
Discovery of Ircinianin Lactones B and C—Two New Cyclic Sesterterpenes from the Marine Sponge Ircinia wistarii

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
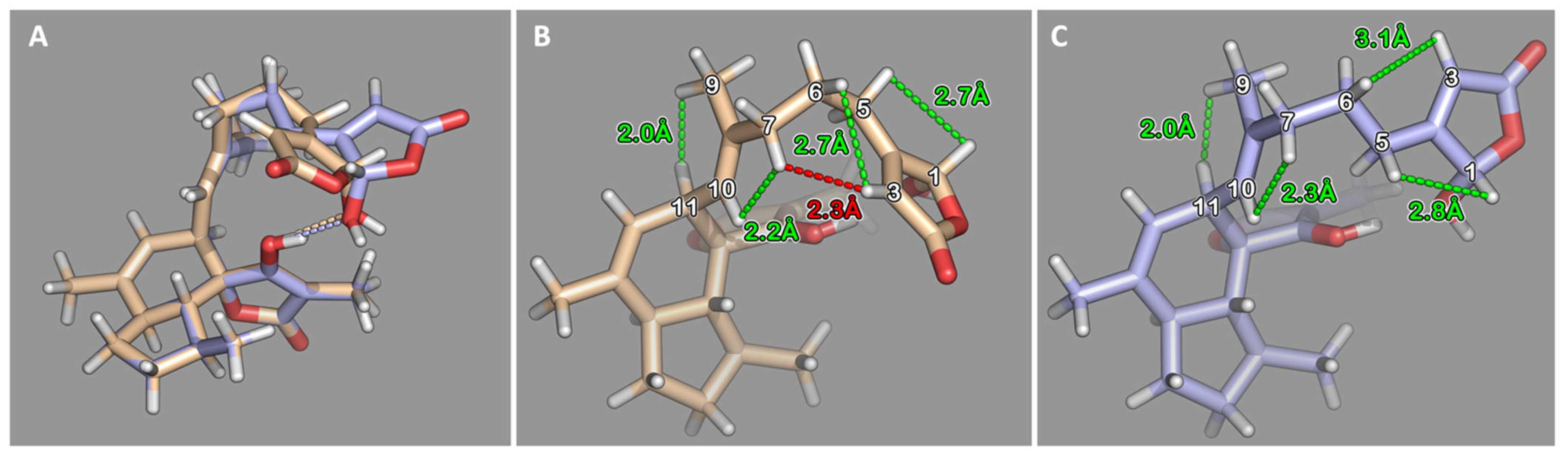
2.1. Isolation and Structure Elucidation
2.2. Biological Evaluation
2.2.1. Antimicrobial Activity
2.2.2. Antiviral Activity
2.2.3. Anthelmintic Activity
2.2.4. Antiprotozoal Activity
2.2.5. Cytotoxicity
2.2.6. Marine Antifouling Activity
2.2.7. Conclusions of the Screening for Biological Activity of Ircinianin (1)
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Computational Methods
3.5. Biological Evaluation
3.5.1. Antibacterial Assay
3.5.2. Antiviral Assay
3.5.3. Anthelmintic Assay
3.5.4. Antiprotozoal Assays
3.5.5. Cytotoxicity Assays
3.5.6. Marine Antifouling Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef] [PubMed]
- Manes, L.V.; Naylor, S.; Crews, P.; Bakus, G.J. Suvanine, a novel sesterterpene from an Ircinia marine sponge. J. Org. Chem. 1985, 50, 284–286. [Google Scholar] [CrossRef]
- Höller, U.; König, G.M.; Wright, A.D. Two New Sesterterpene Tetronic Acids from the Marine Sponge Ircinia oros. J. Nat. Prod. 1997, 60, 832–835. [Google Scholar] [CrossRef]
- Wright, A.E.; McCarthy, P.J.; Schulte, G.K. Sulfircin: A new sesterterpene sulfate from a deep-water sponge of the genus Ircinia. J. Org. Chem. 1989, 54, 3472–3474. [Google Scholar] [CrossRef]
- Barrow, C.J.; Blunt, J.W.; Munro, M.H.G.; Perry, N.B. Oxygenated Furanosesterterpene Tetronic Acids from a Sponge of the Genus Ircinia. J. Nat. Prod. 1988, 51, 1294–1298. [Google Scholar] [CrossRef]
- Cimino, G.; De Stefano, S.; Minale, L.; Fattorusso, E. Ircinin-1 and -2, linear sesterterpenes from the marine sponge Ircinia oros. Tetrahedron 1972, 28, 333–341. [Google Scholar] [CrossRef]
- Faulkner, D.J. Variabilin, an antibiotic from the sponge, Ircinia variabilis. Tetrahedron Lett. 1973, 14, 3821–3822. [Google Scholar] [CrossRef]
- Alfano, G.; Cimino, G.; De Stefano, S. Palinurin, a new linear sesterterpene from a marine sponge. Experientia 1979, 35, 1136–1137. [Google Scholar] [CrossRef]
- Cafieri, F.; Fattorusso, E.; Santacroce, C.; Minale, L. Fasciculatin, a novel sesterterpene from the sponge Ircinia fasciculata. Tetrahedron 1972, 28, 1579–1583. [Google Scholar] [CrossRef]
- Choi, H.J.; Choi, Y.H.; Yee, S.-B.; Im, E.; Jung, J.H.; Kim, N.D. Ircinin-1 induces cell cycle arrest and apoptosis in SK-MEL-2 human melanoma cells. Mol. Carcinog. 2005, 44, 162–173. [Google Scholar] [CrossRef]
- Chianese, G.; Silber, J.; Luciano, P.; Merten, C.; Erpenbeck, D.; Topaloglu, B.; Kaiser, M.; Tasdemir, D. Antiprotozoal Linear Furanosesterterpenoids from the Marine Sponge Ircinia oros. J. Nat. Prod. 2017, 80, 2566–2571. [Google Scholar] [CrossRef] [PubMed]
- Cholbi, R.; Ferrdndiz, M.L.; Terencio, M.C.; Alcaraz, M.J.; Payd, M.; De Rosa, S. Inhibition of phospholipase A2 activities and some inflammatory responses by the marine product ircinin. Naunyn Schmiedeberg's Arch. Pharmacol. 1996, 354, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Balansa, W.; Islam, R.; Fontaine, F.; Piggott, A.M.; Zhang, H.; Webb, T.I.; Gilbert, D.F.; Lynch, J.W.; Capon, R.J. Ircinialactams: Subunit-selective glycine receptor modulators from Australian sponges of the family Irciniidae. Bioorg. Med. Chem. 2010, 18, 2912–2919. [Google Scholar] [CrossRef] [PubMed]
- Hahn, D.; Chin, J.; Kim, H.; Yang, I.; Won, D.H.; Ekins, M.; Choi, H.; Nam, S.-J.; Kang, H. Sesquiterpenoids with PPARδ agonistic effect from a Korean marine sponge Ircinia sp. Tetrahedron Lett. 2014, 55, 4716–4719. [Google Scholar] [CrossRef]
- Buchanan, M.S.; Edser, A.; King, G.; Whitmore, J.; Quinn, R.J. Cheilanthane Sesterterpenes, Protein Kinase Inhibitors, from a Marine Sponge of the Genus Ircinia. J. Nat. Prod. 2001, 64, 300–303. [Google Scholar] [CrossRef]
- Tsoukatou, M.; Hellio, C.; Vagias, C.; Harvala, C.; Roussis, V. Chemical Defense and Antifouling Activity of Three Mediterranean Sponges of the Genus Ircinia. Z. Nat. C 2002, 57, 161–171. [Google Scholar] [CrossRef]
- Epifanio, R.D.A.; Gabriel, R.; Martins, D.L.; Muricy, G. The Sesterterpene Variabilin as a Fish-Predation Deterrent in the Western Atlantic Sponge Ircinia strobilina. J. Chem. Ecol. 1999, 25, 2247–2254. [Google Scholar] [CrossRef]
- Li, K.; Gustafson, K.R. Sesterterpenoids: Chemistry, biology, and biosynthesis. Nat. Prod. Rep. 2021, 38, 1251–1281. [Google Scholar] [CrossRef]
- Shen, Y.-C.; Lo, K.-L.; Lin, Y.-C.; Khalil, A.T.; Kuo, Y.-H.; Shih, P.-S. Novel linear C22-sesterterpenoids from sponge Ircinia formosana. Tetrahedron Lett. 2006, 47, 4007–4010. [Google Scholar] [CrossRef]
- Issa, H.H.; Tanaka, J.; Higa, T. New Cytotoxic Furanosesterterpenes from an Okinawan Marine Sponge, Ircinia sp. J. Nat. Prod. 2003, 66, 251–254. [Google Scholar] [CrossRef]
- Cambie, R.C.; Rickard, C.E.F.; Rutledge, P.S.; Yang, X.-S. Scalarolide and scalarin, sesterterpenes from Cacospongia and Ircinia sponges. Acta Cryst. Sect. C 1999, 55, 112–114. [Google Scholar] [CrossRef]
- Lai, Y.-Y.; Lu, M.-C.; Wang, L.-H.; Chen, J.-J.; Fang, L.-S.; Wu, Y.-C.; Sung, P.-J. New Scalarane Sesterterpenoids from the Formosan Sponge Ircinia felix. Mar. Drugs 2015, 13, 4296–4309. [Google Scholar] [CrossRef] [PubMed]
- Hofheinz, W.; Schönholzer, P. Ircinianin, a Novel Sesterterpene from a Marine Sponge. Helv. Chim. Acta 1977, 60, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Sato, M.-a.; Yoshii, E. Synthesis of (±)-ircinianin, a marine sponge sesterterpene. Tetrahedron Lett. 1986, 27, 3903–3906. [Google Scholar] [CrossRef]
- Uenishi, J.i.; Kawahama, R.; Yonemitsu, O. Total Synthesis of (−)-Ircinianin and (+)-Wistarin. J. Org. Chem. 1997, 62, 1691–1701. [Google Scholar] [CrossRef]
- Majer, T.; Schollmeyer, D.; Koch, P.; Gross, H. (2S,3′S,3a′R,5′R,7a′R)-5′-[(E)-5-(Furan-3-yl)-2-methylpent-1-en-1-yl]-3-hydroxy-3',4,7'-trimethyl-1',2',3',3a',5',7a'-hexahydro-5H-spiro[furan-2,4'-inden]-5-one. IUCrData 2020, 5, x201578. [Google Scholar] [CrossRef]
- Gregson, R.P.; Ouvrier, D.; Wistarin, A. Tetracyclic Furanosesterterpene from the Marine Sponge Ircinia wistarii. J. Nat. Prod. 1982, 45, 412–414. [Google Scholar] [CrossRef]
- Fontana, A.; Fakhr, I.; Mollo, E.; Cimino, G. (−)-Wistarin from the marine sponge Ircinia sp.: The first case of enantiomeric sesterterpenes. Tetrahedron Asymmetry 1999, 10, 3869–3872. [Google Scholar] [CrossRef]
- Coll, J.C.; Kearns, P.S.; Rideout, J.A.; Hooper, J. Ircinianin Sulfate from the Marine Sponge Ircinia (Psammocinia) wistarii. J. Nat. Prod. 1997, 60, 1178–1179. [Google Scholar] [CrossRef]
- Balansa, W.; Islam, R.; Fontaine, F.; Piggott, A.M.; Zhang, H.; Xiao, X.; Webb, T.I.; Gilbert, D.F.; Lynch, J.W.; Capon, R.J. Sesterterpene glycinyl-lactams: A new class of glycine receptor modulator from Australian marine sponges of the genus Psammocinia. Org. Biomol. Chem. 2013, 11, 4695–4701. [Google Scholar] [CrossRef]
- Khushi, S.; Nahar, L.; Salim, A.A.; Capon, R.J. Cacolides: Sesterterpene Butenolides from a Southern Australian Marine Sponge, Cacospongia sp. Mar. Drugs 2018, 16, 456. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Harada, K.; Kubo, M.; Hioki, H.; Fukuyama, Y. Eight New Clerodane Diterpenoids from the Bark of Ptychopetalum olacoides. Nat. Prod. Commun. 2011, 6, 1934578X1100600305. [Google Scholar] [CrossRef]
- Liu, Y.; Hong, J.; Lee, C.-O.; Im, K.S.; Kim, N.D.; Choi, J.S.; Jung, J.H. Cytotoxic Pyrrolo- and Furanoterpenoids from the Sponge Sarcotragus Species. J. Nat. Prod. 2002, 65, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Couperus, P.A.; Clague, A.D.H.; van Dongen, J.P.C.M. 13C Chemical Shifts of some Model Olefins. Org. Magn. Reson. 1976, 8, 426–431. [Google Scholar] [CrossRef]
- Everett, J.R.; Hunt, E.; Tyler, J.W. Ketone–hemiacetal tautomerism in erythromycin A in non-aqueous solutions. An NMR spectroscopic study. J. Chem. Soc. Perkin Transact. 2 1991, 10, 1481–1487. [Google Scholar] [CrossRef]
- Hamada, T.; Harada, D.; Hirata, M.; Yamashita, K.; Palaniveloo, K.; Okamura, H.; Iwagawa, T.; Arima, N.; Iriguchi, T.; de Voogd, N.J.; et al. Manoalide-related Sesterterpene from the Marine Sponge Luffariella variabilis. Nat. Prod. Commun. 2015, 10, 863–864. [Google Scholar] [CrossRef]
- Hog, D.T.; Webster, R.; Trauner, D. Synthetic approaches toward sesterterpenoids. Nat. Prod. Rep. 2012, 29, 752–779. [Google Scholar] [CrossRef]
- Prasad, P.; Zhang, A.; Salim, A.A.; Capon, R.J. Pursuing sesterterpene lactams in Australian Irciniidae sponges. Fitoterapia 2018, 126, 83–89. [Google Scholar] [CrossRef]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef]
- Thakur, N.L.; Anil, A.C. Antibacterial Activity of the Sponge Ircinia ramosa: Importance of its Surface-Associated Bacteria. J. Chem. Ecol. 2000, 26, 57–71. [Google Scholar] [CrossRef]
- Capon, R.; Macleod, J. A New Sesterterpene Tetronic Acid from an Australian Sponge, Ircinia sp. Austral. J. Chem. 1987, 40, 1327–1330. [Google Scholar] [CrossRef]
- Rothberg, I.; Shubiak, P. The structure of some antibiotics from the sponge Ircinia strobilina. Tetrahedron Lett. 1975, 16, 769–772. [Google Scholar] [CrossRef]
- Su, J.H.; Tseng, S.W.; Lu, M.C.; Liu, L.L.; Chou, Y.; Sung, P.J. Cytotoxic C21 and C22 terpenoid-derived metabolites from the sponge Ircinia sp. J. Nat. Prod. 2011, 74, 2005–2009. [Google Scholar] [CrossRef]
- Susumu, K.; Kentaro, Y.; Jasim, U.M.; Toshiyasu, I.; Kiyotake, S.; Katsuhiro, U.; Daisuke, U. Kohamaic Acids A and B, Novel Cytotoxic Sesterterpenic Acids, from the Marine Sponge Ircinia sp. Chem. Lett. 2001, 30, 176–177. [Google Scholar] [CrossRef]
- Almeida Vinagre, P.; Simas, T.; Cruz, E.; Pinori, E.; Svenson, J. Marine Biofouling: A European Database for the Marine Renewable Energy Sector. J. Mar. Sci. Eng. 2020, 8, 495. [Google Scholar] [CrossRef]
- Trepos, R.; Cervin, G.; Hellio, C.; Pavia, H.; Stensen, W.; Stensvåg, K.; Svendsen, J.S.; Haug, T.; Svenson, J. Antifouling Compounds from the Sub-Arctic Ascidian Synoicum pulmonaria: Synoxazolidinones A and C, Pulmonarins A and B, and Synthetic Analogues. J. Nat. Prod. 2014, 77, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Labriere, C.; Elumalai, V.; Staffansson, J.; Cervin, G.; Le Norcy, T.; Denardou, H.; Réhel, K.; Moodie, L.W.K.; Hellio, C.; Pavia, H.; et al. Phidianidine A and Synthetic Analogues as Naturally Inspired Marine Antifoulants. J. Nat. Prod. 2020, 83, 3413–3423. [Google Scholar] [CrossRef]
- Chen, L.; Xia, C.; Qian, P.Y. Optimization of antifouling coatings incorporating butenolide, a potent antifouling agent via field and laboratory tests. Prog. Org. Coat. 2017, 109, 22–29. [Google Scholar] [CrossRef]
- Chiang, H.Y.; Cheng, J.; Liu, X.; Ma, C.; Qian, P.Y. Synthetic Analogue of Butenolide as an Antifouling Agent. Mar. Drugs 2021, 19, 481. [Google Scholar] [CrossRef]
- Babij, N.R.; McCusker, E.O.; Whiteker, G.T.; Canturk, B.; Choy, N.; Creemer, L.C.; Amicis, C.V.D.; Hewlett, N.M.; Johnson, P.L.; Knobelsdorf, J.A.; et al. NMR Chemical Shifts of Trace Impurities: Industrially Preferred Solvents Used in Process and Green Chemistry. Org. Process Res. Dev. 2016, 20, 661–667. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2018.010; 1010 Sherbooke St. West, Suite #910; H3A 2R7. Chemical Computing Group ULC: Montreal, QC, Canada, 2022.
- Halgren, T.A. Merck molecular force field—I—Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field—II—MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field—III—Molecular geometries and vibrational frequencies for MMFF94. J. Comput. Chem. 1996, 17, 553–586. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck molecular force field—IV—Conformational energies and geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field—V—Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry—III—The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculations of Vibrational Absorption and Circular Dicroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigand, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- University of Karlsruhe; Forschungszentrum Karlsruhe GmbH. TURBOMOLE, V7.4.1; TURBOMOLE GmbH: Karlsruhe, Germany, 2007. [Google Scholar]
- Kollwitz, M.; Gauss, J. A direct implementation of the GIAO-MBPT(2) method for calculating NMR chemical shifts—Application to the naphthalenium and anthracenium ions. Chem. Phys. Lett. 1996, 260, 639–646. [Google Scholar] [CrossRef]
- Aryal, N.; Chen, J.; Bhattarai, K.; Hennrich, O.; Handayani, I.; Kramer, M.; Straetener, J.; Wommer, T.; Berscheid, A.; Peter, S.; et al. High Plasticity of the Amicetin Biosynthetic Pathway in Streptomyces sp. SHP 22-7 Led to the Discovery of Streptcytosine P and Cytosaminomycins F and G and Facilitated the Production of 12F-Plicacetin. J. Nat. Prod. 2022, 85, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.B.; Cockerill, F.R.; Bradford, P.A.; Eliopoulos, G.M.; Hindler, J.A.; Jenkins, S.G.; Lewis II, J.S.; Limbago, B.; Miller, L.A.; Nicolau, D.P.; et al. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 10th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015; Volume 35. [Google Scholar]
- Böffert, R.; Businger, R.; Preiß, H.; Ehmann, D.; Truffault, V.; Simon, C.; Ruetalo, N.; Hamprecht, K.; Müller, P.; Wehkamp, J.; et al. The human a-defensin-derived peptide HD5(1-9) inhibits cellular attachment and entry of human cytomegalovirus. Antivir. Res. 2020, 177, 104779. [Google Scholar] [CrossRef] [PubMed]
- Große, M.; Ruetalo, N.; Layer, M.; Hu, D.; Businger, R.; Rheber, S.; Setz, C.; Rauch, P.; Auth, J.; Fröba, M.; et al. Quinine inhibits infection of human cell lines with SARS-CoV-2. Viruses 2021, 13, 647. [Google Scholar] [CrossRef] [PubMed]
- Hübner, M.P.; Townson, S.; Gokool, S.; Tagboto, S.; Maclean, M.J.; Verocai, G.G.; Wolstenholme, A.J.; Frohberger, S.J.; Hoerauf, A.; Specht, S.; et al. Evaluation of the in vitro susceptibility of various filarial nematodes to emodepside. Int. J. Parasitol. Drugs Drug Resist. 2021, 17, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Schiefer, A.; Hübner, M.P.; Krome, A.; Lämmer, C.; Ehrens, A.; Aden, T.; Koschel, M.; Neufeld, H.; Chaverra-Muñoz, L.; Jansen, R.; et al. Corallopyronin A for short-course anti-wolbachial, macrofilaricidal treatment of filarial infections. PLoS Negl. Trop. Dis. 2020, 14, e0008930. [Google Scholar] [CrossRef] [PubMed]
- Baltz, T.; Baltz, D.; Giroud, C.; Crockett, J. Cultivation in a semi-defined medium of animal infective forms of Trypanosoma brucei, T. equiperdum, T. evansi, T. rhodesiense and T. gambiense. EMBO J. 1985, 4, 1273–1277. [Google Scholar] [CrossRef]
- Räz, B.; Iten, M.; Grether-Buhler, Y.; Kaminsky, R.; Brun, R. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 1997, 68, 139–147. [Google Scholar] [CrossRef]
- Huber, W.; Koella, J.C. A comparison of the three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993, 55, 257–261. [Google Scholar] [CrossRef]
- Buckner, F.S.; Verlinde, C.L.; La Flamme, A.C.; Van Voorhis, W.C. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrob. Agents Chemother. 1996, 40, 2592–2597. [Google Scholar] [CrossRef]
- Cunningham, I. New culture medium for maintenance of tsetse tissues and growth of trypanosomatids. J. Protozool. 1977, 24, 325–329. [Google Scholar] [CrossRef]
- Desjardins, R.E.; Canfield, C.J.; Haynes, J.D.; Chulay, J.D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Matile, H.; Pink, J.R.L. Plasmodium falciparum malaria parasite cultures and their use in immunology. In Immunological Methods; Lefkovits, I., Pernis, B., Eds.; Academic Press: San Diego, CA, USA, 1990. [Google Scholar] [CrossRef]
- Ponnudurai, T.; Leeuwenberg, A.D.; Meuwissen, J.H. Chloroquine sensitivity of isolates of Plasmodium falciparum adapted to in vitro culture. Trop. Geogr. Med. 1981, 33, 50–54. [Google Scholar]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Page, B.; Page, M.; Noel, C. A new fluorimetric assay for cytotoxicity measurements in-vitro. Int. J. Oncol. 1993, 3, 473–476. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Gogal, R.M.; Walsh, J.E. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: An alternative to [3H] thymidine incorporation assay. J. Immunol. Methods 1994, 170, 211–224. [Google Scholar] [CrossRef]
- Grant, T.M.; Rennison, D.; Cervin, G.; Pavia, H.; Hellio, C.; Foulon, V.; Brimble, M.A.; Cahill, P.; Svenson, J. Towards eco-friendly marine antifouling biocides–Nature inspired tetrasubstituted 2,5-diketopiperazines. Sci. Total Environ. 2022, 812, 152487. [Google Scholar] [CrossRef]
- Cahill, P.L.; Atalah, J.; Selwood, A.I.; Kuhajek, J.M. Metamorphosis of the invasive ascidian Ciona savignyi: Environmental variables and chemical exposure. PeerJ 2016, 4, e1739. [Google Scholar] [CrossRef]
- ASTM. Standard Guide for Conducting Static Short-Term Chronic Toxicity Tests Starting with Embryos of Four Species of Saltwater Bivalve Molluscs; ASTM International: West Conshohocken, PA, USA, 2021. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Irciniacin (1) | Ircinianin lactone B (7) | Ircinianin lactone C (8) | ||||
|---|---|---|---|---|---|---|
| Position | δH, Mult. (J in Hz) | δC, Type A | δH, Mult. (J in Hz) | δC, Type A | δH, Mult. (J in Hz) | δC, Type A |
| 1 | 7.38, t (1.7) | 144.0, CH | 5.84, br q (1.2) | 104.4, CH | 6.02, d (2.3) | 101.1, CH |
| 2 | 6.30, d (0.9) | 112.1, CH | 6.98, br quin (1.2) | 144.65, CH 144.72 E | - | 172.7, C |
| 3 | - | 126.5, C | - | 139.03, C 139.08 E | 5.90, d (2.3) | 117.9, CH |
| 4 | 7.26, m | 140.3, CH | - | 173.5, C | - | 173.8, C |
| 5 | 2.41, br t (7.5) F | 25.3, CH2 | 2.26, br t (7.8) | 25.5, CH2 | 2.43, m F | 28.2, CH2 |
| 6 | 1.68, m * | 29.5, CH2 | 1.67, m * | 26.44, CH2 26.47 E | 1.76, m * | 25.9, CH2 |
| 7 | 2.04, m | 40.5, CH2 | 2.07, dd (7.9, 7.2) | 40.2, CH2 | 2.11, m | 40.4, CH2 |
| 8 | - | 136.6, C | - | 135.75, C 135.78 E | - | 135.9, C |
| 9 | 1.57, d (1.3) | 16.3, CH3 | 1.57, d (1.3) | 16.06, CH3 16.08 E | 1.59, d | 16.2, CH3 |
| 10 | 5.11, dd (10.3, 1.1) | 125.0, CH | 5.12, m | 125.41, CH 125.44 E | 5.15, d (10.3) | 125.7, CH |
| 11 | 3.08, dm (10.3) | 48.7, CH C | 3.06, dm (10.3) | 48.6, CH C | 3.08, dm (10.1) | 48.7, CH C |
| 12 | 5.03, m | 123.6, CH | 5.01, m | 123.3, CH | 5.03, m | 123.4, CH |
| 13 | - | 137.1, C | - | 137.0, C | - | 137.2, C |
| 14 | 1.71, m | 20.8, CH3 D | 1.70, d (1.4) | 20.56, CH3 B | 1.71, m | 20.68, CH3 D |
| 15 | 2.42, m F | 46.2, CH | 2.40, m | 46.0, CH | 2.42, m F | 46.2, CH |
| 16 | a. 1.89, m b. 1.34, m G | 27.3, CH2 | a. 1.89, m b. 1.32, m F | 27.1, CH2 | a. 1.89, m b. 1.34, m G | 27.3, CH2 |
| 17 | a. 2.00, m b. 1.32, m G | 33.6, CH2 | a. 2.00, m b. 1.29, m F | 33.4, CH2 | a. 2.02, m b. 1.31, m G | 33.6, CH2 |
| 18 | 1.65, m | 33.2, CH | 1.63, m * | 33.1, CH | 1.64, m * | 33.2, CH |
| 19 | 0.92, d (6.3) | 20.7, CH3 D | 0.92, d (6.2) | 20.62, CH3 D | 0.93, d (6.2) | 20.74, CH3 D |
| 20 | 1.61, m * | 52.0, CH | 1.61, m * | 51.7, CH | 1.62, m * | 51.8, CH |
| 21 | - | 86.9, C | - | 86.7, C | - | 86.8, C |
| 22 | - | 179.2, C B | - | 179.3, C B | - | 179.1, C B |
| 23 | - | 97.5, C | - | 97.3, C | - | 97.5, C |
| 24 | - | 177.7, C B | - | 177.6, C B | - | 177.7, C B |
| 25 | 1.64, br s | 6.1, CH3 | 1.63, br s | 6.0, CH3 | 1.65, br s | 6.1, CH3 |
| 1′ | 3.52, br s | 57.13, CH3 57.16 E | ||||
| Substance | P. falciparum | T. brucei rhodesiense | T. cruzi | L. donovani |
|---|---|---|---|---|
| Ircinianin (1) | 25.4 | 82.8 | 190.9 | 16.6 |
| positive controls | 0.006 a | 0.020 b | 3.36 c | 0.486 d |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majer, T.; Bhattarai, K.; Straetener, J.; Pohlmann, J.; Cahill, P.; Zimmermann, M.O.; Hübner, M.P.; Kaiser, M.; Svenson, J.; Schindler, M.; et al. Discovery of Ircinianin Lactones B and C—Two New Cyclic Sesterterpenes from the Marine Sponge Ircinia wistarii. Mar. Drugs 2022, 20, 532. https://doi.org/10.3390/md20080532
Majer T, Bhattarai K, Straetener J, Pohlmann J, Cahill P, Zimmermann MO, Hübner MP, Kaiser M, Svenson J, Schindler M, et al. Discovery of Ircinianin Lactones B and C—Two New Cyclic Sesterterpenes from the Marine Sponge Ircinia wistarii. Marine Drugs. 2022; 20(8):532. https://doi.org/10.3390/md20080532
Chicago/Turabian StyleMajer, Thomas, Keshab Bhattarai, Jan Straetener, Justus Pohlmann, Patrick Cahill, Markus O. Zimmermann, Marc P. Hübner, Marcel Kaiser, Johan Svenson, Michael Schindler, and et al. 2022. "Discovery of Ircinianin Lactones B and C—Two New Cyclic Sesterterpenes from the Marine Sponge Ircinia wistarii" Marine Drugs 20, no. 8: 532. https://doi.org/10.3390/md20080532