
Semisynthesis and Cytotoxic Evaluation of an Ether Analogue Library Based on a Polyhalogenated Diphenyl Ether Scaffold Isolated from a Lamellodysidea Sponge
 , , , and
, , , and
Abstract
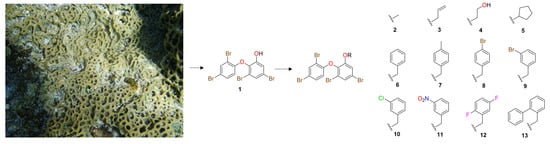
:
1. Introduction
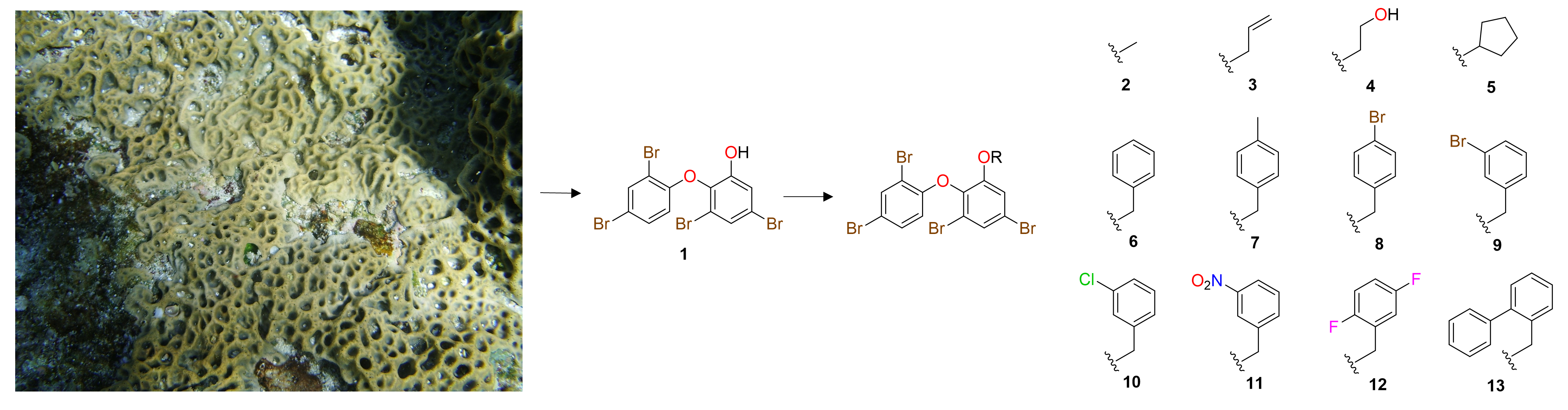
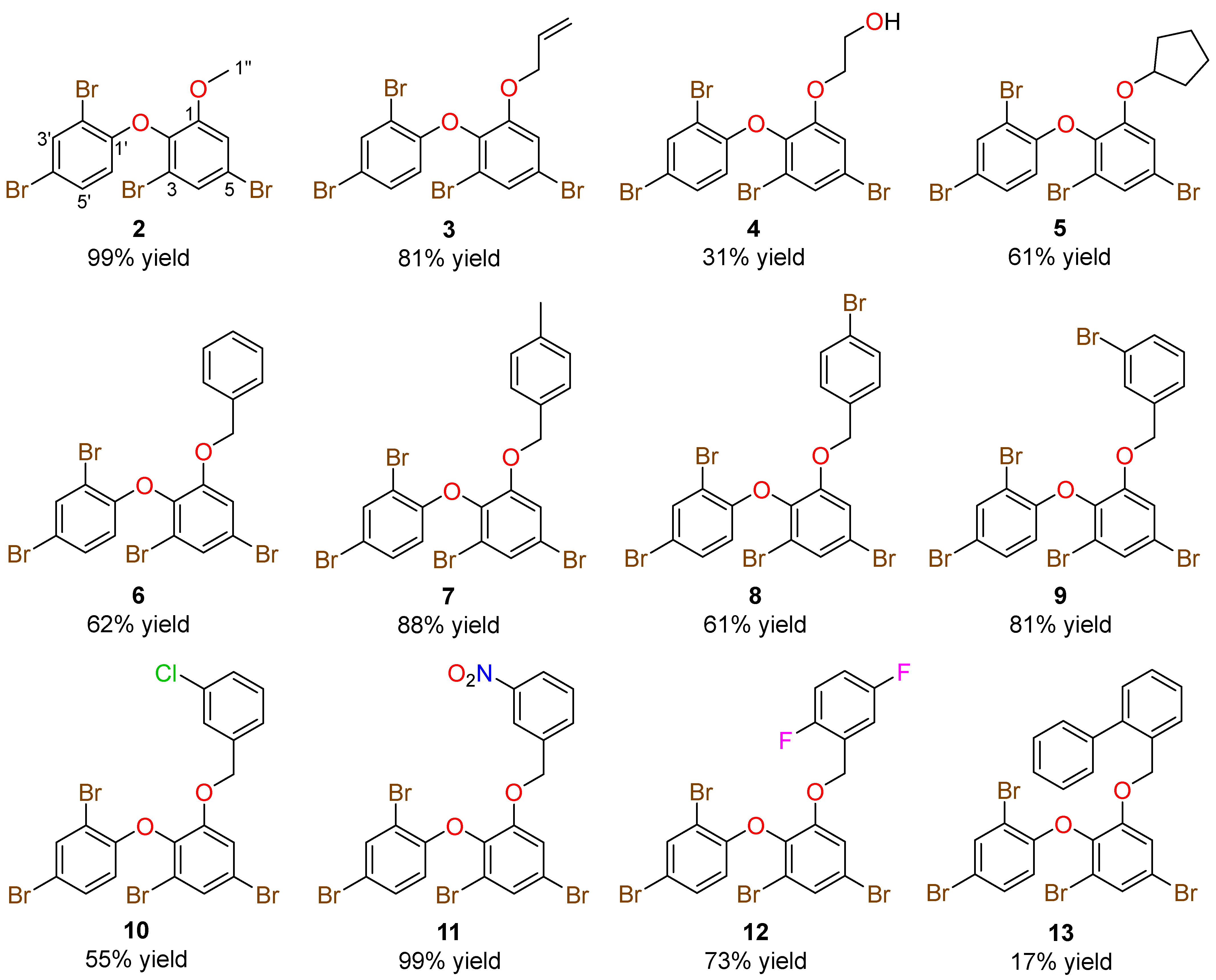
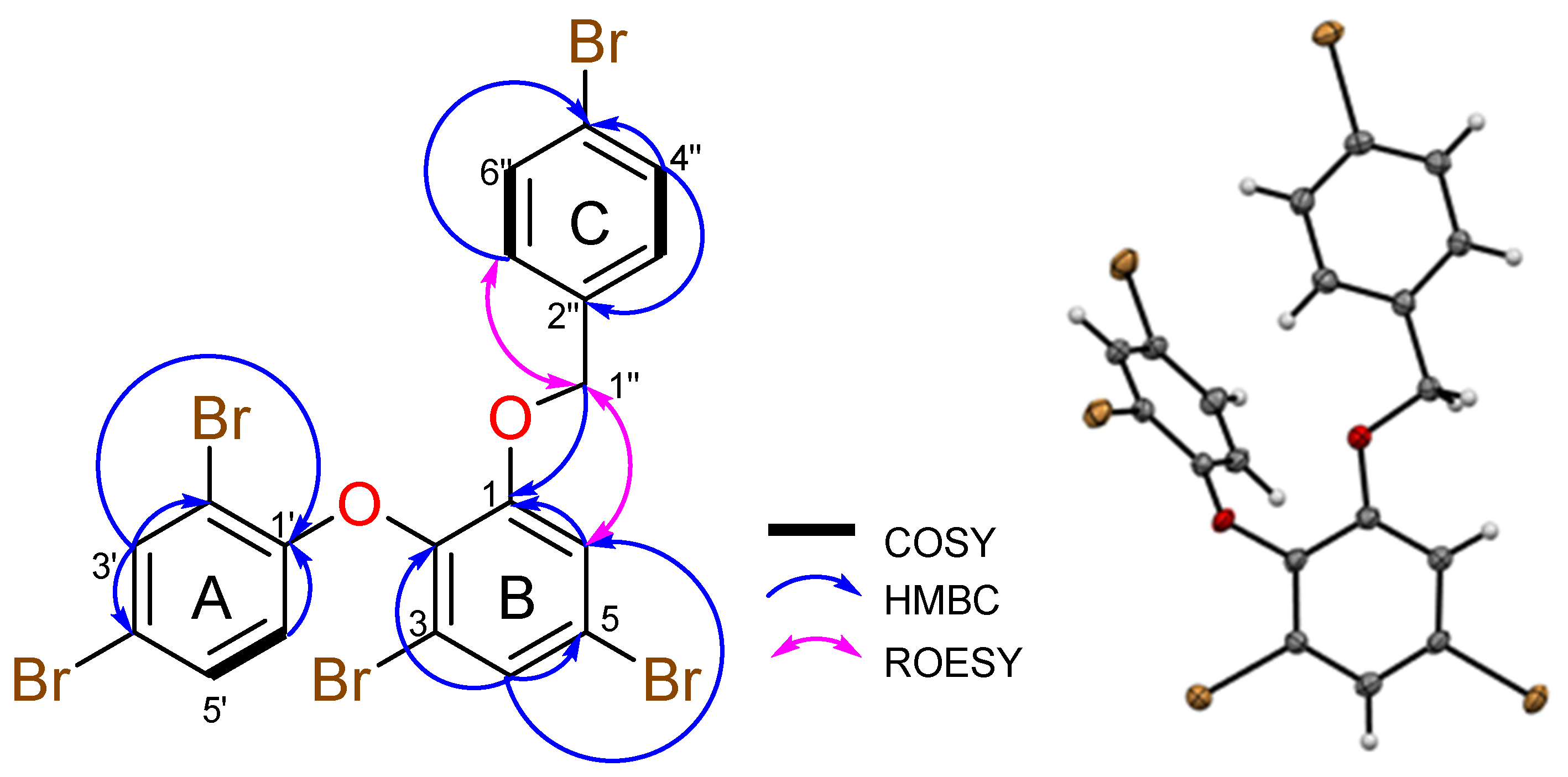
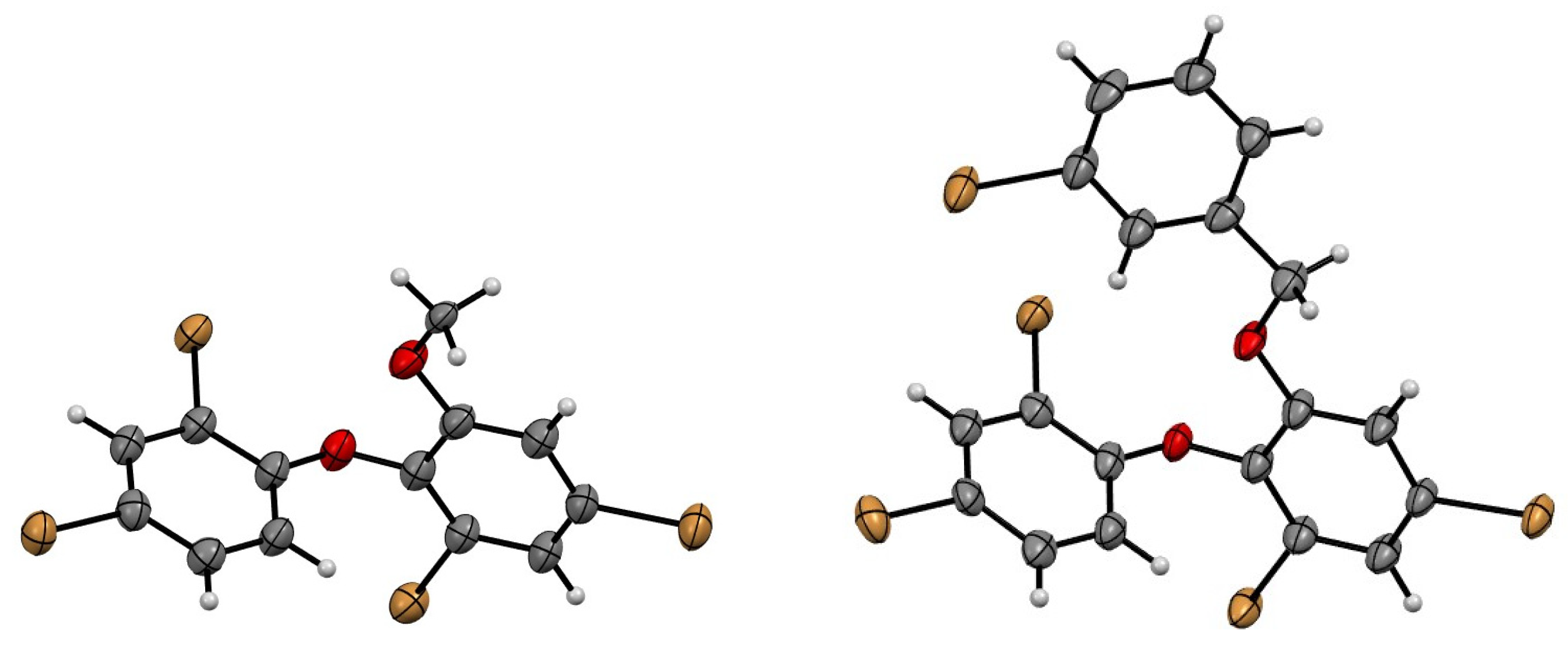
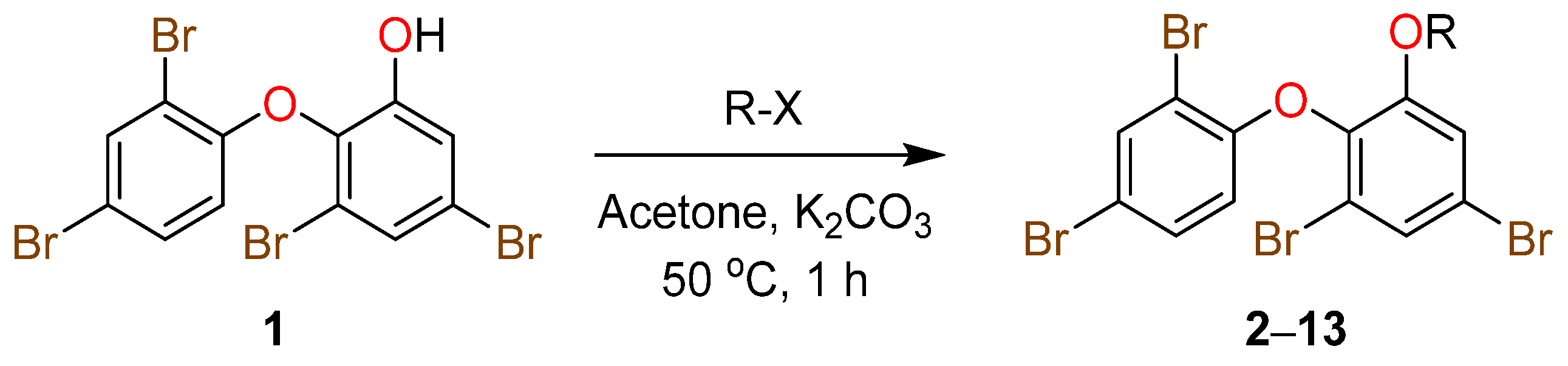
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental
3.2. Animal Material
3.3. Extraction and Isolation of O-PHDE 1
3.4. General Preparation of Ether Derivatives from O-PHDE Scaffold 1
3.5. X-ray Crystallography Analysis of Compounds 2, 8, and 9
3.6. Experimental Data for Natural Product 1 and Semisynthetic Compounds 2–13
3.7. Cancer Cell Cytotoxicity Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schorn, M.A.; Jordan, P.A.; Podell, S.; Blanton, J.M.; Agarwal, V.; Biggs, J.S.; Allen, E.E.; Moore, B.S. Comparative Genomics of Cyanobacterial Symbionts Reveals Distinct, Specialized Metabolism in Tropical Dysideidae Sponges. mBio 2019, 10, e00821-19. [Google Scholar] [CrossRef] [PubMed]
- Unson, M.D.; Holland, N.D.; Faulkner, D.J. A brominated secondary metabolite synthesized by the cyanobacterial symbiont of a marine sponge and accumulation of the crystalline metabolite in the sponge tissue. Mar. Biol. 1994, 119, 1–11. [Google Scholar] [CrossRef]
- Liu, H.; Lohith, K.; Rosario, M.; Pulliam, T.H.; O’Connor, R.D.; Bell, L.J.; Bewley, C.A. Polybrominated Diphenyl Ethers: Structure Determination and Trends in Antibacterial Activity. J. Nat. Prod. 2016, 79, 1872–1876. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Canning, C.B.; Bhargava, K.; Sun, X.; Zhu, W.; Zhou, N.; Zhang, Y.; Zhou, K. Polybrominated diphenyl ethers with potent and broad spectrum antimicrobial activity from the marine sponge Dysidea. Bioorg. Med. Chem. Lett. 2015, 25, 2181–2183. [Google Scholar] [CrossRef] [PubMed]
- Becerro, M.A.; Starmer, J.A.; Paul, V.J. Chemical defenses of cryptic and aposematic Gastropterid molluscs feeding on their host sponge Dysidea granulosa. J. Chem. Ecol. 2006, 32, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Pennings, S.C.; Pablo, S.R.; Paul, V.J.; Duffy, E. Effects of sponge secondary metabolites in different diets on feeding by three groups of consumers. J. Exp. Mar. Biol. Ecol. 1994, 180, 137–149. [Google Scholar] [CrossRef]
- Faisal, M.R.; Kellermann, M.Y.; Rohde, S.; Putra, M.Y.; Murniasih, T.; Risdian, C.; Mohr, K.I.; Wink, J.; Praditya, D.F.; Steinmann, E.; et al. Ecological and Pharmacological Activities of Polybrominated Diphenyl Ethers (PBDEs) from the Indonesian Marine Sponge Lamellodysidea herbacea. Mar. Drugs 2021, 19, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; El Gamal, A.A.; Yamanaka, K.; Poth, D.; Kersten, R.D.; Schorn, M.; Allen, E.E.; Moore, B.S. Biosynthesis of polybrominated aromatic organic compounds by marine bacteria. Nat. Chem. Biol. 2014, 10, 640–647. [Google Scholar] [CrossRef]
- Agarwal, V.; Li, J.; Rahman, I.; Borgen, M.; Aluwihare, L.I.; Biggs, J.S.; Paul, V.J.; Moore, B.S. Complexity of Naturally Produced Polybrominated Diphenyl Ethers Revealed via Mass Spectrometry. Environ. Sci. Technol. 2015, 49, 1339–1346. [Google Scholar] [CrossRef]
- Calcul, L.; Chow, R.; Oliver, A.G.; Tenney, K.; White, K.N.; Wood, A.W.; Fiorilla, C.; Crews, P. NMR Strategy for Unraveling Structures of Bioactive Sponge-Derived Oxy-polyhalogenated Diphenyl Ethers. J. Nat. Prod. 2009, 72, 443–449. [Google Scholar] [CrossRef]
- NatureBank. Available online: https://www.griffith.edu.au/institute-drug-discovery/unique-resources/naturebank (accessed on 13 October 2023).
- Ekins, M.G.; (Queensland Museum, South Brisbane, QLD 4101, Australia); Ramage, K.S.; (Griffith Institute for Drug Discovery, School of Environment and Science, Griffith University, Brisbane, QLD 4111, Australia). Personal communication, 2023.
- Carté, B.K.; Faulkner, D.J. Polybrominated Diphenyl Ethers from Dysidea herbacea, Dysidea chlorea and Phyllospongia foliascens. Tetrahedron 1981, 37, 2335–2339. [Google Scholar] [CrossRef]
- Kapojos, M.M.; Abdjul, D.B.; Yamazaki, H.; Kirikoshi, R.; Takahashi, O.; Rotinsulu, H.; Wewengkang, D.S.; Sumilat, D.A.; Ukai, K.; Namikoshi, M. Protein tyrosine phosphatase 1B inhibitory polybromobiphenyl ethers and monocyclofarnesol-type sesquiterpenes from the Indonesian marine sponge Lamellodysidea cf. herbacea. Phytochem. Lett. 2018, 24, 10–14. [Google Scholar] [CrossRef]
- Fu, X.; Schmitz, F.J.; Govindan, M.; Abbas, S.A.; Hanson, K.M.; Horton, P.A.; Crews, P.; Laney, M.; Schatzman, R.C. Enzyme inhibitors: New and known polybrominated phenols and diphenyl ethers from four Indo-Pacific Dysidea sponges. J. Nat. Prod. 1995, 58, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Johnson, R.K.; Hecht, S.M. Polybrominated diphenyl ethers from a sponge of the Dysidea genus that inhibit Tie2 kinase. Bioorg. Med. Chem. 2005, 13, 657–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Skildum, A.; Stromquist, E.; Rose-Hellekant, T.; Chang, L.C. Bioactive Polybrominated Diphenyl Ethers from the Marine Sponge Dysidea sp. J. Nat. Prod. 2008, 71, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Shin, D.; Kamiya, K.; Ishida, R.; Setiawan, A.; Kotoku, N.; Kobayashi, M. Marine spongean polybrominated diphenyl ethers, selective growth inhibitors against the cancer cells adapted to glucose starvation, inhibits mitochondrial complex II. J. Nat. Med. 2017, 71, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Huxley, C.; Wibowo, M.; Lum, K.Y.; Gordon, S.; D’Hyon, S.; Guan, H.; Wang, X.; Chen, Y.; Si, M.; Wang, M.; et al. Synthesis of bilocularin A carbamate derivatives and their evaluation as leucine transport inhibitors in prostate cancer cells. Phytochemistry 2020, 179, 112478–112485. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bidgood, C.L.; Levrier, C.; Gunter, J.H.; Nelson, C.C.; Sadowski, M.C.; Davis, R.A. Synthesis of a Unique Psammaplysin F Library and Functional Evaluation in Prostate Cancer Cells by Multiparametric Quantitative Single Cell Imaging. J. Nat. Prod. 2020, 83, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Herath, H.; Preston, S.; Jabbar, A.; Garcia-Bustos, J.; Taki, A.C.; Addison, R.S.; Hayes, S.; Beattie, K.D.; McGee, S.L.; Martin, S.D.; et al. Identification of Fromiamycalin and Halaminol A from Australian Marine Sponge Extracts with Anthelmintic Activity against Haemonchus contortus. Mar. Drugs 2019, 17, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Teuten, E.L.; Xu, L.; Reddy, C.M. Two abundant bioaccumulated halogenated compounds are natural products. Science 2005, 307, 917–920. [Google Scholar] [CrossRef]
- Tietjen, I.; Cassel, J.; Register, E.T.; Zhou, X.Y.; Messick, T.E.; Keeney, F.; Lu, L.D.; Beattie, K.D.; Rali, T.; Tebas, P. The natural stilbenoid (–)-hopeaphenol inhibits cellular entry of SARS-CoV-2 USA-WA1/2020, B. 1.1. 7, and B. 1.351 variants. Antimicrob. Agents Chemother. 2021, 65, e00772-21. [Google Scholar] [CrossRef] [PubMed]
- Varricchio, A.; Khan, S.; Price, Z.K.; Davis, R.A.; Ramesh, S.A.; Yool, A.J. Pharmacological Inhibition of Membrane Signaling Mechanisms Reduces the Invasiveness of U87-MG and U251-MG Glioblastoma Cells In Vitro. Cancers 2023, 15, 1027–1047. [Google Scholar] [CrossRef] [PubMed]
- Aragao, D.; Aishima, J.; Cherukuvada, H.; Clarken, R.; Clift, M.; Cowieson, N.P.; Ericsson, D.J.; Gee, C.L.; Macedo, S.; Mudie, N.; et al. MX2: A high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron Rad. 2018, 25, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0–New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Farrugia, L. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Carrasco-Pozo, C.; Tan, K.N.; Rodriguez, T.; Avery, V.M. The Molecular Effects of Sulforaphane and Capsaicin on Metabolism upon Androgen and Tip60 Activation of Androgen Receptor. Int. J. Mol. Sci. 2019, 20, 5384–5405. [Google Scholar] [CrossRef] [PubMed]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Evaluation of chemotherapeutics in a three-dimensional breast cancer model. J. Cancer Res. Clin. Oncol. 2015, 141, 951–959. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1H mult. (J in Hz, int.) | 13C, mult. | COSY | HMBC | ROESY |
|---|---|---|---|---|---|
| 1 | - | 151.9, C | - | - | - |
| 2 | - | 139.8, C | - | - | - |
| 3 | - | 117.9, C | - | - | - |
| 4 | 7.63 d (2.1, 1H) | 127.1, CH | 6 | 1 w, 2, 5, 6 | - |
| 5 | - | 119.1, C | - | - | - |
| 6 | 7.56 d (2.1, 1H) | 117.8, CH | 4 | 1, 2, 4, 5 | 1″ |
| 1′ | - | 152.7, C | - | - | - |
| 2′ | - | 111.8, C | - | - | - |
| 3′ | 7.90 d (2.4, 1H) | 135.2, CH | 5′ | 1′, 2′, 4′, 5′ | - |
| 4′ | - | 114.4, C | - | - | - |
| 5′ | 7.41 dd (8.8, 2.4, 1H) | 131.7, CH | 3′, 6′ | 1′, 2′ w, 3′, 4′ | 6′ |
| 6′ | 6.53 d (8.8, 1H) | 116.3, CH | 5′ | 1′, 2′, 3′ w, 4′ | 5′ |
| 1″ | 5.12 s (2H) | 69.8, CH2 | - | 1, 2″ w, 3″, 7″ | 6, 3″, 7″ |
| 2″ | - | 135.2, C | - | - | - |
| 3″ | 7.05 m (1H) | 129.3, CH | 4″ | 1″, 5″, 7″ | 1″, 4″ |
| 4″ | 7.48 m (1H) | 131.2, CH | 3″ | 2″, 3″, 5″, 6″, 7″ w | 3″ |
| 5″ | - | 121.2, C | - | - | - |
| 6″ | 7.48 m (1H) | 131.2, CH | 7″ | 2″, 3″ w, 4″, 5″, 7″ | 7″ |
| 7″ | 7.05 m (1H) | 129.3, CH | 6″ | 1″, 3″, 5″ | 1″, 6″ |
| Compound | Average % Inhibition at 50 µM ± SD | |||
|---|---|---|---|---|
| DU145 | LNCaP | MCF-7 | MDA-MB-231 | |
| 1 | 39 ± 5 | 63 ± 1 | 63 ± 0 | 65 ± 10 |
| 2 | IA a | IA | IA | IA |
| 3 | 21 ± 1 | 22 ± 9 | 64 ± 1 | 55 ± 4 |
| 4 | IA | IA | IA | IA |
| 5 | 11 ± 1 | 4 ± 0 | 55 ± 1 | 17 ± 2 |
| 6 | IA | IA | IA | IA |
| 7 | IA | IA | IA | IA |
| 8 | IA | IA | 1 ± 1 | IA |
| 9 | 13 ± 4 | 9 ± 1 | 62 ± 4 | 8 ± 11 |
| 10 | 14 ± 3 | 3 ± 1 | 47 ± 0 | 7 ± 10 |
| 11 | IA | 13 ± 1 | 26 ± 2 | IA |
| 12 | IA | 2 ± 1 | IA | IA |
| 13 | IA | IA | IA | IA |
| Puromycin IC50 (µM) | 0.24 ± 0.01 | 0.20 ± 0.00 | 0.21 ± 0.08 | 0.15 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramage, K.S.; Lock, A.; White, J.M.; Ekins, M.G.; Kiefel, M.J.; Avery, V.M.; Davis, R.A. Semisynthesis and Cytotoxic Evaluation of an Ether Analogue Library Based on a Polyhalogenated Diphenyl Ether Scaffold Isolated from a Lamellodysidea Sponge. Mar. Drugs 2024, 22, 33. https://doi.org/10.3390/md22010033
Ramage KS, Lock A, White JM, Ekins MG, Kiefel MJ, Avery VM, Davis RA. Semisynthesis and Cytotoxic Evaluation of an Ether Analogue Library Based on a Polyhalogenated Diphenyl Ether Scaffold Isolated from a Lamellodysidea Sponge. Marine Drugs. 2024; 22(1):33. https://doi.org/10.3390/md22010033
Chicago/Turabian StyleRamage, Kelsey S., Aaron Lock, Jonathan M. White, Merrick G. Ekins, Milton J. Kiefel, Vicky M. Avery, and Rohan A. Davis. 2024. "Semisynthesis and Cytotoxic Evaluation of an Ether Analogue Library Based on a Polyhalogenated Diphenyl Ether Scaffold Isolated from a Lamellodysidea Sponge" Marine Drugs 22, no. 1: 33. https://doi.org/10.3390/md22010033