
Progress in Isoindolone Alkaloid Derivatives from Marine Microorganism: Pharmacology, Preparation, and Mechanism

 , ,
, ,
Abstract
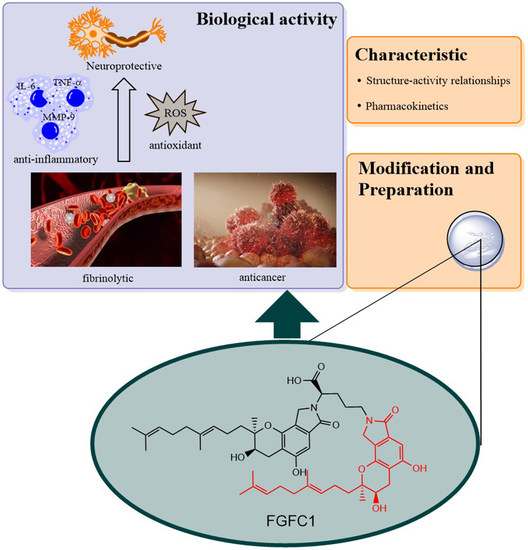
:
1. Introduction
2. Pharmacological Activity
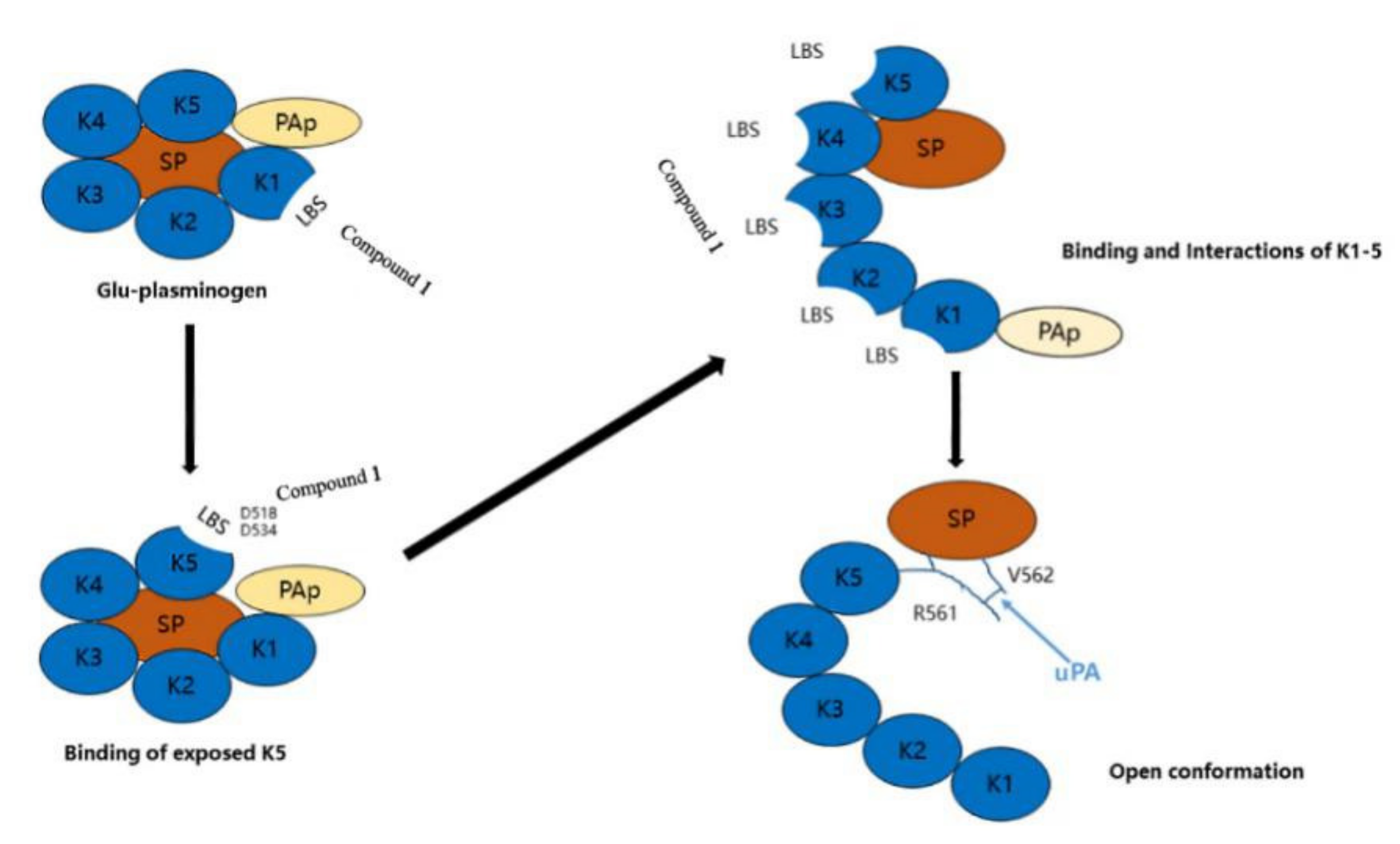
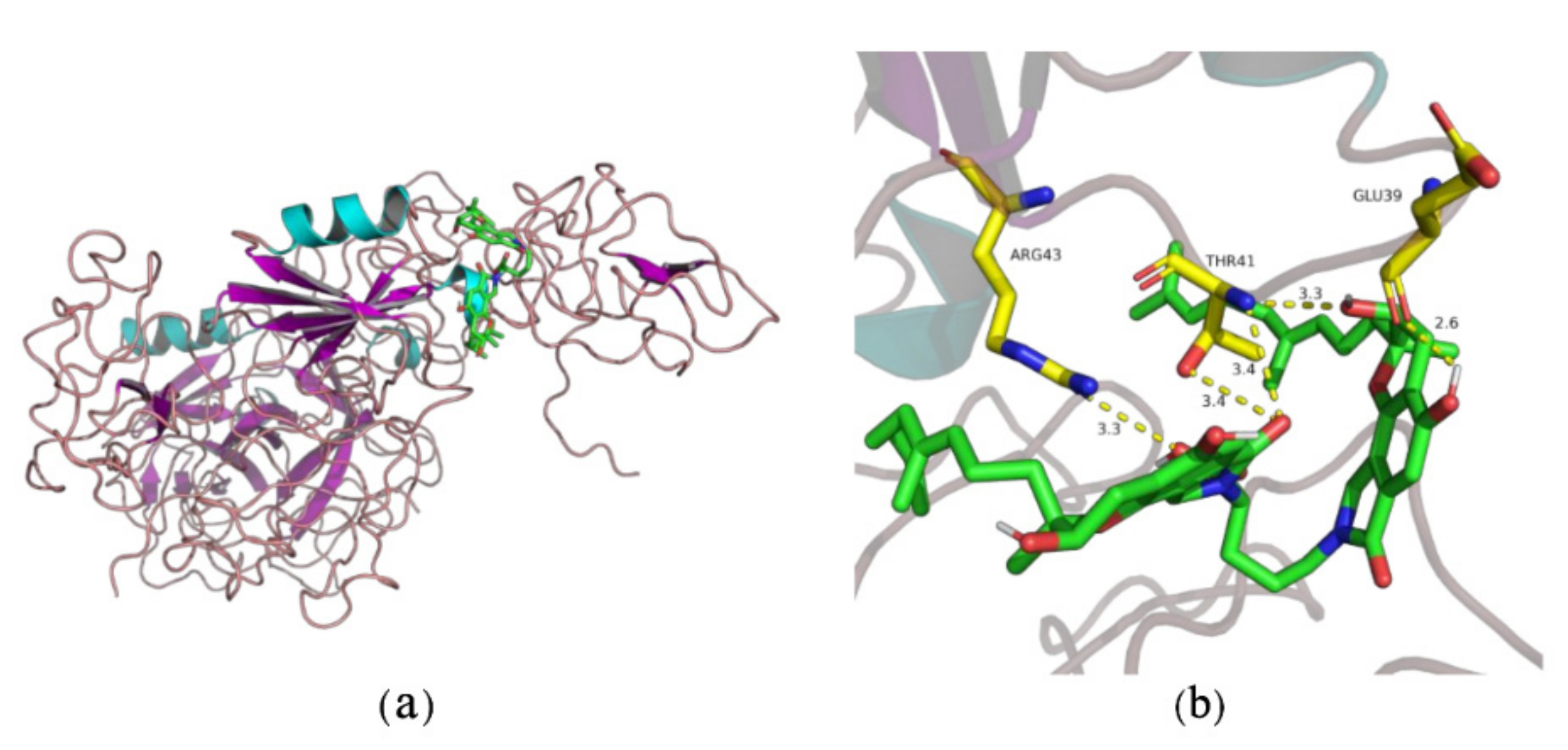
2.1. Thrombolytic Activity
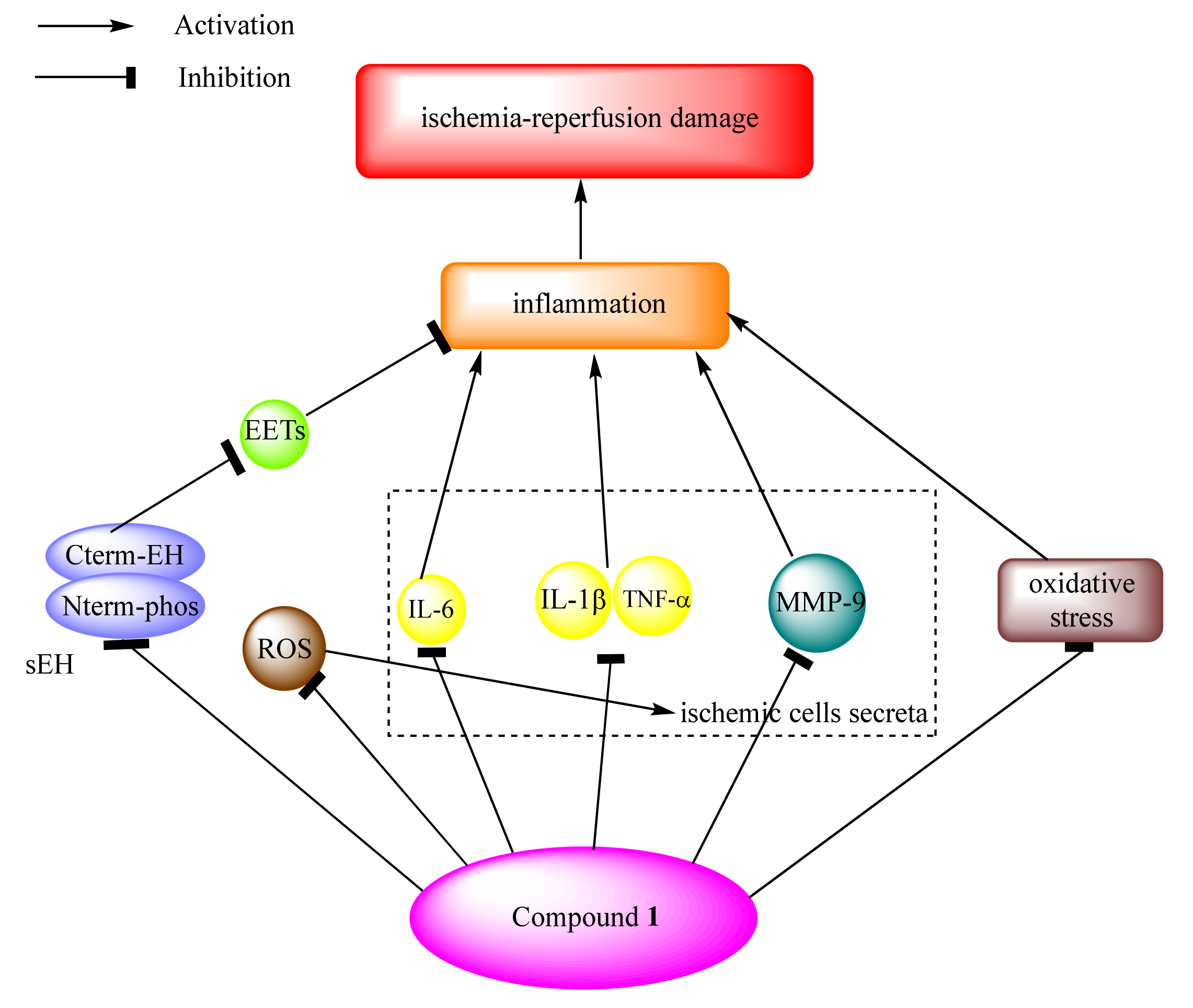
2.2. Effects on Inflammation and Oxidant Related Damage: In Reperfusion of Occluded Vessels
2.3. Neuroprotective Activity
2.4. Effects on IgA Nephropathy and Acute Kidney Injury
2.5. Effects on Cancer: Non-Small Cell Lung Cancer
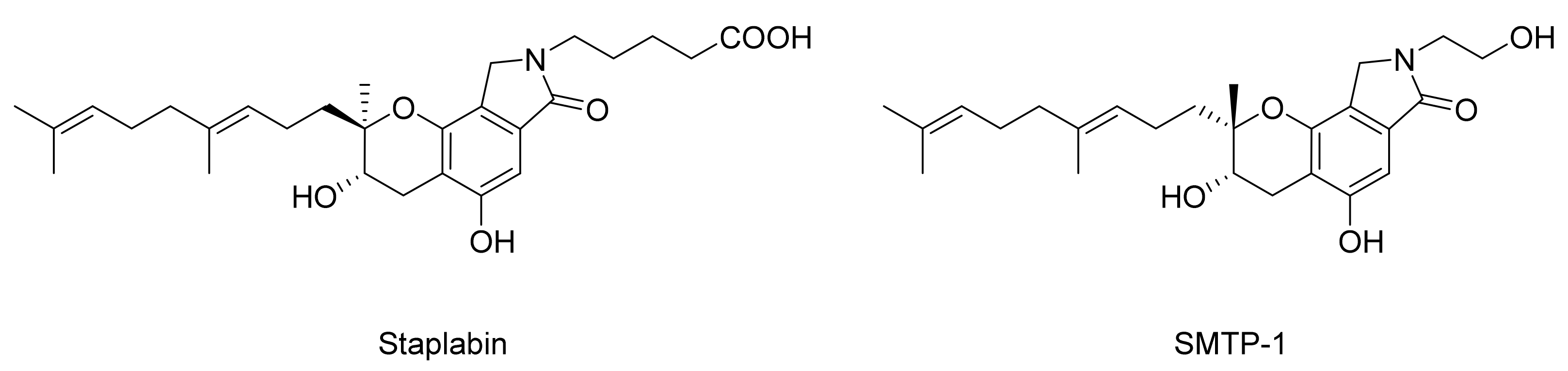
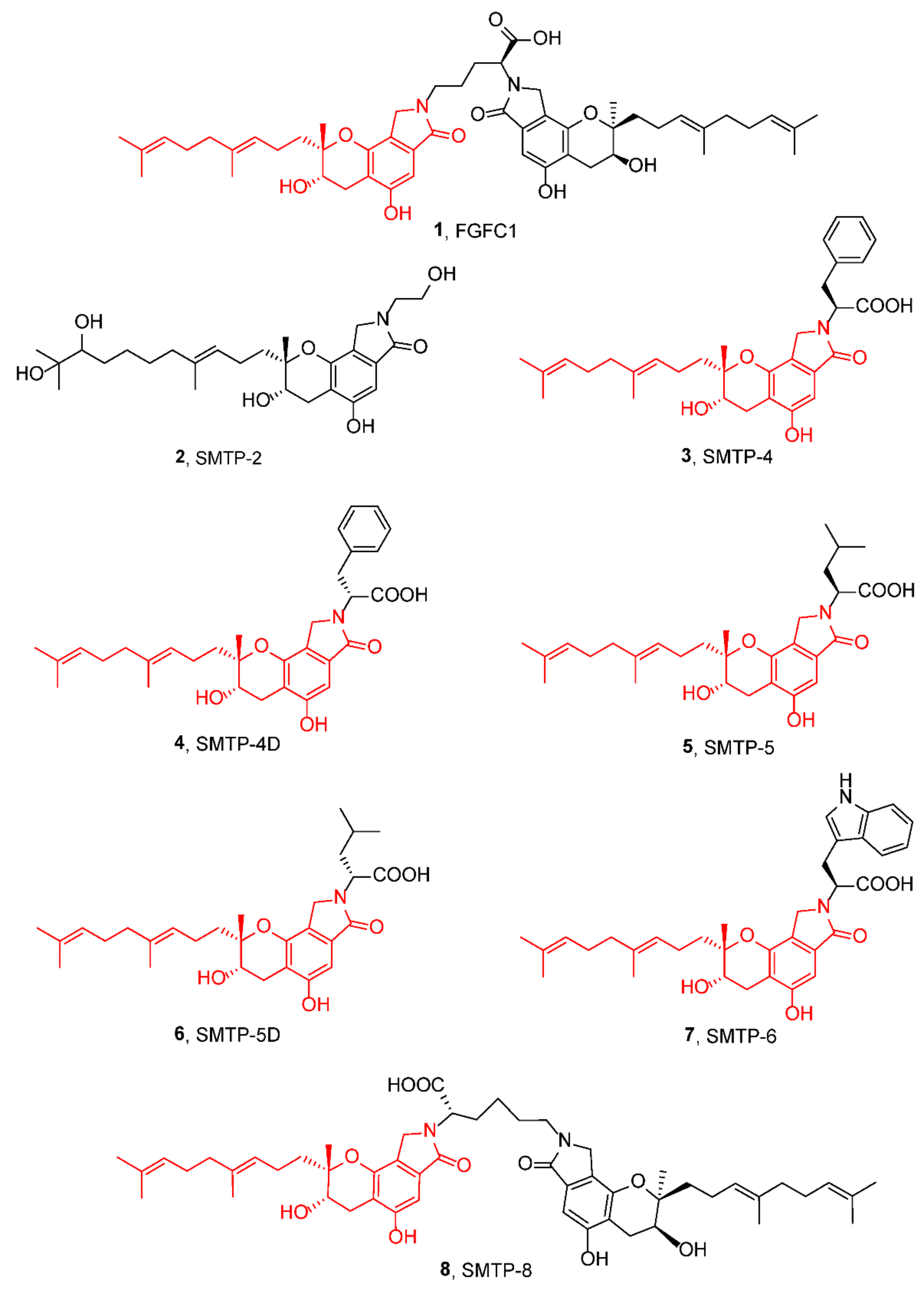
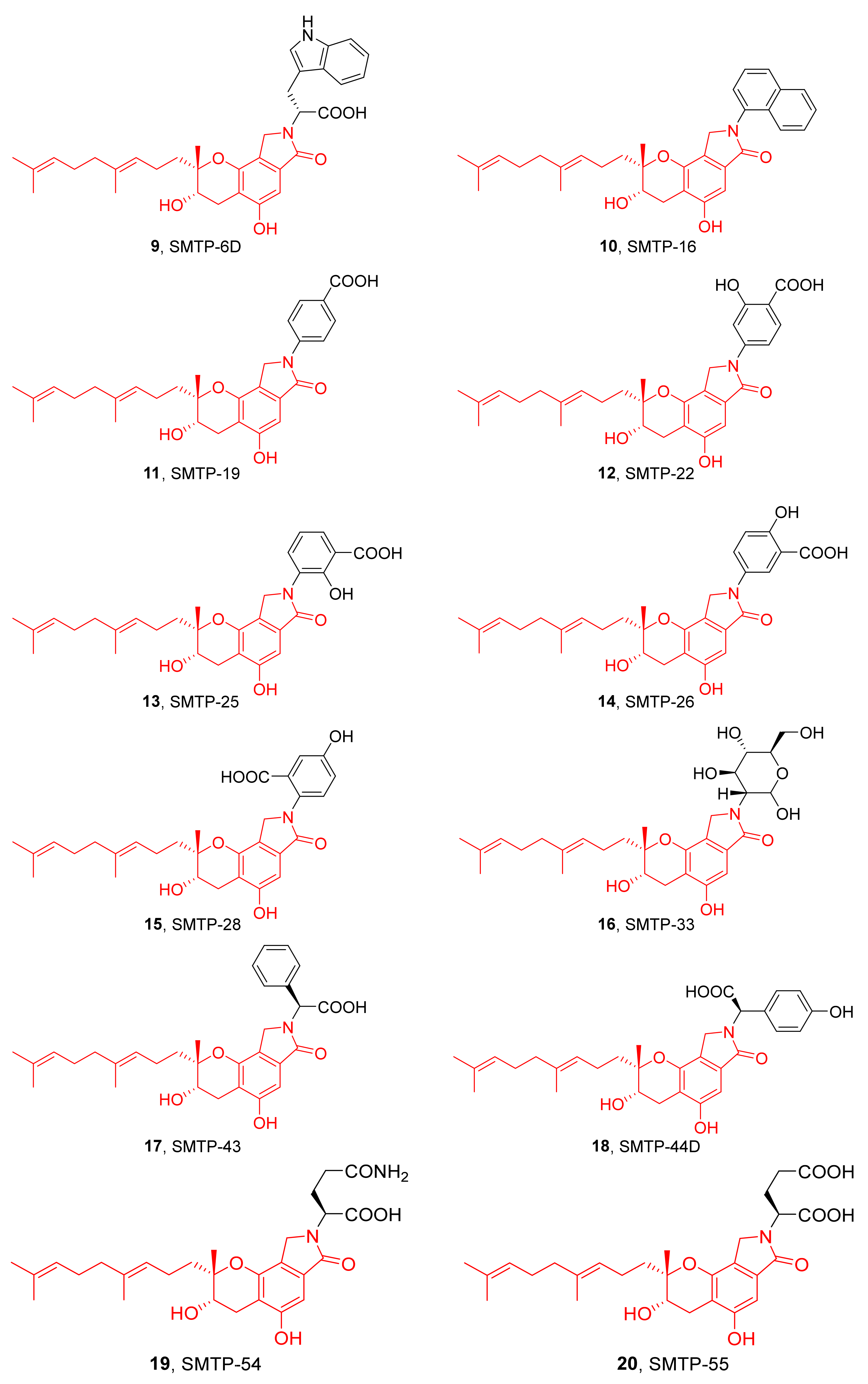
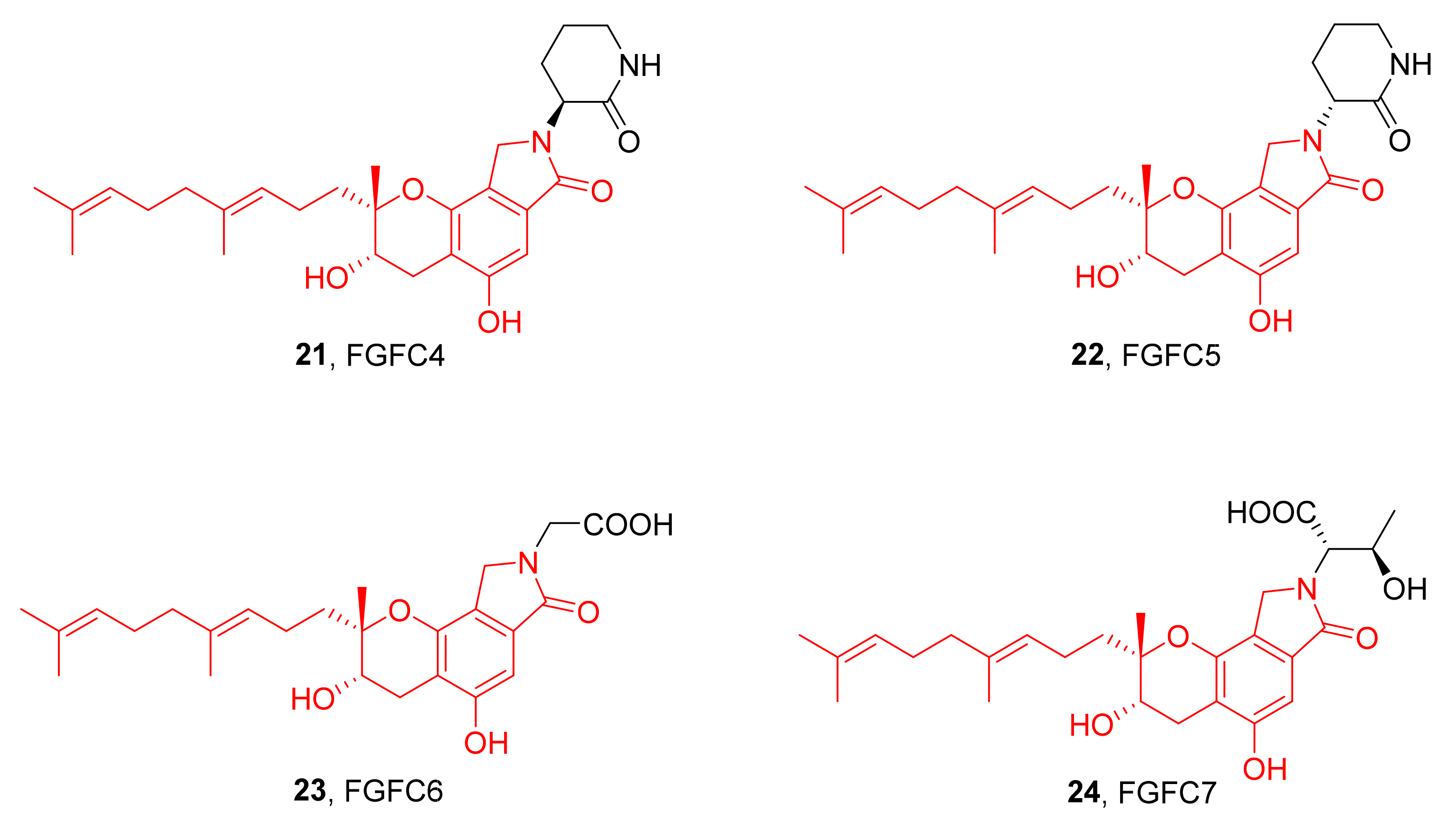
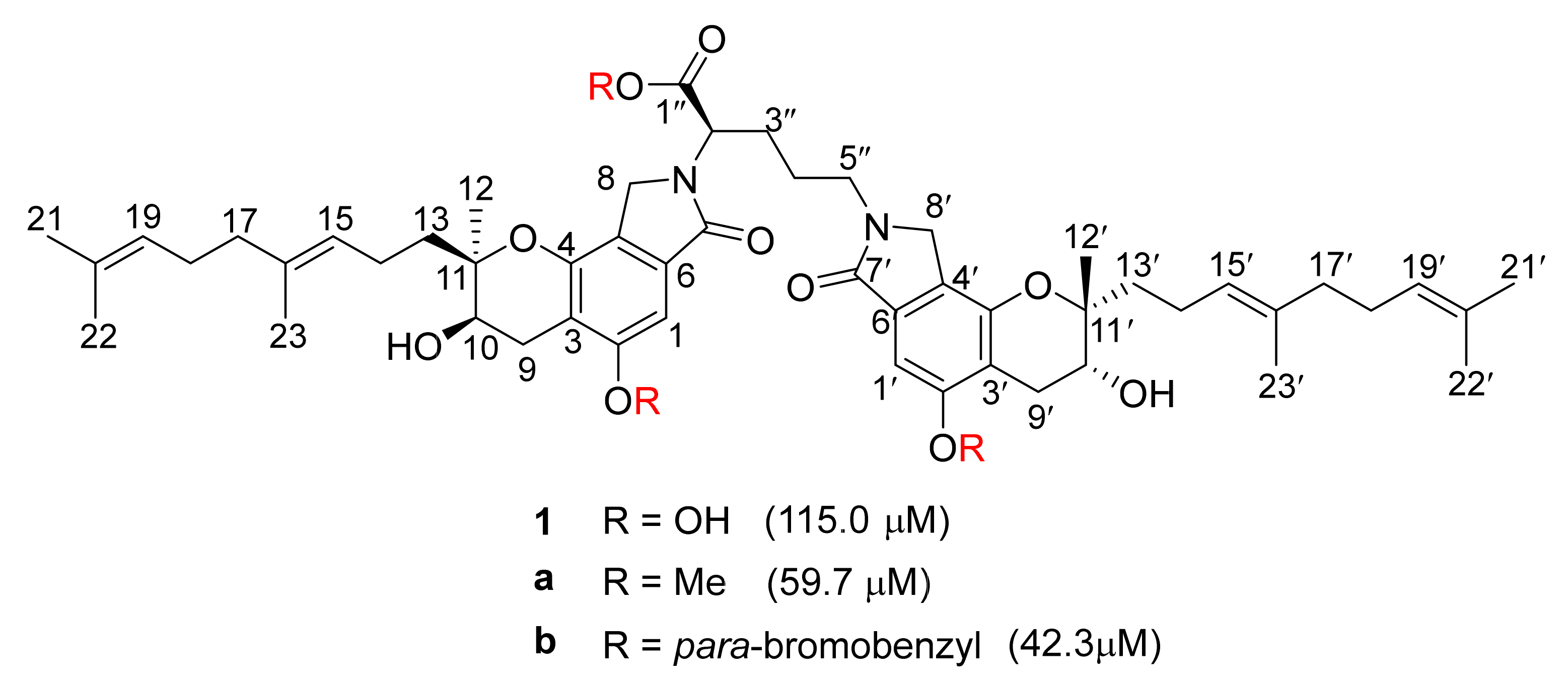
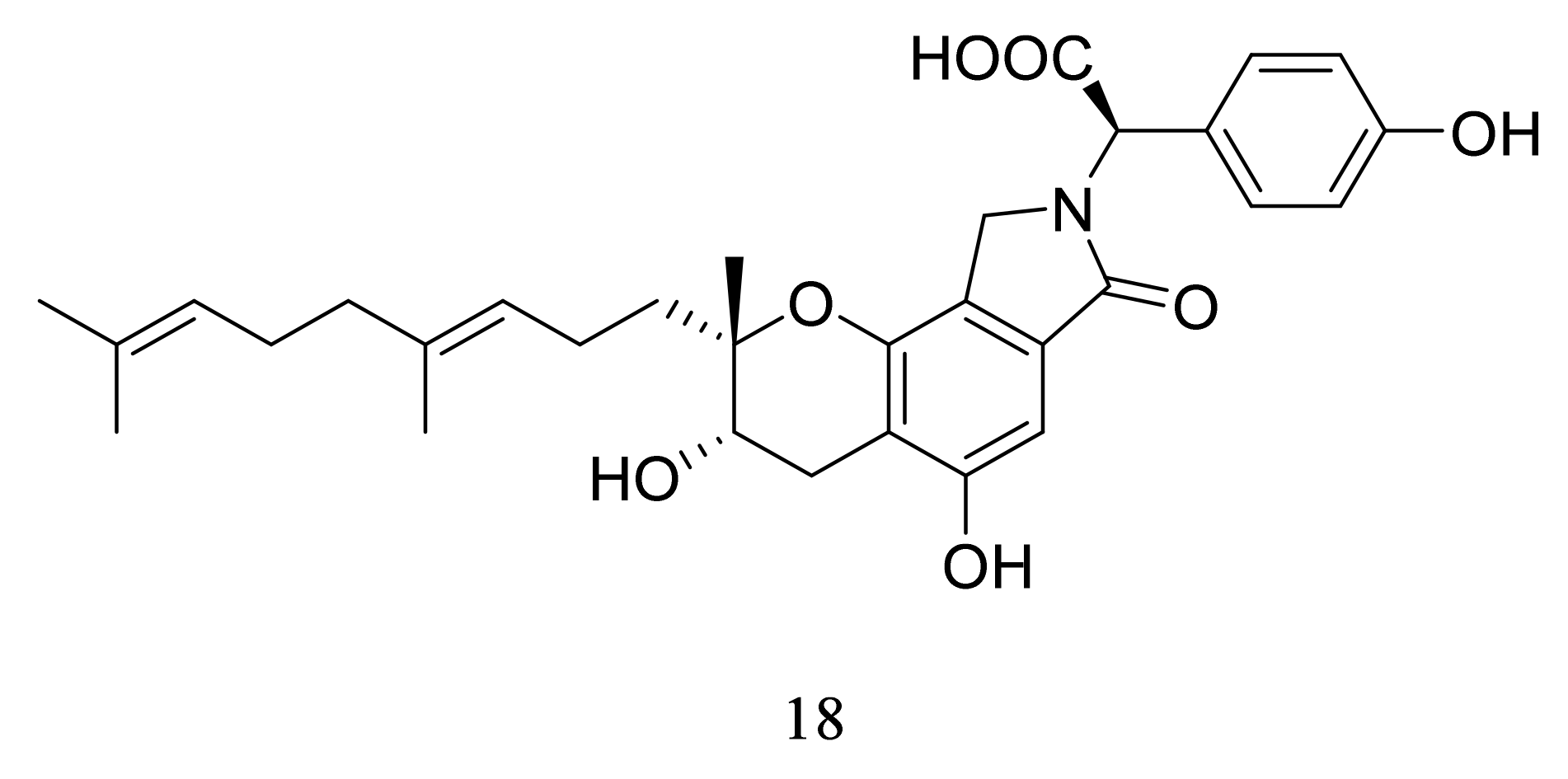
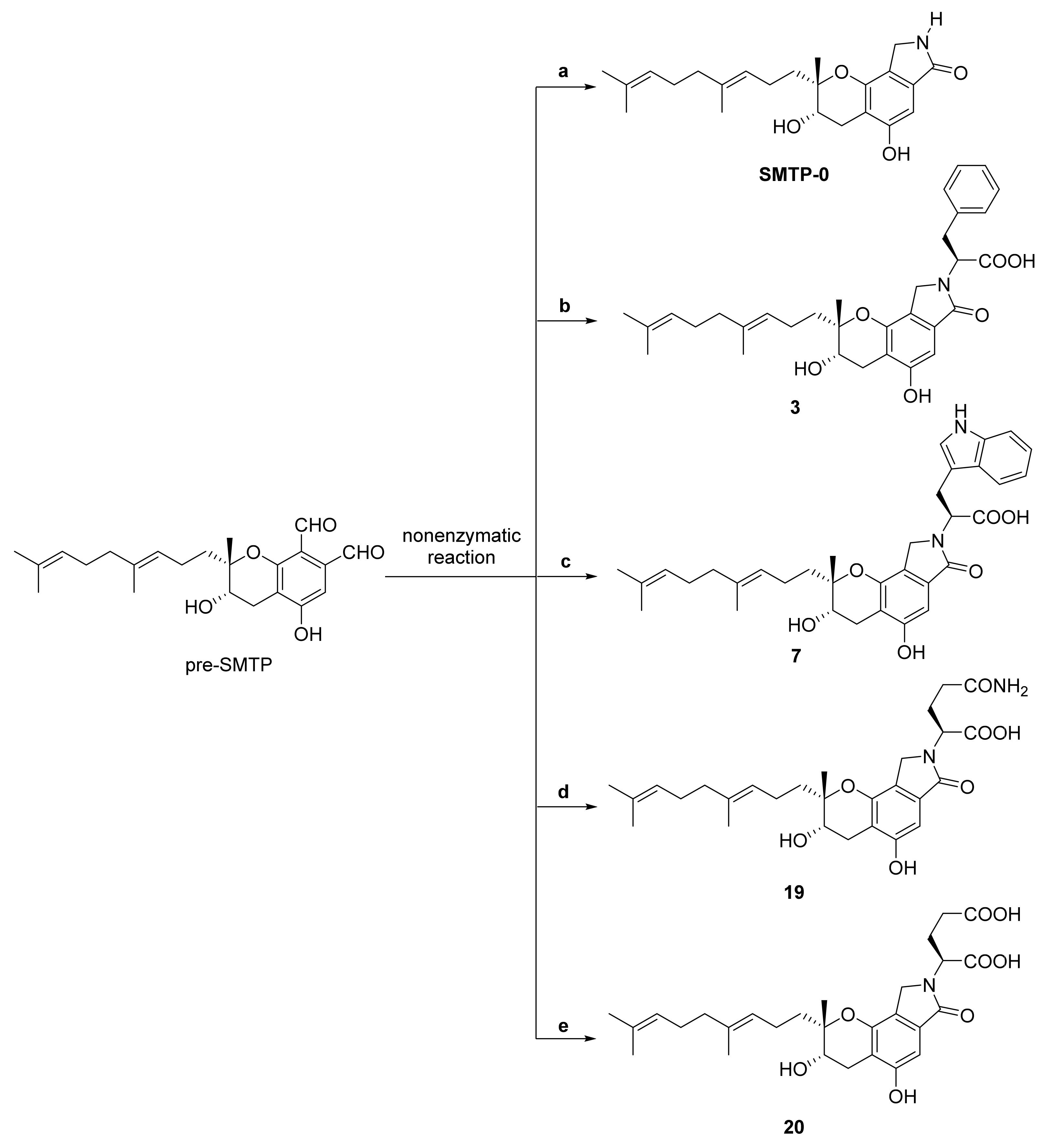
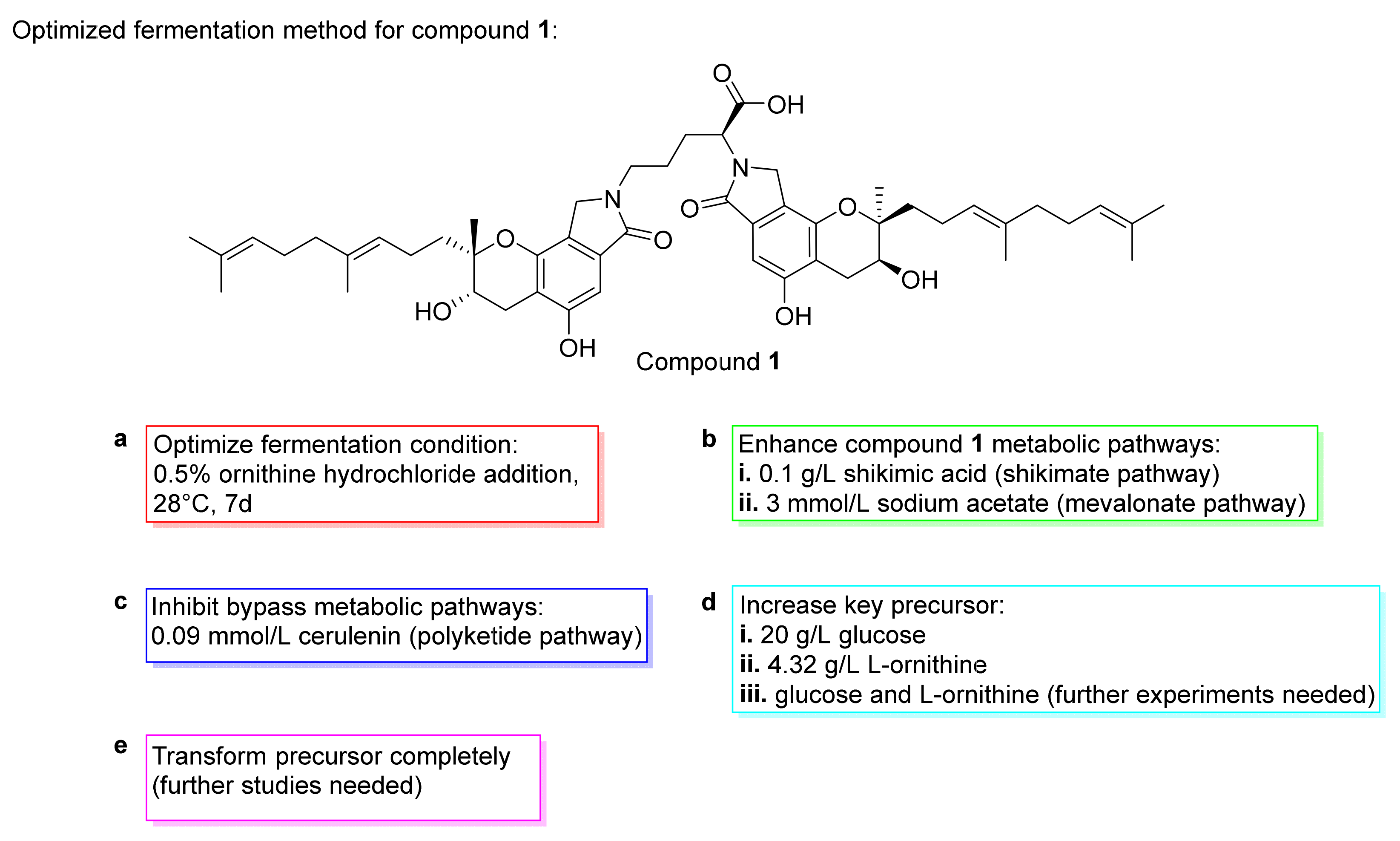
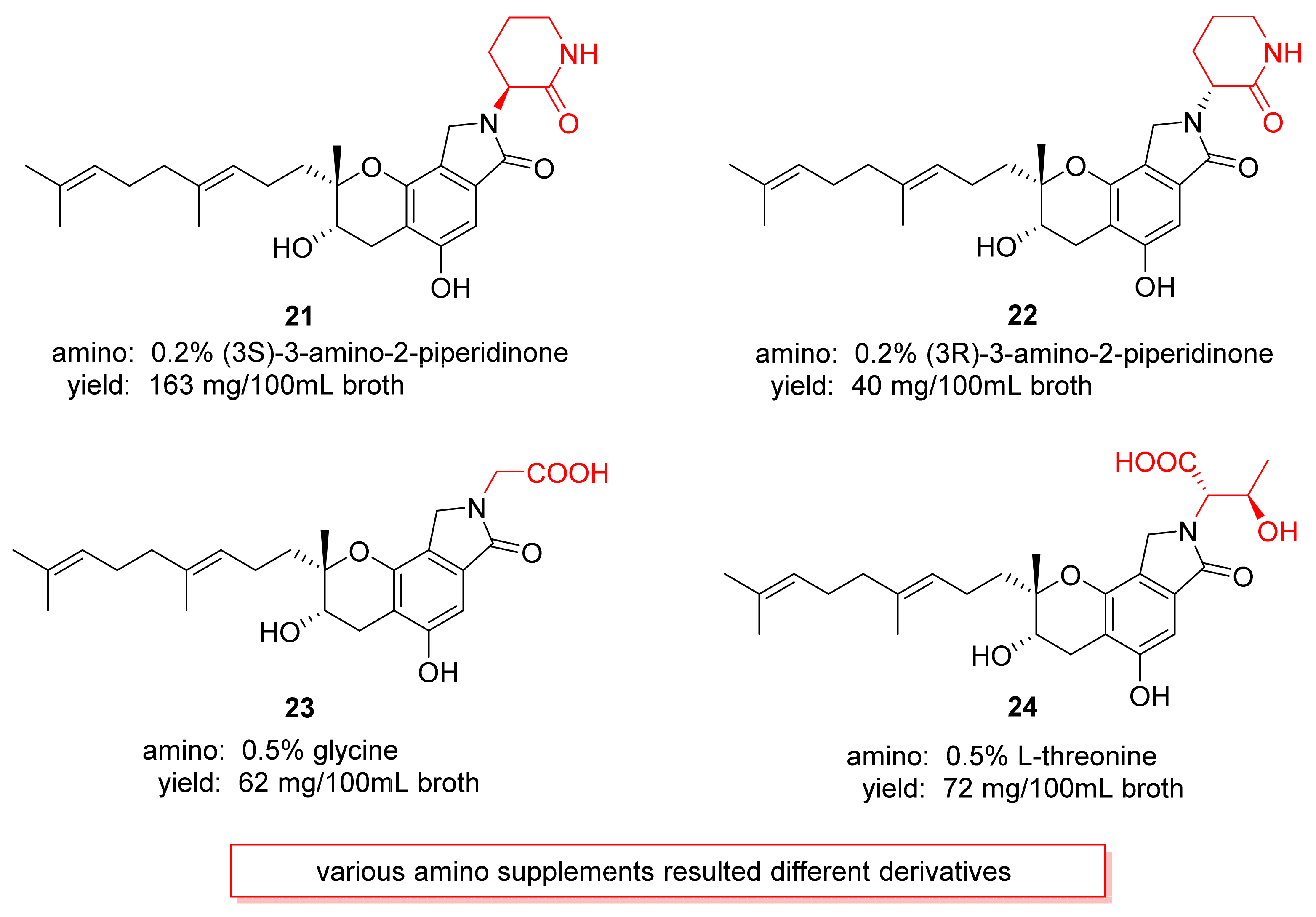
3. Preparation of Compound 1 and Staplabin Congeners
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hasumi, K.; Suzuki, E. Impact of SMTP targeting plasminogen and soluble epoxide hydrolase on thrombolysis, inflammation, and ischemic stroke. Int. J. Mol. Sci. 2021, 22, 954. [Google Scholar] [CrossRef] [PubMed]
- Hasumi, K.; Yamamichi, S.; Harada, T. Small-molecule modulators of zymogen activation in the fibrinolytic and coagulation systems. FEBS J. 2010, 277, 3675–3687. [Google Scholar] [CrossRef]
- Rijken, D.C.; Lijnen, H.R. New insights into the molecular mechanisms of the fibrinolytic system. J. Thromb. Haemost. 2009, 7, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Dabiri, G.; Damstetter, E.; Ebot, E.B.; Powers, J.G.; Phillips, T. Coagulation disorders and their cutaneous presentations: Pathophysiology. J. Am. Acad. Dermatol. 2016, 74, 783–792. [Google Scholar] [CrossRef]
- Booth, N.A. Fibrinolysis and thrombosis. Bailliere’s Best Pract. Res. Clin. Haematol. 1999, 12, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Kumar, A.; Kumar, M.; Singh, S.; Jain, M.; Dikshit, M. Synthesis of novel 3-carboxamide-benzocoumarin derivatives as orally active antithrombotic agents. Bioorg. Med. Chem. Lett. 2011, 21, 7034–7040. [Google Scholar] [CrossRef] [PubMed]
- Waluyo, D.; Prabandari, E.E.; Pramisandi, A.; Hidayati, D.N.; Chrisnayanti, E.; Puspitasari, D.J.; Dewi, D.; Suryani; Kristiningrum; Oktaviani, A.N.; et al. Exploring natural microbial resources for the discovery of anti-malarial compounds. Parasitol. Int. 2021, 85, 102432. [Google Scholar] [CrossRef]
- Jing, L.; Luohao, L.; Runqing, Z.; Vuanghao, L.; Qianqian, S.; Lizhen, F. Natural products from the genus Daldinia and their bioactivities. Med. Res. 2021, 5, 210005. [Google Scholar]
- Xiaojing, S.; Hong, W.; Jiao, C.; Zhubin, Z.; Fanqiu, N. Chemical constituents and bioactivities of Aconitum episcopale. Med. Res. 2021, 5, 210001. [Google Scholar]
- Romano, G.; Costantini, M.; Sansone, C.; Lauritano, C.; Ruocco, N.; Ianora, A. Marine microorganisms as a promising and sustainable source of bioactive molecules. Mar. Environ. Res. 2017, 128, 58–69. [Google Scholar] [CrossRef]
- Hui, H.; Junwen, W.; Xueyan, L.; Wenhui, W.; Kejin, S.; Chaoyan, Z. Renoprotective effect of sulphate polysaccharide from brown algae on ethylene glycol-induced renal damage in rats. Med. Res. 2020, 4, 190010. [Google Scholar]
- Junwen, W.; Xueyan, L.; Chaoyan, Z. Recent advances on bioactivity of seaweed polysaccharides. Med. Res. 2019, 3, 200003. [Google Scholar]
- Malve, H. Exploring the ocean for new drug developments: Marine pharmacology. J. Pharm. BioAllied Sci. 2016, 8, 83–91. [Google Scholar] [CrossRef]
- Phuphanich, S.; Maria, B.; Braeckman, R.; Chamberlain, M. A pharmacokinetic study of intra-CSF administered encapsulated cytarabine (DepoCyt) for the treatment of neoplastic meningitis in patients with leukemia, lymphoma, or solid tumors as part of a phase III study. J. Neuro-Oncol. 2007, 81, 201–208. [Google Scholar] [CrossRef]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudêncio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Br. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef] [Green Version]
- Martinez, A. Marine-derived drugs in neurology. Curr. Opin. Investig. Drugs 2007, 8, 525–530. [Google Scholar]
- Kohyama, T.; Hasumi, K.; Hamanaka, A.; Endo, A. SMTP-1 and -2, novel analogs of staplabin produced by Stachybotrys microspora IFO30018. J. Antibiot. 1997, 50, 172–174. [Google Scholar] [CrossRef] [Green Version]
- Takayasu, R.; Hasumi, K.; Shinohara, C.; Endo, A. Enhancement of fibrin binding and activation of plasminogen by staplabin through induction of a conformational change in plasminogen. FEBS Lett. 1997, 18, 58–62. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, C.; Hasumi, K.; Hatsumi, W.; Endo, A. Saplabin, a novel fungal triprenyl phenol which stimulates the binding of plasminogen to fibrin and U937 Cells. J. Antibiot. 1996, 49, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Ohyama, S.; Hasumi, K. Activation of fibrinolysis by SMTP-7 and -8, novel staplabin analogs with a pseudosymmetric structure. J. Antibiot. 2000, 53, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Koide, H.; Hu, W.; Nishimura, N.; Narasaki, R.; Kitano, Y.; Hasumi, K. Structure-activity relationships of 11 new congeners of the SMTP plasminogen modulator. J. Antibiot. 2010, 63, 589–593. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Kimura, Y.; Ohata, H.; Hashimoto, T.; Shibata, K.; Hasumi, K.; Honda, K. Distinct effects of tissue-type plasminogen activator and SMTP-7 on cerebrovascular inflammation following thrombolytic reperfusion. Stroke 2011, 42, 1097–1104. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Narasaki, R.; Nishimura, N.; Hasumi, K. SMTP (Stachybotrys microspora triprenyl phenol) enhances clot clearance in a pulmonary embolism model in rats. Thromb. J. 2012, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Kitano, Y.; Hasumi, K. SMTP-4D, -5D, -6D, -7D and -8D, a new series of the non-lysine-analog plasminogen modulators with a D-amino acid moiety. J. Antibiot. 2003, 56, 832–837. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Narasaki, R.; Ohyama, S.; Hasumi, K. Selective production of staplabin and SMTPs in cultures of Stachybotrys microspora fed with precursor amines. J. Antibiot. 2001, 54, 962–966. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart disease and stroke statistics—2017 update: A report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, X.; Chen, X.; Wei, Y. Neuronal injuries in cerebral infarction and ischemic stroke: From mechanisms to treatment (review). Int. J. Mol. Med. 2022, 49, 15. [Google Scholar] [CrossRef]
- Hashimoto, T.; Shibata, K.; Nobe, K.; Hasumi, K.; Honda, K. A novel embolic model of cerebral infarction and evaluation of Stachybotrys microspora triprenyl phenol-7 (SMTP-7), a novel fungal triprenyl phenol metabolite. J. Pharmacol. Sci. 2010, 114, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, K.; Hashimoto, T.; Nobe, K.; Hasumi, K.; Honda, K. A novel finding of a low-molecular-weight compound, SMTP-7, having thrombolytic and anti-inflammatory effects in cerebral infarction of mice. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2010, 382, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, H.; Nishimura, N.; Suzuki, E.; Zhuang, J.; Hasegawa, K.; Takamatsu, H.; Honda, K.; Hasumi, K. SMTP-7, a novel small-molecule thrombolytic for ischemic stroke: A study in rodents and primates. J. Cereb. Blood Flow Metab. 2014, 34, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Nishimura, N.; Yoshikawa, T.; Kunikiyo, Y.; Hasegawa, K.; Hasumi, K. Efficacy of SMTP-7, a small-molecule anti-inflammatory thrombolytic, in embolic stroke in monkeys. Pharmacol. Res. Perspect. 2018, 6, e00448. [Google Scholar] [CrossRef] [Green Version]
- Ito, A.; Niizuma, K.; Shimizu, H.; Fujimura, M.; Hasumi, K.; Tominaga, T. SMTP-7, a new thrombolytic agent, decreases hemorrhagic transformation after transient middle cerebral artery occlusion under warfarin anticoagulation in mice. Brain Res. 2014, 1578, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Wu, W.; Zhu, Q.; Fu, S.; Wang, X.; Hong, S.; Guo, R.; Bao, B. Identification and fibrinolytic evaluation of an isoindolone derivative isolated from a rare marine fungus Stachybotrys longispora FG216. Chin. J. Chem. 2015, 33, 1089–1095. [Google Scholar] [CrossRef]
- Yan, T.; Wu, W.; Su, T.; Chen, J.; Zhu, Q.; Zhang, C.; Wang, X.; Bao, B. Effects of a novel marine natural product: Pyrano indolone alkaloid fibrinolytic compound on thrombolysis and hemorrhagic activities in vitro and in vivo. Arch. Pharmacal. Res. 2015, 8, 1530–1540. [Google Scholar] [CrossRef]
- Gao, C.; Shen, Q.; Tang, P.; Cao, Y.; Lin, H.; Li, B.; Sun, P.; Bao, B.; Wu, W. In vitro study of the fibrinolytic activity via single chain urokinase-type plasminogen activator and molecular docking of FGFC1. Molecules 2021, 26, 1816. [Google Scholar] [CrossRef]
- Hasumi, K.; Ohyama, S.; Kohyama, T.; Ohsaki, Y.; Takayasu, R.; Endo, A. Isolation of SMTP-3, 4, 5 and -6, novel analogs of staplabin, and their effects on plasminogen activation and fibrinolysis. J. Antibiot. 1998, 51, 1059–1068. [Google Scholar] [CrossRef] [Green Version]
- Koide, H.; Hasegawa, K.; Nishimura, N.; Narasaki, R.; Hasumi, K. A new series of the SMTP plasminogen modulators with a phenylamine-based side chain. J. Antibiot. 2012, 65, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Shibata, K.; Hashimoto, T.; Hasumi, K.; Honda, K.; Nobe, K. Evaluation of the effects of a new series of SMTPs in the acetic acid-induced embolic cerebral infarct mouse model. Eur. J. Pharmacol. 2018, 818, 221–227. [Google Scholar] [CrossRef]
- Akamatsu, Y.; Saito, A.; Fujimura, M.; Shimizu, H.; Mekawy, M.; Hasumi, K.; Tominaga, T. Stachybotrys microspora triprenyl phenol-7, a novel fibrinolytic agent, suppresses superoxide production, matrix metalloproteinase-9 expression, and thereby attenuates ischemia/reperfusion injury in rat brain. Neurosci. Lett. 2011, 503, 110–114. [Google Scholar] [CrossRef]
- Hashimoto, T.; Shibata, K.; Ohata, H.; Hasumi, K.; Honda, K. Altered gene expression in an embolic stroke model after thrombolysis with tissue plasminogen activator and Stachybotrys microspora triprenyl phenol-7. J. Pharmacol. Sci. 2014, 125, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Ohta, Y.; Shang, J.; Li, X.; Liu, X.; Shi, X.; Feng, T.; Yamashita, T.; Sato, K.; Takemoto, M.; et al. Reduction of ischemia reperfusion-related brain hemorrhage by Stachybotrys microspora triprenyl phenol-7 in mice with antioxidant effects. J. Stroke Cerebrovasc. Dis. 2018, 27, 3521–3528. [Google Scholar] [CrossRef]
- Koyanagi, K.; Narasaki, R.; Yamamichi, S.; Suzuki, E.; Hasumi, K. Mechanism of the action of SMTP-7, a novel small-molecule modulator of plasminogen activation. Blood Coagul. Fibrinolysis 2014, 25, 316–321. [Google Scholar] [CrossRef]
- Guo, R.H.; Duan, D.; Hong, S.T.; Zhou, Y.; Wang, F.; Wang, S.J.; Wu, W.H.; Bao, B. A marine fibrinolytic compound FGFC1 stimulating enzymatic kinetic parameters of a reciprocal activation system based on a single chain urokinase-type plasminogen activator and plasminogen. Process Biochem. 2018, 68, 190–196. [Google Scholar] [CrossRef]
- Rahman, M.N.; Becker, L.; Petrounevitch, V.; Hill, B.C.; Jia, Z.; Koschinsky, M.L. Comparative analyses of the lysine binding site properties of apolipoprotein(a) kringle IV types 7 and 10. Biochemistry 2002, 41, 1149–1155. [Google Scholar] [CrossRef]
- Xue, Y.; Bodin, C.; Olsson, K. Crystal structure of the native plasminogen reveals an activation-resistant compact conformation. J. Thromb. Haemostasis. 2012, 10, 1385–1396. [Google Scholar] [CrossRef]
- Law, R.H.P.; Abu-Ssaydeh, D.; Whisstock, J.C. New insights into the structure and function of the plasminogen/plasmin system. Curr. Opin. Struct. Biol. 2013, 23, 836–841. [Google Scholar] [CrossRef]
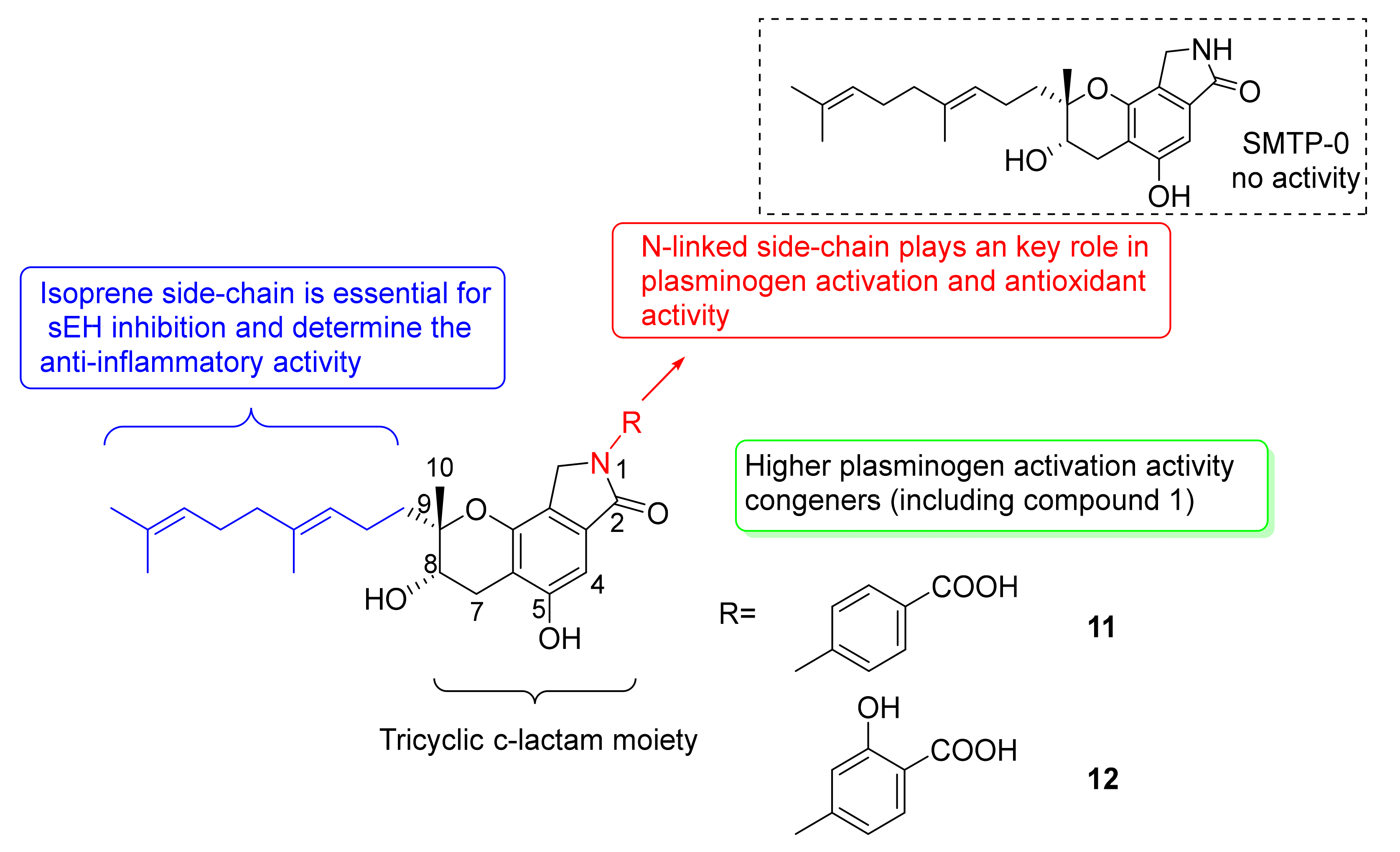
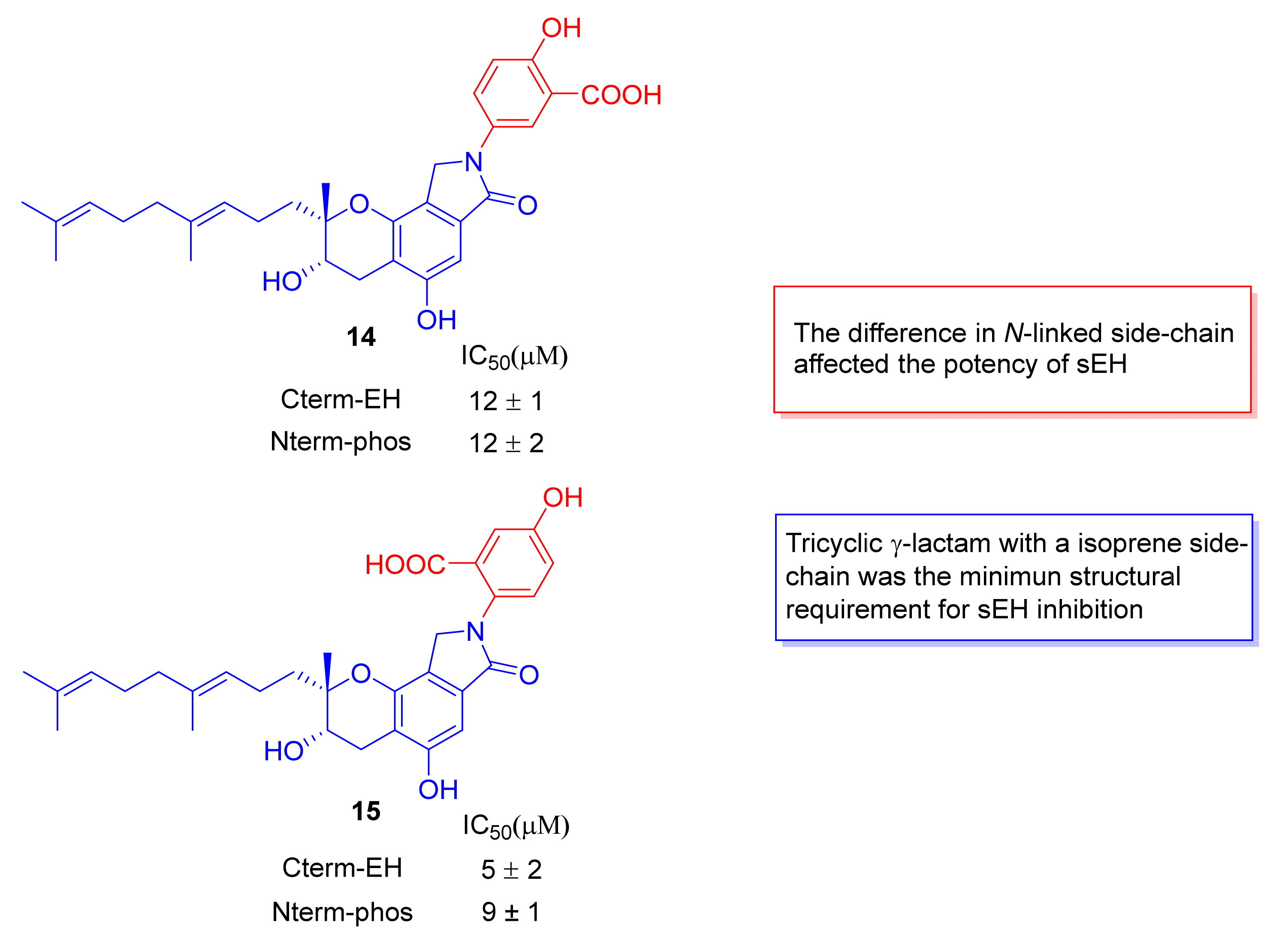
- Otake, S.; Ogawa, N.; Kitano, Y.; Hasumi, K.; Suzuki, E. Isoprene side-chain of SMTP is essential for soluble epoxide hydrolase inhibition and cellular localization. Nat. Prod. Commun. 2016, 11, 223–227. [Google Scholar] [CrossRef] [Green Version]
- Hasumi, K.; Hasegawa, K.; Kitano, Y. Isolation and absolute configuration of SMTP-0, a simplest congener of the SMTP family nonlysine-analog plasminogen modulators. J. Antibiot. 2007, 60, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Yellepeddi, V.; Rower, J.; Liu, X.; Kumar, S.; Rashid, J.; Sherwin, C.M.T. State-of-the-art review on physiologically based pharmacokinetic modeling in pediatric drug development. Clin. Pharmacokinet. 2019, 58, 1–13. [Google Scholar] [CrossRef]
- Su, T.; Wu, W.; Yan, T.; Zhang, C.; Zhu, Q.; Bao, B. Pharmacokinetics and tissue distribution of a novel marine fibrinolytic compound in Wistar rat following intravenous administrations. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 942, 77–82. [Google Scholar] [CrossRef]
- Ma, Z.; Guo, R.; Elango, J.; Bao, B.; Wu, W. Evaluation of marine diindolinonepyrane in vitro and in vivo: Permeability characterization in Caco-2 cells monolayer and pharmacokinetic properties in Beagle dogs. Mar. Drugs 2019, 17, 651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, H.S.; Zhang, H.F.; Dong, Y.L.; Chen, S.Y.; Wang, M.Y.; Dong, W.H.; Xing, J.F. Absorption and transportation characteristics of scutellarin and scutellarein across Caco-2 monolayer model. Zhongxiyi Jiehe Xuebao 2010, 8, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, H.; Sheng, R.; Fu, Z.; Fan, J.; Wu, W.; Tu, Q.; Guo, R. Synthesis and bioactivities of marine pyran-isoindolone derivatives as potential antithrombotic agents. Mar. Drugs 2021, 19, 218. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. Does inflammation contribute to thrombotic events? Haemostasis 2000, 30, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Irving, P.M.; Pasi, K.J.; Rampton, D.S. Thrombosis and inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2005, 3, 617–628. [Google Scholar] [CrossRef]
- Wakefield, T.W.; Myers, D.D.; Henke, P.K. Mechanisms of venous thrombosis and resolution. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 387–391. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.R.; Hammock, B.D. Soluble epoxide hydrolase: Gene structure, expression and deletion. Gene 2013, 526, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Morisseau, C.; Hammock, B.D. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 37–58. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.C.; Hammock, B.D. Discovery of inhibitors of soluble epoxide hydrolase: A target with multiple potential therapeutic indications. J. Med. Chem. 2012, 55, 1789–1808. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.W.; Morisseau, C.; Hammock, B.D. Epoxide hydrolases: Their roles and interactions with lipid metabolism. Prog. Lipid Res. 2005, 44, 1–51. [Google Scholar] [CrossRef]
- Thomson, S.J.; Askari, A.; Bishop-Bailey, D. Anti-inflammatory effects of epoxyeicosatrienoic acids. Int. J. Vasc. Med. 2012, 2012, 605101. [Google Scholar] [CrossRef]
- Ulu, A.; Harris, T.R.; Morisseau, C.; Miyabe, C.; Inoue, H.; Schuster, G.; Dong, H.; Iosif, A.M.; Liu, J.Y.; Weiss, R.H.; et al. Anti-inflammatory effects of ω-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-II-dependent hypertension. J. Cardiovasc. Pharmacol. 2013, 62, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, N.; Suzuki, E.; Ishikawa, M.; Shirafuji, T.; Hasumi, K. Soluble epoxide hydrolase as an anti-inflammatory target of the thrombolytic stroke drug SMTP-7. J. Biol. Chem. 2014, 289, 35826–35838. [Google Scholar] [CrossRef] [Green Version]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Rosell, A.; Cuadrado, E.; Ortega-Aznar, A.; Hernández-Guillamon, M.; Lo, E.H.; Montaner, J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 2008, 39, 1121–1126. [Google Scholar] [CrossRef] [Green Version]
- Shibata, K.; Hashimoto, T.; Nobe, K.; Hasumi, K.; Honda, K. Neuroprotective mechanisms of SMTP-7 in cerebral infarction model in mice. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 384, 103–108. [Google Scholar] [CrossRef]
- Adibhatla, R.M.; Hatcher, J.F. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic. Biol. Med. 2006, 40, 376–387. [Google Scholar] [CrossRef]
- Huang, Y.; Ohta, Y.; Shang, J.; Morihara, R.; Nakano, Y.; Fukui, Y.; Liu, X.; Shi, X.; Feng, T.; Yamashita, T.; et al. Antineuroinflammatory effect of SMTP-7 in ischemic mice. J. Stroke Cerebrovasc. Dis. 2018, 27, 3084–3094. [Google Scholar] [CrossRef]
- Shi, X.; Ohta, Y.; Shang, J.; Morihara, R.; Nakano, Y.; Fukui, Y.; Liu, X.; Feng, T.; Huang, Y.; Sato, K.; et al. Neuroprotective effects of SMTP-44D in mice stroke model in relation to neurovascular unit and trophic coupling. J. Neurosci. Res. 2018, 96, 1887–1899. [Google Scholar] [CrossRef]
- Shinouchi, R.; Shibata, K.; Hashimoto, T.; Jono, S.; Hasumi, K.; Nobe, K. SMTP-44D improves diabetic neuropathy symptoms in mice through its antioxidant and anti-inflammatory activities. Pharmacol. Res. Perspect. 2020, 8, e00648. [Google Scholar] [CrossRef]
- Pop-Busui, R.; Boulton, A.J.; Feldman, E.L.; Bril, V.; Freeman, R.; Malik, R.A.; Sosenko, J.M.; Ziegler, D. Diabetic neuropathy: A position statement by the American Diabetes Association. Diabetes Care 2017, 40, 136–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesfaye, S.; Boulton, A.J.; Dickenson, A.H. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care 2013, 36, 2456–2465. [Google Scholar] [CrossRef] [Green Version]
- Tomino, Y. IgA nephropathy: Lessons from an animal model, the ddY mouse. J. Nephrol. 2008, 21, 463–467. [Google Scholar] [PubMed]
- Mubarak, M. IgA nephropathy: An update on pathogenesis and classification. J. Coll. Physicians Surg. Pak. 2011, 21, 230–233. [Google Scholar] [PubMed]
- Kemmochi, S.; Hayashi, H.; Taniai, E.; Hasumi, K.; Sugita-Konishi, Y.; Kumagai, S.; Mitsumori, K.; Shibutani, M. Protective effect of Stachybotrys microspora triprenyl phenol-7 on the deposition of IgA to the glomerular mesangium in nivalenol-induced IgA nephropathy using BALB/c mice. J. Toxicol. Pathol. 2012, 25, 149–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuelo, J.G. Normotensive ischemic acute renal failure. N. Engl. J. Med. 2007, 357, 797–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L.; Acute Kidney Injury Advisory Group of The American Society of Nephrology. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [Green Version]
- Shibata, K.; Hashimoto, T.; Hasumi, K.; Nobe, K. Potent efficacy of Stachybotrys microspora triprenyl phenol-7, a small molecule having anti-inflammatory and antioxidant activities, in a mouse model of acute kidney injury. Eur. J. Pharmacol. 2021, 910, 174496. [Google Scholar] [CrossRef]
- Ross, R. Successful growth of tumours. Nature 1989, 339, 16–17. [Google Scholar] [CrossRef]
- Gately, S.; Twardowski, P.; Stack, M.S.; Cundiff, D.L.; Grella, D.; Castellino, F.J.; Enghild, J.; Kwaan, H.C.; Lee, F.; Kramer, R.A.; et al. The mechanism of cancer-mediated conversion of plasminogen to the angiogenesis inhibitor angiostatin. Proc. Natl. Acad. Sci. USA 1997, 94, 10868–10872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanabal, M.; Sethuraman, N. Endogenous angiogenesis inhibitors as therapeutic agents: Historical perspective and future direction. Recent Pat. Anti-Cancer Drug Discov. 2006, 1, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, S.; Harada, T.; Chikanishi, T.; Miura, Y.; Hasumi, K. Nonlysine-analog plasminogen modulators promote autoproteolytic generation of plasmin(ogen) fragments with angiostatin-like activity. Eur. J. Biochem. 2004, 271, 809–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.D.; Fidler-Benaoudia, M.; Keegan, T.H.; Hipp, H.S.; Jemal, A.; Siegel, R.L. Cancer statistics for adolescents and young adults, 2020. CA-Cancer J. Clin. 2020, 70, 443–459. [Google Scholar] [CrossRef]
- Chan, S.K.; Gullick, W.J.; Hill, M.E. Mutations of the epidermal growth factor receptor in non-small cell lung cancer—Search and destroy. Eur. J. Cancer 2006, 42, 17–23. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, B.; Feng, J.; Wu, H.; Duan, N.; Zhu, Y.; Zhao, Y.; Shen, S.; Zhang, K.; Wu, W.; et al. FGFC1 selectively inhibits erlotinib-resistant non-small cell lung cancer via elevation of ROS mediated by the EGFR/PI3K/Akt/mTOR pathway. Front. Pharmacol. 2022, 12, 764699. [Google Scholar] [CrossRef]
- Joseph, B.; Marchetti, P.; Formstecher, P.; Kroemer, G.; Lewensohn, R.; Zhivotovsky, B. Mitochondrial dysfunction is an essential step for killing of non-small cell lung carcinomas resistant to conventional treatment. Oncogene 2017, 36, 4818. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Li, S.; Zhang, B.; Duan, N.; Zhou, R.; Yan, S.; Elango, J.; Liu, N.; Wu, W. FGFC1 exhibits anti-cancer activity via inhibiting NF-κB signaling pathway in EGFR-mutant NSCLC cells. Mar. Drugs 2022, 20, 76. [Google Scholar] [CrossRef]
- Nishimura, Y.; Suzuki, E.; Hasegawa, K.; Nishimura, N.; Kitano, Y.; Hasumi, K. Pre-SMTP, a key precursor for the biosynthesis of the SMTP plasminogen modulators. J. Antibiot. 2012, 65, 483–485. [Google Scholar] [CrossRef] [Green Version]
- Do Minh, T.; Johnson, A.L.; Jones, J.E.; Senise, P.P., Jr. Reactions of phthalaldehyde with ammonia and amines. J. Org. Chem. 1977, 42, 4217–4221. [Google Scholar] [CrossRef]
- Su, T.; Bao, B.; Yan, T.; Zhang, C.; Bu, Y.; Wu, W. Response surface methodology to optimize marine microbe culture for producing fungi fibrinolytic compound. Chin. J. Biotechnol. 2013, 29, 857–861. [Google Scholar]
- Wang, M.; He, H.; Na, K.; Cai, M.; Zhou, X.; Zhao, W.; Zhang, Y. Designing novel glucose/ornithine replenishment strategies by biosynthetic and bioprocess analysis to improve fibrinolytic FGFC1 production by the marine fungus Stachybotrys longispora. Process Biochem. 2015, 50, 2012–2018. [Google Scholar] [CrossRef]
- Yin, Y.; Fu, Q.; Wu, W.; Cai, M.; Zhou, X.; Zhang, Y. Producing novel fibrinolytic isoindolinone derivatives in marine fungus Stachybotrys longispora FG216 by the rational supply of amino compounds according to its biosynthesis pathway. Mar. Drugs 2017, 15, 214. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Antithrombotic Effects | Efficacy (10 mg/kg) |
|---|---|
| consistent clot clearance | 43.3 ± 40.5% |
| thrombotic middle cerebral artery occlusion recanalization | 32.5-fold |
| neurologic deficit amelioration | 29% |
| cerebral infarct reduction | 46% |
| cerebral hemorrhage decrease | 51% |
| infarct, edema and clot sizes reduction | 65%, 37%, and 55%, respectively |
| Fibrinolysis Activities (10 mg/kg) | Infarction Area Size Reduction | Neurological Score Reduction | Edema Percentage Reduction |
|---|---|---|---|
| Compound 1 | 4.9 ± 1.1% | 1.7 ± 0.4% | 5.8 ± 1.0% |
| Congener 12 | 4.4 ± 0.5% | 1.7 ± 0.4% | 4.6 ± 1.0% |
| Congener 17 | 5.7 ± 1.2% | 1.5 ± 0.5% | 3.3 ± 1.4% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hang, S.; Chen, H.; Wu, W.; Wang, S.; Fang, Y.; Sheng, R.; Tu, Q.; Guo, R. Progress in Isoindolone Alkaloid Derivatives from Marine Microorganism: Pharmacology, Preparation, and Mechanism. Mar. Drugs 2022, 20, 405. https://doi.org/10.3390/md20060405
Hang S, Chen H, Wu W, Wang S, Fang Y, Sheng R, Tu Q, Guo R. Progress in Isoindolone Alkaloid Derivatives from Marine Microorganism: Pharmacology, Preparation, and Mechanism. Marine Drugs. 2022; 20(6):405. https://doi.org/10.3390/md20060405
Chicago/Turabian StyleHang, Sijin, Hui Chen, Wenhui Wu, Shiyi Wang, Yiwen Fang, Ruilong Sheng, Qidong Tu, and Ruihua Guo. 2022. "Progress in Isoindolone Alkaloid Derivatives from Marine Microorganism: Pharmacology, Preparation, and Mechanism" Marine Drugs 20, no. 6: 405. https://doi.org/10.3390/md20060405