects of anti-epileptic drug therapy on bone mineral density in ambulatory epileptic children. Brain Dev. 1994, 16, 382–385. [Google Scholar] [CrossRef]
- Pack, A.M. The Association Between Antiepileptic Drugs and Bone Disease. Epilepsy Curr. Am. Epilepsy Soc. 2003, 3, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Pack, A. Bone health in people with epilepsy: Is it impaired and what are the risk factors? Seizure 2008, 17, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Pack, A.M.; Gidal, B.; Vazquez, B. Bone disease associated with antiepileptic drugs. Cleve Clin. J. Med. 2004, 71, S42–S48. [Google Scholar] [CrossRef]
- Sheth, R.D. Metabolic concerns associated with antiepileptic medications. Neurology 2004, 63, S24–S29. [Google Scholar] [CrossRef]
- Verrotti, A.; Greco, R.; Morgese, G.; Chiarelli, F. Increased bone turnover in epileptic patients treated with carbamazepine. Ann. Neurol. 2000, 47, 385–388. [Google Scholar] [CrossRef]
- Verrotti, A.; Greco, R.; Latini, G.; Morgese, G.; Chiarelli, F. Increased bone turnover in prepubertal, pubertal, and postpubertal patients receiving carbamazepine. Epilepsia 2002, 43, 1488–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillon, R.; Reynaert, J.; Claes, J.H.; Lissens, W.; De Moor, P. The effect of anticonvulsant therapy on serum levels of 25-hydroxy-vitamin D, calcium, and parathyroid hormone. J. Clin. Endocrinol. Metab. 1975, 41, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Dent, C.E.; Richens, A.; Rowe, D.J.; Stamp, T.C. Osteomalacia with long-term anticonvulsant therapy in epilepsy. Br. Med. J. 1970, 4, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Stamp, T.C.; Round, J.M.; Rowe, D.J.; Haddad, J.G. Plasma levels and therapeutic effect of 25- hydroxycholecalciferol in epileptic patients taking anticonvulsant drugs. Br. Med. J. 1972, 4, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Andress, D.L.; Ozuna, J.; Tirschwell, D.; Grande, L.; Johnson, M.; Jacobson, A.F.; Spain, W. Antiepileptic drug-induced bone loss in young male patients who have seizures. Arch. Neurol. 2002, 59, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Perucca, E. Clinically relevant drug interactions with antiepileptic drugs. Br. J. Clin. Pharmacol. 2006, 61, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Tsukahara, H.; Kimura, K.; Todoroki, Y.; Ohshima, Y.; Hiraoka, M.; Shigematsu, Y.; Tsukahara, Y.; Miura, M.; Mayumi, M. Bone mineral status in ambulatory pediatric patients on long-term anti-epileptic drug therapy. Pediatr. Int. 2002, 44, 247–253. [Google Scholar] [CrossRef]
- Verrotti, A.; Coppola, G.; Parisi, P.; Mohn, A.; Chiarelli, F. Bone and calcium metabolism and antiepileptic drugs. Clin. Neurol. Neurosurg. 2010, 112, 1–10. [Google Scholar] [CrossRef]
- Ecevit, C.; Aydogan, A.; Kavakli, T.; Altinoz, S. Effect of carbamazepine and valproate on bone mineral density. Pediatric. Neurol. 2004, 31, 279–282. [Google Scholar] [CrossRef]
- Kafali, G.; Erselcan, T.; Tanzer, F. Effect of antiepileptic drugs on bone mineral density in children between ages 6 and 12 years. Clin. Pediatr. 1999, 38, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Sheth, R.D.; Wesolowski, C.A.; Jacob, J.C.; Penney, S.; Hobbs, G.R.; Riggs, J.E.; Bodensteiner, J.B. Effect of carbamazepine and valproate on bone mineral density. J. Pediatr. 1995, 127, 256–262. [Google Scholar] [CrossRef]
- Sato, Y.; Kondo, I.; Ishida, S.; Motooka, H.; Takayama, K.; Tomita, Y.; Maeda, H.; Satoh, K. Decreased bone mass and increased bone turnover with valproate therapy in adults with epilepsy. Neurology 2001, 57, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Oner, N.; Kaya, M.; Karasalihoglu, S.; Karaca, H.; Celtik, C.; Tutunculer, F. Bone mineral metabolism changes in epileptic children receiving valproic acid. J. Paediatr. Child Health 2004, 40, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Wang, S.Y.; Salter, D.M.; Wang, C.C.; Chen, S.J.; Fan, H.C. The impact of the use of antiepileptic drugs on the growth of children. BMC Pediatr. 2013, 13, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Legido, A.; De Luca, F. Effects of valproic acid on longitudinal bone growth. J. Child Neurol. 2004, 19, 26–30. [Google Scholar] [CrossRef]
- Jambalganiin, U.; Tsolmongyn, B.; Koide, N.; Odkhuu, E.; Naiki, Y.; Komatsu, T.; Yoshida, T.; Yokochi, T. A novel mechanism for inhibition of lipopolysaccharide-induced proinflammatory cytokine production by valproic acid. Int. Immunopharmacol. 2014, 20, 181–187. [Google Scholar] [CrossRef]
- Tan, T.Y.; Lu, C.H.; Chuang, H.Y.; Lin, T.K.; Liou, C.W.; Chang, W.N.; Chuang, Y.C. Long-term antiepileptic drug therapy contributes to the acceleration of atherosclerosis. Epilepsia 2009, 50, 1579–1586. [Google Scholar] [CrossRef]
- Van Beneden, K.; Geers, C.; Pauwels, M.; Mannaerts, I.; Verbeelen, D.; van Grunsven, L.A.; Van den Branden, C. Valproic acid attenuates proteinuria and kidney injury. J. Am. Soc. Nephrol. JASN 2011, 22, 1863–1875. [Google Scholar] [CrossRef]
- Arbez, J.; Lamarthee, B.; Gaugler, B.; Saas, P. Histone deacetylase inhibitor valproic acid affects plasmacytoid dendritic cells phenotype and function. Immunobiology 2014, 219, 637–643. [Google Scholar] [CrossRef]
- Ichiyama, T.; Okada, K.; Lipton, J.M.; Matsubara, T.; Hayashi, T.; Furukawa, S. Sodium valproate inhibits production of TNF-alpha and IL-6 and activation of NF-kappaB. Brain Res. 2000, 857, 246–251. [Google Scholar] [CrossRef]
- Eyal, S.; Yagen, B.; Sobol, E.; Altschuler, Y.; Shmuel, M.; Bialer, M. The activity of antiepileptic drugs as histone deacetylase inhibitors. Epilepsia 2004, 45, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Pack, A.M.; Morrell, M.J.; Marcus, R.; Holloway, L.; Flaster, E.; Done, S.; Randall, A.; Seale, C.; Shane, E. Bone mass and turnover in women with epilepsy on antiepileptic drug monotherapy. Ann. Neurol. 2005, 57, 252–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheth, R.D.; Harden, C.L. Screening for bone health in epilepsy. Epilepsia 2007, 48, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Stephen, L.J.; McLellan, A.R.; Harrison, J.H.; Shapiro, D.; Dominiczak, M.H.; Sills, G.J.; Brodie, M.J. Bone density and antiepileptic drugs: A case-controlled study. Seizure 1999, 8, 339–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestergaard, P. Epilepsy, osteoporosis and fracture risk—A meta-analysis. Acta Neurol. Scand. 2005, 112, 277–286. [Google Scholar] [CrossRef]
- Patsalos, P.N.; Berry, D.J.; Bourgeois, B.F.; Cloyd, J.C.; Glauser, T.A.; Johannessen, S.I.; Leppik, I.E.; Tomson, T.; Perucca, E. Antiepileptic drugs—Best practice guidelines for therapeutic drug monitoring: A position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia 2008, 49, 1239–1276. [Google Scholar] [CrossRef]
- Shim, K.S. Pubertal growth and epiphyseal fusion. Ann. Pediatr. Endocrinol. Metab. 2015, 20, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Barna, M.; Niswander, L. Visualization of cartilage formation: Insight into cellular properties of skeletal progenitors and chondrodysplasia syndromes. Dev. Cell 2007, 12, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Zuscik, M.J.; Hilton, M.J.; Zhang, X.; Chen, D.; O’Keefe, R.J. Regulation of chondrogenesis and chondrocyte differentiation by stress. J. Clin. Investig. 2008, 118, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Samsa, W.E.; Zhou, X.; Zhou, G. Signaling pathways regulating cartilage growth plate formation and activity. Semin. Cell Dev. Biol. 2017, 62, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozhemyakina, E.; Lassar, A.B.; Zelzer, E. A pathway to bone: Signaling molecules and transcription factors involved in chondrocyte development and maturation. Development 2015, 142, 817–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, A.; Wezeman, F.H. Developmental toxicity of valproic acid during embryonic chick vertebral chondrogenesis. Spine 2000, 25, 2158–2164. [Google Scholar] [CrossRef] [PubMed]
- Aulthouse, A.L.; Hitt, D.C. The teratogenic effects of valproic acid in human chondrogenesis in vitro. Teratology 1994, 49, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, R.F.; Oberdorf, J.A.; Sienko, S.; Aiona, M.D.; Boston, B.A.; Connelly, K.J.; Bahney, C.; LaRouche, J.; Almubarak, S.M.; Coleman, D.T.; et al. A degradation fragment of type X collagen is a real-time marker for bone growth velocity. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef] [Green Version]
- Amin, A.R.; Dave, M.; Attur, M.; Abramson, S.B. COX-2, NO, and cartilage damage and repair. Curr. Rheumatol. Rep. 2000, 2, 447–453. [Google Scholar] [CrossRef]
- Arasapam, G.; Scherer, M.; Cool, J.C.; Foster, B.K.; Xian, C.J. Roles of COX-2 and iNOS in the bony repair of the injured growth plate cartilage. J. Cell. Biochem. 2006, 99, 450–461. [Google Scholar] [CrossRef]
- Welting, T.J.; Caron, M.M.; Emans, P.J.; Janssen, M.P.; Sanen, K.; Coolsen, M.M.; Voss, L.; Surtel, D.A.; Cremers, A.; Voncken, J.W.; et al. Inhibition of cyclooxygenase-2 impacts chondrocyte hypertrophic differentiation during endochondral ossification. Eur. Cells Mater. 2011, 22, 420–436. [Google Scholar] [CrossRef]
- Pucci, B.; Adams, C.S.; Fertala, J.; Snyder, B.C.; Mansfield, K.D.; Tafani, M.; Freeman, T.; Shapiro, I.M. Development of the terminally differentiated state sensitizes epiphyseal chondrocytes to apoptosis through caspase-3 activation. J. Cell. Physiol. 2007, 210, 609–615. [Google Scholar] [CrossRef]
- Furumatsu, T.; Asahara, H. Histone acetylation influences the activity of Sox9-related transcriptional complex. Acta Med. Okayama 2010, 64, 351–357. [Google Scholar] [PubMed]
- Roberts, S.B.; Wootton, E.; De Ferrari, L.; Albagha, O.M.; Salter, D.M. Epigenetics of osteoarticular diseases: Recent developments. Rheumatol. Int. 2015, 35, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Blanco, A.; Segura-Pacheco, B.; Perez-Cardenas, E.; Taja-Chayeb, L.; Cetina, L.; Candelaria, M.; Cantu, D.; Gonzalez-Fierro, A.; Garcia-Lopez, P.; Zambrano, P.; et al. Histone acetylation and histone deacetylase activity of magnesium valproate in tumor and peripheral blood of patients with cervical cancer. A phase I study. Mol. Cancer 2005, 4, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, R.B.; Matsuda, K.; Oh, J.; Barbosa, A.C.; Yang, X.; Meadows, E.; McAnally, J.; Pomajzl, C.; Shelton, J.M.; Richardson, J.A.; et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 2004, 119, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Otero, M.; Favero, M.; Dragomir, C.; Hachem, K.E.; Hashimoto, K.; Plumb, D.A.; Goldring, M.B. Human chondrocyte cultures as models of cartilage-specific gene regulation. Methods Mol. Biol. 2012, 806, 301–336. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mRNA | Sequences | Product Size (Base Pairs) | Accession No. |
|---|---|---|---|
| Rat | |||
| GADPH | 5′-GAACGGGAAGCTCACTGGC-3′ | 70 | S67722 |
| 5′-GCATGTCAGATCCACAACGG-3′ | |||
| COX-2 | 5′-CCCTGAAACCTTACACATCGTTT-3′ | 90 | X02231 |
| 5′-TGGCATCGATGTCATGGTAGA-3′ | |||
| β-actin | 5′-GGAGATTACTGCCCTGGCTCCTA-3′ | 150 | S67722 |
| 5′-GACTCATCGTACTCCTGCTTGCTG-3′ | |||
| Col2a1 | 5′-TCCTAAGGGTGCCAATGGTGA-3′ | 112 | X02231 |
| 5′-GGACCAACTTTGCCTTGAGGAC-3′ | |||
| Col10a1 | 5′-TTCACAAAGAGCGGACAGAGA-3′ | 143 | S67722 |
| 5′-TCAAATGGGATGGGAGCA-3′ | |||
| ACAN | 5′-TCCGCTGGTCTGATGGACAC-3′ | 101 | X02231 |
| 5′-CCAGATCATCACTACGCAGTCCTC-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, H.-C.; Wang, S.-Y.; Peng, Y.-J.; Lee, H.-S. Valproic Acid Impacts the Growth of Growth Plate Chondrocytes. Int. J. Environ. Res. Public Health 2020, 17, 3675. https://doi.org/10.3390/ijerph17103675
Fan H-C, Wang S-Y, Peng Y-J, Lee H-S. Valproic Acid Impacts the Growth of Growth Plate Chondrocytes. International Journal of Environmental Research and Public Health. 2020; 17(10):3675. https://doi.org/10.3390/ijerph17103675
Chicago/Turabian StyleFan, Hueng-Chuen, Shih-Yu Wang, Yi-Jen Peng, and Herng-Sheng Lee. 2020. "Valproic Acid Impacts the Growth of Growth Plate Chondrocytes" International Journal of Environmental Research and Public Health 17, no. 10: 3675. https://doi.org/10.3390/ijerph17103675