
A Comprehensive Computational NMR Analysis of Organic Polyarsenicals including the Marine Sponge-Derived Arsenicins A–D and Their Synthetic Analogs

Abstract
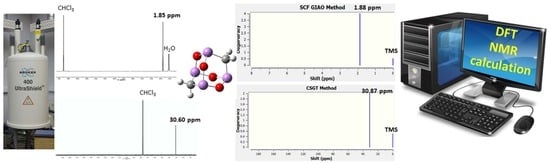
:
1. Introduction
2. Results and Discussion
2.1. DFT-Calculated vs. Experimental 1H-NMR Chemical Shifts of Polyarsenicals 1–12: Evaluation of Functionals and Basis Sets
2.2. Calculation of 1H,1H Coupling Constants
2.3. DFT-Calculated vs. Experimental 13C-NMR Chemical Shifts Polyarsenicals 1–12: Evaluation of Functionals and Basis Sets
2.4. Comprehensive Evaluation of 1H- and 13C- Chemical Shifts
3. Materials and Methods
Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molinski, T.F. Microscale methodology for structure elucidation of natural products. Curr. Opin. Biotechnol. 2010, 21, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational Prediction of 1H and 13C Chemical Shifts: A Useful Tool for Natural Product, Mechanistic, and Synthetic Organic Chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497. [Google Scholar] [CrossRef]
- Mancini, I.; Guella, G.; Frostin, M.; Hnawia, E.; Laurent, D.; Debitus, C.; Pietra, F. On the First Polyarsenic Organic Compound from Nature: Arsenicin A from the New Caledonian Marine Sponge Echinochalina bargibanti. Chem. A Eur. J. 2006, 12, 8989–8994. [Google Scholar] [CrossRef] [PubMed]
- Guella, G.; Mancini, I.; Mariotto, G.; Rossi, B.; Viliani, G. Vibrational analysis as a powerful tool in structure elucidation of polyarsenicals: A DFT-based investigation of arsenicin A. Phys. Chem. Chem. Phys. 2009, 11, 2420–2427. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Rae, A.D.; Salem, G.; Weir, M.L.; Willis, A.C.; Wild, S.B. Arsenicin A, A Natural Polyarsenical: Synthesis and Crystal Structure. Organometallics 2010, 29, 32–33. [Google Scholar] [CrossRef]
- Lu, D.; Coote, M.L.; Ho, J.; Kilah, N.L.; Lin, C.Y.; Salem, G.; Weir, M.L.; Willis, A.C.; Wild, S.B.; Dilda, P.J. Resolution and Improved Synthesis of (±)-Arsenicin A: A Natural Adamantane-Type Tetraarsenical Possessing Strong Anti-Acute Promelocytic Leukemia Cell Line Activity. Organometallics 2012, 31, 1808–1816. [Google Scholar] [CrossRef]
- Mancini, I.; Planchestainer, M.; Defant, A. Synthesis and In-vitro Anticancer Evaluation of Polyarsenicals Related to the Marine Sponge Derived Arsenicin A. Sci. Rep. 2017, 7, 11548–11556. [Google Scholar] [CrossRef]
- Marx, M.B.L.; Pritzkow, H.; Keppler, B.K. Struktur von S-9,10-Dimethyl-1,3,5,7-tetraarsa-2,4,6,8-tetraoxaadamantan und 9,10-Diethyl-1,3,5,7-tetraarsa-2,4,6,8-tetraoxaadamantan. Z. Für Anorg. Und Allg. Chem. 1996, 622, 1097–1100. [Google Scholar] [CrossRef]
- Vigna, J.; Sighel, D.; Rosatti, E.F.; Defant, A.; Pancher, M.; Sidarovich, V.; Quattrone, A.; Mancini, I. Expanding the Chemical Space of Arsenicin A-C Related Polyarsenicals and Evaluation of Some Analogs as Inhibitors of Glioblastoma Stem Cell Growth. Mar. Drugs 2023, 21, 186. [Google Scholar] [CrossRef]
- Tähtinen, P.; Saielli, G.; Guella, G.; Mancini, I.; Bagno, A. Computational NMR Spectroscopy of Organoarsenicals and the Natural Polyarsenic Compound Arsenicin A. Chem. A Eur. J. 2008, 14, 10445–10452. [Google Scholar] [CrossRef] [PubMed]
- Tähtinen, P.; Guella, G.; Saielli, G.; Debitus, C.; Hnawia, E.; Mancini, I. New Sulfur-Containing Polyarsenicals from the New Caledonian Sponge Echinochalina bargibanti. Mar. Drugs 2018, 16, 382. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Arulmozhiraja, S.; Coote, M.L.; Rae, A.D.; Salem, G.; Willis, A.C.; Wild, S.B.; Benhenda, S.; Breitenbach, V.L.; De Thé, H.; et al. Sulfur Derivatives of the Natural Polyarsenical Arsenicin a: Biologically Active, Organometallic Arsenic-Sulfur Cages Related to the Minerals Realgar and Uzonite. Organometallics 2015, 34, 829–840. [Google Scholar] [CrossRef]
- Paul, N.P.; Galván, A.E.; Yoshinaga-Sakurai, K.; Rosen; Yoshinaga, M. Arsenic in medicine: Past, present and future. Biometals 2023, 36, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Thomas Bally, T.; Rablen, P.R. Quantum-Chemical Simulation of 1H NMR Spectra. 2.Comparison of DFT-Based Procedures for Computing Proton-Proton Coupling Constants in Organic Molecules. J. Org. Chem. 2011, 76, 4818–4830. [Google Scholar] [CrossRef]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient implementation of the Gauge-Independent Atomic Orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Li, Y.; Gao, H.; Zhang, J.; Li, S.; Zhou, W. Comparison of GIAO and CSGT for calculating 13C and 15N nuclear magnetic resonance chemical shifts of substituent neutral 5-aminotetrazole and 5-nitrotetrazole compounds. Magn. Reson. Chem. 2012, 50, 16–21. [Google Scholar] [CrossRef]
- Toomsalu, E.; Burk, P. Critical test of some computational methods for prediction of NMR 1H and 13C chemical shifts. J. Mol. Model. 2015, 21, 244–264. [Google Scholar] [CrossRef]
- Bursch, M.; Mewes, J.-M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef]
- Jan Vıcha, J.; Novotny, J.; Komorovsky, S.; Straka, M.; Kaupp, M.; Marek, R. Relativistic Heavy-Neighbor-Atom Effects on NMR Shifts: Concepts and Trends Across the Periodic Table. Chem. Rev. 2020, 120, 7065–7103. [Google Scholar] [CrossRef]
- Betz, R.; Klüfers, P.; Reichvilser, M.M.; Roeßner, F.W. The structures of methylenebis(dichloroarsane) and methylenebisarsonic acid—A combined theoretical and experimental study. Z. Für Anorg. Und Allg. Chem. 2008, 634, 696–700. [Google Scholar] [CrossRef]
- Hesse, M.; Meier, H.; Zeeh, B. Nuclear Magnetic Resonance Spectroscopy NMR Spectra and Molecular Structure. In Spectroscopic Methods in Organic Chemistry, 2nd ed.; Thieme: Stuttgart, Germany, 2008; pp. 89–104. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-Functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. 4. A new dynamical correlation functional and implications for exactexchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Handy, N.C.; Cohen, A. Left-Right correlation energy. J. Mol. Phys. 2001, 99, 403–412. [Google Scholar] [CrossRef]
- Hoe, W.M.; Cohen, A.J.; Handy, N.C. Assessment of a new local exchange functional OPTX. Chem. Phys. Lett. 2001, 341, 319–328. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation-energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 mode. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical metageneralized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- London, F. The quantic theory of inter-atomic currents in aromatic combinations. J. Phys. Radium 1937, 8, 397–409. [Google Scholar] [CrossRef]
- McWeeny, R. Perturbation Theory for Fock-Dirac Density Matrix. Phys. Rev. 1962, 126, 1028–1034. [Google Scholar] [CrossRef]
- Ditchfield, R. Molecular Orbital Theory of Magnetic Shielding and Magnetic Susceptibility. J. Chem. Phys. 1972, 56, 5688–5691. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. 1. Gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| O-Polyarsenicals | |||||
|---|---|---|---|---|---|
| TPSSh | |||||
| Comp. | Expt 2 | aug-cc-pVDZ | aug-cc-pVTZ | x2c-TZVPPAll-s | |
| 1 | 2J = 13.8 | 2J | −15.3 | −13.2 | −13.9 |
| 4J = 0.9 | 4J | 1.1 | 1.6 | 1.6 | |
| 2 | n.d. | 2J | −15.0 | −13.0 | −13.5 |
| 3 | 2J = 13.8 | 2J | −15.2 | −13.1 | −13.7 |
| 4 | 2J = 13.8 | 2J | −15.2 | −13.1 | −13.7 |
| 5 | JCH,Me = 7.9 | JCH,Me | 7.1 | 8.1 | 8.3 |
| S-polyarsenicals | |||||
| OLYP | |||||
| aug-cc-pVDZ | aug-cc-pVTZ | x2c-TZVPPAll-s | |||
| 6 | 2J = 12.4 | J14,15 | −13.6 | −12.0 | −12.6 |
| 2J = 13.5 | J12,13 | −14.2 | −12.9 | −13.5 | |
| 2J = 13.8 | J10,11 | −14.4 | −13.1 | −13.7 | |
| 4J = 1.7 | J10,12 | 0.76 | 1.1 | 1.1 | |
| 7 | 2J = 12.8 | J14,15 | −13.8 | −12.3 | −12.9 |
| 2J = 13.7 | J12,13 | −14.3 | −13.0 | −13.6 | |
| 2J = 13.8 | J10,11 | −14.4 | −13.0 | −13.6 | |
| 4J = 1.7 | J13,15 | 0.70 | 1.1 | 1.1 | |
| 4J = 1.9 | J10,12 | 0.92 | 1.4 | 1.3 | |
| 8 | 2J = 12.3 | 2J | −14.2 | −13.5 | −13.4 |
| 9 | 2J = 12.3 | 2J | −13.3 | −11.8 | −12.5 |
| 10 | JCH,Me = 7.9 | JCH,Me | 6.0 | 6.4 | 6.7 |
| 11 | JCH,Me = 7.2 | JCH,Me | 6.3 | 7.3 | 7.6 |
| 12 | JCH,Me = 7.2 | JCH,Me | 5.6 | 6.4 | 6.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Defant, A.; Mancini, I. A Comprehensive Computational NMR Analysis of Organic Polyarsenicals including the Marine Sponge-Derived Arsenicins A–D and Their Synthetic Analogs. Mar. Drugs 2023, 21, 511. https://doi.org/10.3390/md21100511
Defant A, Mancini I. A Comprehensive Computational NMR Analysis of Organic Polyarsenicals including the Marine Sponge-Derived Arsenicins A–D and Their Synthetic Analogs. Marine Drugs. 2023; 21(10):511. https://doi.org/10.3390/md21100511
Chicago/Turabian StyleDefant, Andrea, and Ines Mancini. 2023. "A Comprehensive Computational NMR Analysis of Organic Polyarsenicals including the Marine Sponge-Derived Arsenicins A–D and Their Synthetic Analogs" Marine Drugs 21, no. 10: 511. https://doi.org/10.3390/md21100511