
The Polysaccharidic Nature of the Skeleton of Marennine as Determined by NMR Spectroscopy
 , ,
, ,
Abstract
:1. Introduction
2. Results
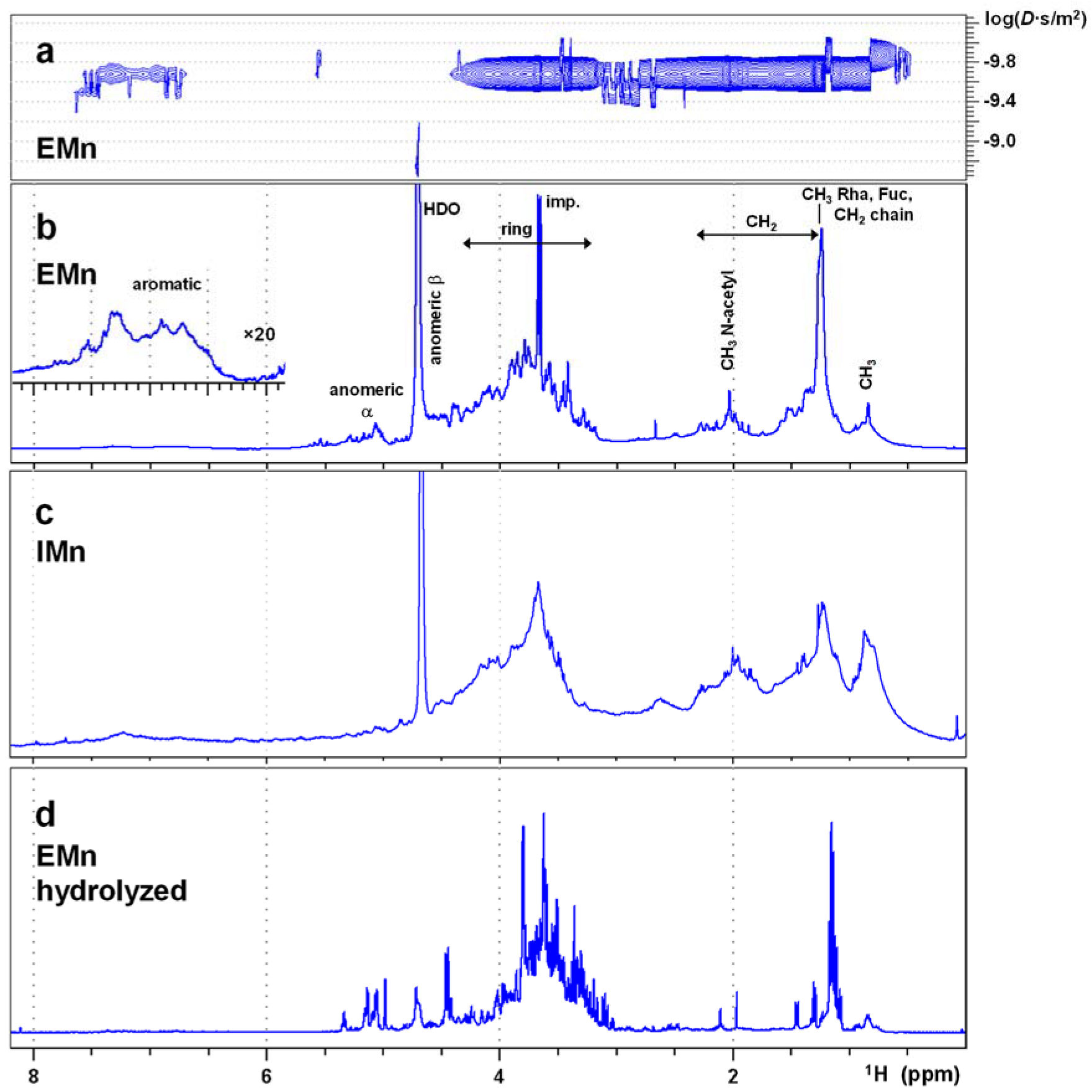
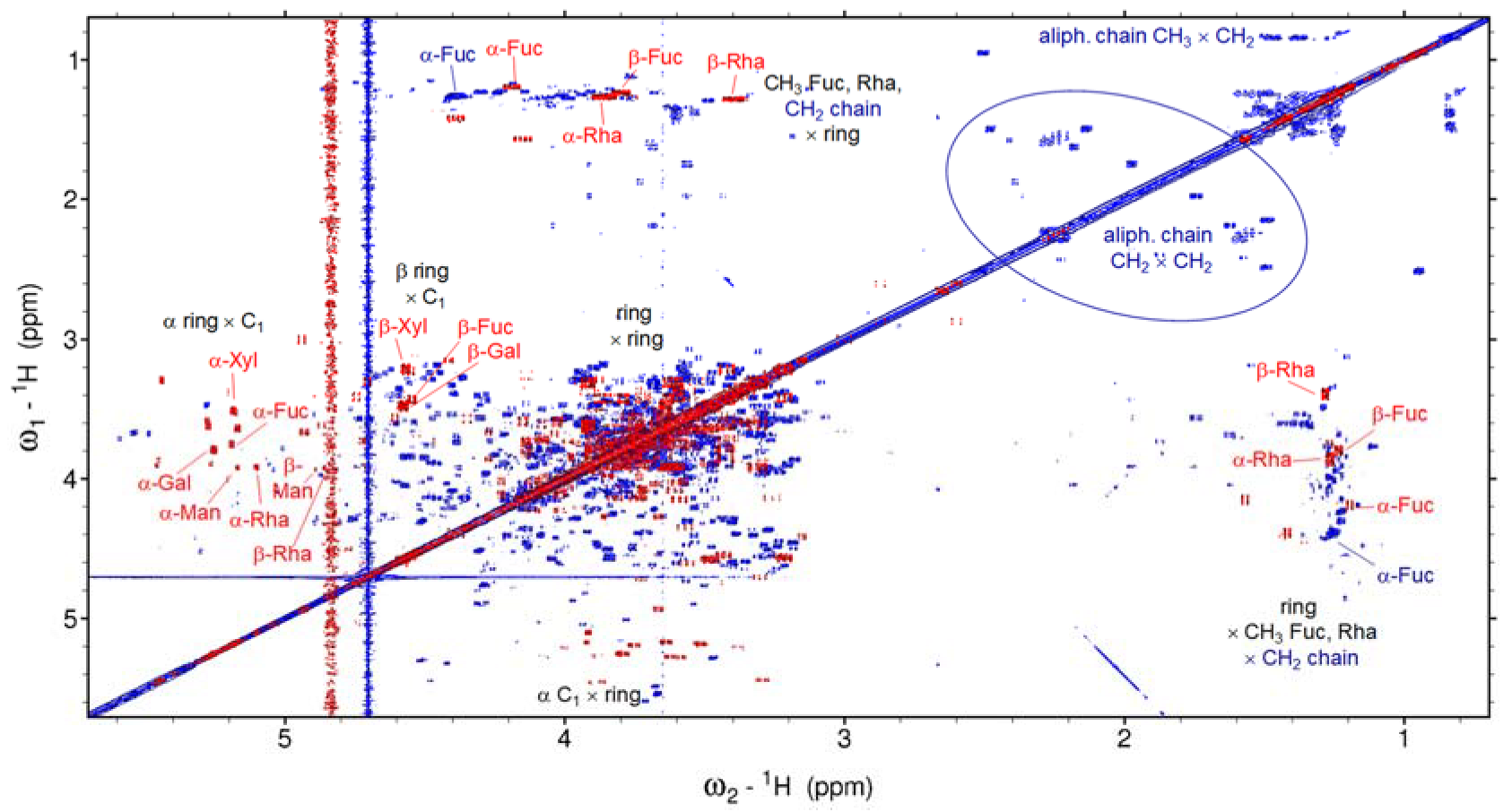
2.1. NMR of the Extracellular Form of Marennine (EMn)
2.2. NMR of the Intracellular Form of Marennine (IMn)
2.3. NMR of EMn after Acidic Hydrolysis
3. Discussion
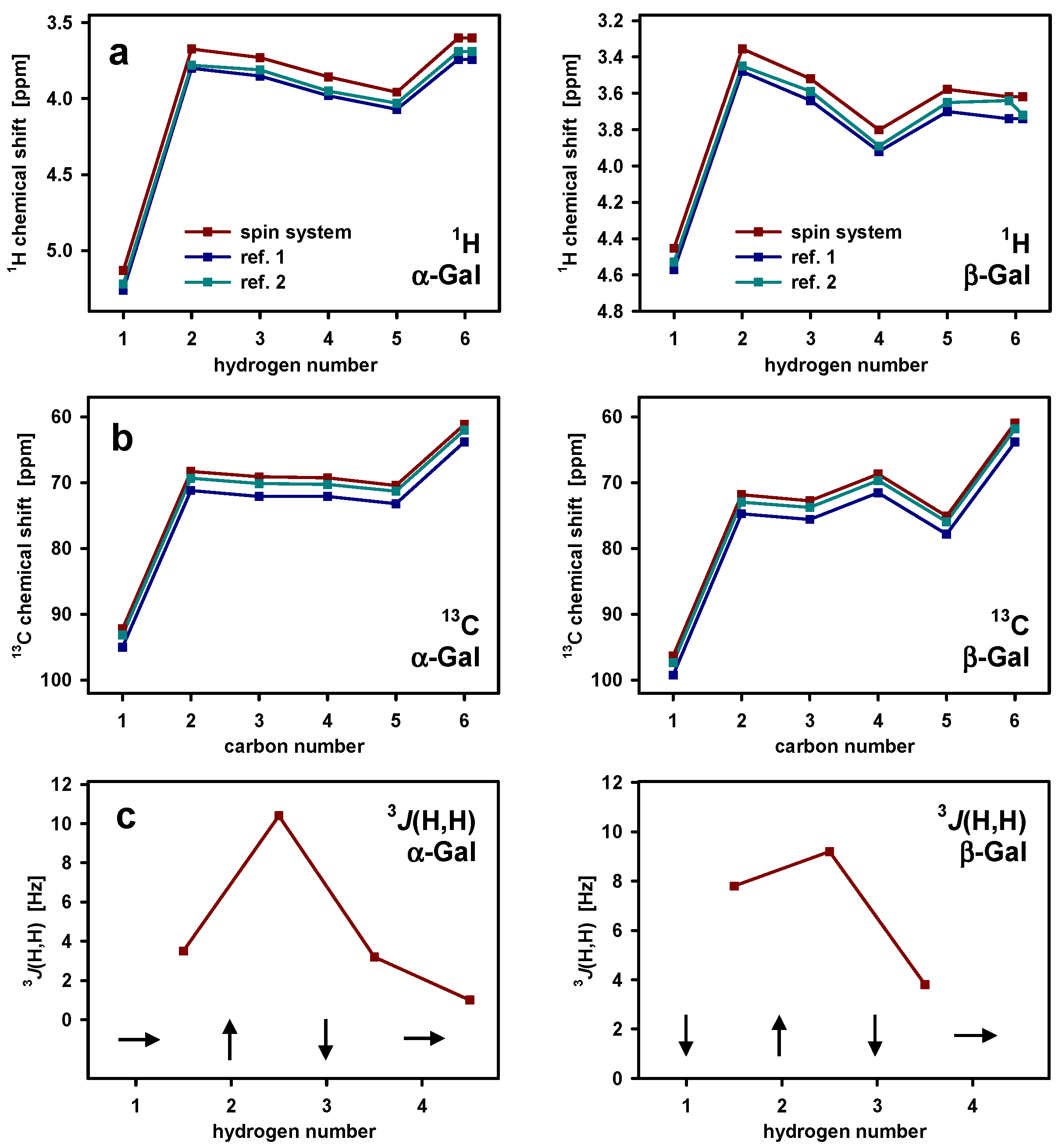
3.1. Extracellular Polysaccharide
3.2. Variability
3.3. Additional Tests
3.4. Chromophore
4. Materials and Methods
4.1. Extraction and Purification of Marennine
4.2. Hydrolysis
4.3. NMR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sprat, T. The history of the generation and ordering green oysters, commonly called Colchester-oysters. In The History of the Royal Society; Martyn: London, UK, 1667. [Google Scholar]
- Gaillon, B. Des huîtres vertes et des causes de cette coloration. J. Phys. Chim. Hist. Nat. Arts 1820, 91, 222–225. [Google Scholar]
- Bory de Saint-Vincent, J.B. Navicules. In Dictionnaire Classique d’Histoire Naturelle; Rey et Gravier/Baudouin: Paris, France, 1824; pp. 562–565. [Google Scholar]
- Simonsen, R. The Diatom Plankton of the Indian Ocean Expedition of RV “Meteor”; Deutsche Forschungsgemeinschaft: Bonn, Germany, 1974; Volume 19, pp. 46–49. [Google Scholar]
- Lankester, E.R. On green oysters. Q. J. Microsc. Sci. 1886, 26, 71–94. [Google Scholar] [CrossRef] [Green Version]
- Pouvreau, J.-B.; Morançais, M.; Fleury, F.; Rosa, P.; Thion, L.; Cahingt, B.; Zal, F.; Fleurence, J.; Pondaven, P. Preliminary characterization of the blue-green pigment “marennine” from the marine tychopelagic diatom Haslea ostrearia (Gaillon/Bory) Simonsen. J. Appl. Phycol. 2006, 18, 757–767. [Google Scholar] [CrossRef]
- Prasetiya, F.; Decottignies, P.; Barillé, L.; Gastineau, R.; Jacquette, B.; Figiel, A.; Morançais, M.; Tremblay, R.; Mouget, J.-L.; Cognie, B. Cell size-based, passive selection of the blue diatom Haslea ostrearia by the oyster Crassostrea gigas. J. Mollus. Stud. 2017, 83, 145–152. [Google Scholar] [CrossRef]
- Gastineau, R.; Prasetiya, F.; Falaise, C.; Cognie, B.; Decottignies, P.; Morançais, M.; Méléder, V.; Davidovich, N.; Turcotte, F.; Tremblay, R.; et al. Marennine-like pigments: Blue diatom or green oyster cult? In Blue Technologies: Production and Use of Marine Molecules; Wiley: Weinheim, Germany, 2018; pp. 529–551. [Google Scholar]
- Bocat, L. Sur la marennine de la diatomée bleue; comparaison avec la phycocyanine. C. R. Soc. Biol. 1907, 62, 1073–1075. [Google Scholar]
- Newsome, A.G.; Culver, C.A.; van Breemen, R.B. Nature’s palette: The search for natural blue colorants. J. Agr. Food. Chem. 2014, 62, 6498–6511. [Google Scholar] [CrossRef] [PubMed]
- Gastineau, R.; Turcotte, F.; Pouvreau, J.-B.; Morançais, M.; Fleurence, J.; Windarto, E.; Prasetiya, F.; Arsad, S.; Jaouen, P.; Babin, M.; et al. Marennine, promising blue pigments from a widespread Haslea diatom species complex. Mar. Drugs 2014, 12, 3161–3189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gastineau, R.; Pouvreau, J.-B.; Hellio, C.; Morançais, M.; Fleurence, J.; Gaudin, P.; Bourgougnon, N.; Mouget, J.-L. Biological activities of purified marennine, the blue pigment responsible for the greening of oysters. J. Agr. Food Chem. 2012, 60, 3599–3605. [Google Scholar] [CrossRef]
- Falaise, C.; Francois, C.; Travers, M.-A.; Morga, B.; Haure, J.; Tremblay, R.; Turcotte, F.; Pasetto, P.; Gastineau, R.; Hardivillier, Y.; et al. Antimicrobial compounds from eukaryotic microalgae against human pathogens and diseases in aquaculture. Mar. Drugs 2016, 14, 159. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, F.; Mouget, J.-L.; Genard, B.; Lemarchand, K.; Deschênes, J.S.; Tremblay, R. Prophylactic effect of Haslea ostrearia supernatant culture containing the pigment marennine to stabilize bivalve hatchery production. Aquat. Living Resour. 2016, 29, 401. [Google Scholar] [CrossRef] [Green Version]
- Bizio, B. Ricerche sopra il coloramento in verde delle branchie delle ostriche. In Memorie dell’Imperial Regio Instituto Veneto di Scienze Lettere ed Arti; Antonelli: Venice, Italy, 1845; Volume 2. [Google Scholar]
- Coste, V. Voyage d’Exploration sur le Littoral de la France Et de l’Italie; Imprimerie Impériale: Paris, France, 1861. [Google Scholar]
- Ryder, J.A. On the cause of the greening of oysters. With a supplementary note on the coloration of the blood corpuscles of the oysters. In Report of Commission of Fish and Fisheries for 1882; Governement Printing Office: Washington, DC, USA, 1884. [Google Scholar]
- Robert, J.M.; Hallet, J.N. Absorption spectrum in vivo of the blue pigment “marennine” of the pennate diatom Navicula ostrearia Bory. J. Exp. Bot. 1981, 32, 341–345. [Google Scholar] [CrossRef]
- Bachrach, E. Le bleuissement des diatomées et le verdissement des huîtres. Revue critique. Rev. Trav. Inst. Pêches Marit. 1935, 8, 112–123. [Google Scholar]
- Genevès, L.; Choussy, M.; Barbier, M.; Neuville, D.; Daste, P. Ultrastructure et composition pigmentaire comparées des chromatophores de la diatomée Navicula ostrearia (Gaillon) Bory normale et bleue. C. R. Acad. Sci. Sér. D Sci. Nat. 1976, 282, 449–452. [Google Scholar]
- Ranson, G. Le verdissement des huîtres. Sciences 1937, 8, 13–24. [Google Scholar]
- Neuville, D.; Daste, P.H. Production de pigment bleu par la diatomée Navicula ostrearia (Gaillon) Bory maintenue en culture uni-algale sur un milieu synthétique carencé en azote nitrique. C. R. Acad. Sci. Paris 1972, 274, 2030–2033. [Google Scholar]
- Vandanjon, L.; Jaouen, P.; Rossignol, N.; Quéméneur, F.; Robert, J.M. Concentration and desalting by membrane processes of a natural pigment produced by the marine diatom Haslea ostrearia Simonsen. J. Biotechnol. 1999, 70, 393–402. [Google Scholar] [CrossRef]
- Branda, S.; Vik, A.; Freidmen, L.; Kolter, R. Biofilms: The matrix revisited. Trends Microbiol. 2005, 13, 20–26. [Google Scholar] [CrossRef]
- Decho, A.W.; Gutierrez, T. Microbial Extracellular Polymeric Substances (EPSs) in ocean systems. Front. Microbiol. 2017, 8, 922. [Google Scholar] [CrossRef] [PubMed]
- Underwood, G.H.; Paterson, D.M. The importance of extracellular carbohydrate production by marine epipelic diatoms. Adv. Bot. Res. 2003, 40, 183–240. [Google Scholar]
- Shniukova, E.I.; Zolotareva, E.K. Diatom exopolysaccharides: A review. Int. J. Algae. 2015, 17, 50–67. [Google Scholar] [CrossRef]
- Qi, M.; Zheng, C.; Wu, W.; Yu, G.; Wang, P. Exopolysaccharides from marine microbes: Source, structure and application. Mar. Drugs 2022, 20, 512. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, W.; Arnold, A.A.; Turcotte, F.; Saint-Louis, R.; Deschênes, J.-S.; Genard, B.; Marcotte, I.; Tremblay, R. Extraction improvement of the bioactive blue-green pigment “marennine” from diatom Haslea ostrearia’s blue water: A solid-phase method based on graphitic matrices. Mar. Drugs 2020, 18, 653. [Google Scholar] [CrossRef]
- Furevi, A.; Ruda, A.; Angles d’Ortoli, T.; Mobarak, H.; Ståhle, J.; Hamark, C.; Fontana, C.; Engström, O.; Apostolica, P.; Widmalm, G. Complete 1H and 13C NMR chemical shift assignments of mono- to tetrasaccharides as basis for NMR chemical shift predictions of oligo- and polysaccharides using the computer program CASPER. Carbohydr. Res. 2022, 513, 108528. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, Y.; Robert, J.-M.; Rincé, Y.; Ginsburger-Vogel, T. The cytoplasmic fine structure of the diatom Haslea ostrearia (Bacillariophyceae) in relation to marennine production. Phycologia 1998, 37, 84–91. [Google Scholar] [CrossRef]
- Shi, H.; Wan, Y.; Li, O.; Zhang, X.; Xie, M.; Nie, S.; Yin, J. Two-step hydrolysis method for monosaccharide composition analysis of natural polysaccharides rich in uronic acids. Food Hydrocoll. 2020, 101, 105524. [Google Scholar] [CrossRef]
- Jansson, P.-E.; Kenne, L.; Widmalm, G. Computer-assisted structural analysis of polysaccharides with an extended version of CASPER using 1H and 13C N.M.R. data. Carbohyd. Res. 1989, 188, 169–191. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. [Google Scholar] [CrossRef]
- Einbu, A.; Vårum, K.M. Depolymerization and de-N-acetylation of chitin oligomers in hydrochloric acid. Biomacromolecules 2007, 8, 309–314. [Google Scholar] [CrossRef]
- Beattie, A.; Hirst, E.L.; Percival, E. Studies on the metabolism of the Chrysophyceae. Comparative structural investigations on leucosin (chrysolaminarin) separated from diatoms and laminarin from the brown algae. Biochem. J. 1961, 79, 531–537. [Google Scholar] [CrossRef]
- Haug, A.; Myklestad, S. Polysaccharides of marine diatoms with special reference to Chaetoceros species. Mar. Biol. 1972, 34, 217–222. [Google Scholar] [CrossRef]
- Urbani, R.; Sist, P.; Pletikapic, G.; Misic Radic, T.; Svetličić, V.; Zutic, V. Diatom polysaccharides: Extracellular production, isolation and molecular characterization. In The Complex World of Polysaccharides; Karunaratne, D.N., Ed.; InTechOpen: London, UK, 2012; pp. 345–370. [Google Scholar]
- Gügi, B.; Le Costaouec, T.; Burel, C.; Lerouge, P.; Helbert, W.; Bardor, M. Diatom-specific oligosaccharide and polysaccharide structures help to unravel biosynthetic capabilities in diatoms. Mar. Drugs 2015, 13, 5993–6018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierre, G.; Graber, M.; Rafiliposon, A.R.; Dupuy, C.; Orvain, F.; De Crignis, M.; Maugard, T. Biochemical composition and changes of extracellular polysaccharides (ECPS) produced during microphytobenthic biofilm development (Marennes-Oléron, France). Microb. Ecol. 2012, 63, 157–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raize, O.; Argaman, Y.; Yannai, S. Mechanisms of biosorption of different heavy metals by brown marine macroalgae. Biotechnol. Bioeng. 2004, 87, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Lu, F.; Wei, X.; Zhao, R. Fucoidan: Structure and bioactivity. Molecules 2008, 13, 1671–1695. [Google Scholar] [CrossRef] [Green Version]
- Grachev, A.A.; Gerbst, A.G.; Ustyuzhanina, N.E.; Shashkov, A.S.; Usov, A.I.; Nifantiev, N.E. NMR Investigation of the influence of sulfate groups at C-2 and C-4 on the conformational behavior of fucoidan fragments with homo-(1→3)-linked backbone. J. Carbohydr. Chem. 2006, 25, 315–330. [Google Scholar] [CrossRef]
- Clément, M.-J.; Tissot, B.; Chevolot, L.; Adjadj, E.; Du, Y.; Curmi, P.A.; Daniel, R. NMR characterization and molecular modeling of fucoidan showing the importance of oligosaccharide branching in its anticomplementary activity. Glycobiology 2010, 20, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, R. Natural polysaccharides and their interactions with dye molecules: Applications in effluent treatment. Environ. Sci. Technol. 2004, 38, 4905–4909. [Google Scholar] [CrossRef]
- Yoshida, Z.-I.; Osawa, E. Hydrogen bonding of phenol to electrons of aromatics, polyolefins, heteroaromatics, fulvenes, and azulenes. J. Am. Chem. Soc. 1966, 88, 4019–4026. [Google Scholar] [CrossRef]
- Francezon, N.; Herbaut, M.; Bardeau, J.-F.; Cougnon, C.; Bélanger, W.; Tremblay, R.; Jacquette, B.; Dittmer, J.; Pouvreau, J.-B.; Mouget, J.-L.; et al. Electrochromic Properties and Electrochemical Behavior of Marennine, a Bioactive Blue-Green Pigment Produced by the Marine Diatom Haslea ostrearia. Mar. Drugs 2021, 19, 231. [Google Scholar] [CrossRef]
- Pouvreau, J.-B.; Morançais, M.; Massé, G.; Rosa, P.; Robert, J.-M.; Fleurence, J.; Pondaven, P. Purification of the blue-green pigment “marennine” from the marine tychopelagic diatom Haslea ostrearia (Gaillon/Bory) Simonsen. J. Appl. Phycol. 2006, 18, 769–781. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
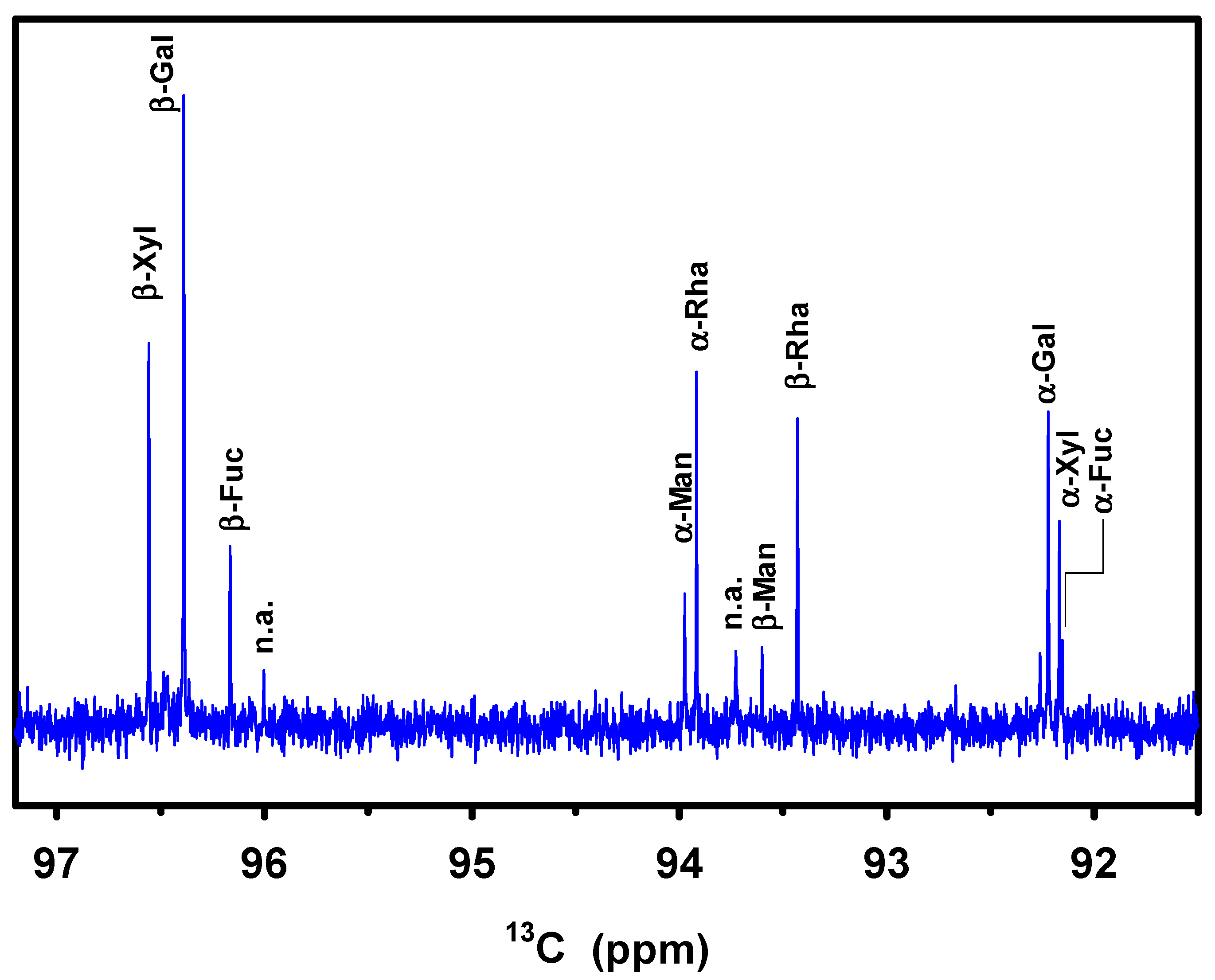
| Type | 13C C1 (ppm) | Relative Intensity | % Conformation 1 (Tabular Value) | Fraction 2, α and β |
|---|---|---|---|---|
| α-Galp | 92.22 | 0.44 | 31 (32) % | 36% |
| β-Galp | 96.39 | 1.00 | 69 (64) % | |
| α-Xylp | 92.17 | 0.27 | 34 (35) % | 19% |
| β-Xylp | 96.56 | 0.51 | 66 (65) % | |
| α-Manp | 93.97 | 0.20 | 62 (67) % | 8% |
| β-Manp | 93.60 | 0.12 | 38 (33) % | |
| α-Rhap | 93.92 | 0.45 | 55 (60) % | 20% |
| β-Rhap | 93.43 | 0.36 | 45 (40) % | |
| α-Fucp | 92.15 | 0.11 | 33 (21) % | 8% |
| β-Fucp | 96.16 | 0.23 | 67 (67) % |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zebiri, I.; Jacquette, B.; Francezon, N.; Herbaut, M.; Latigui, A.; Bricaud, S.; Tremblay, R.; Pasetto, P.; Mouget, J.-L.; Dittmer, J. The Polysaccharidic Nature of the Skeleton of Marennine as Determined by NMR Spectroscopy. Mar. Drugs 2023, 21, 42. https://doi.org/10.3390/md21010042
Zebiri I, Jacquette B, Francezon N, Herbaut M, Latigui A, Bricaud S, Tremblay R, Pasetto P, Mouget J-L, Dittmer J. The Polysaccharidic Nature of the Skeleton of Marennine as Determined by NMR Spectroscopy. Marine Drugs. 2023; 21(1):42. https://doi.org/10.3390/md21010042
Chicago/Turabian StyleZebiri, Ilhem, Boris Jacquette, Nellie Francezon, Mickaël Herbaut, Amina Latigui, Sullivan Bricaud, Réjean Tremblay, Pamela Pasetto, Jean-Luc Mouget, and Jens Dittmer. 2023. "The Polysaccharidic Nature of the Skeleton of Marennine as Determined by NMR Spectroscopy" Marine Drugs 21, no. 1: 42. https://doi.org/10.3390/md21010042