
Taeanamides A and B, Nonribosomal Lipo-Decapeptides Isolated from an Intertidal-Mudflat-Derived Streptomyces sp.

 , , , , , and
, , , , , and
Abstract
:1. Introduction
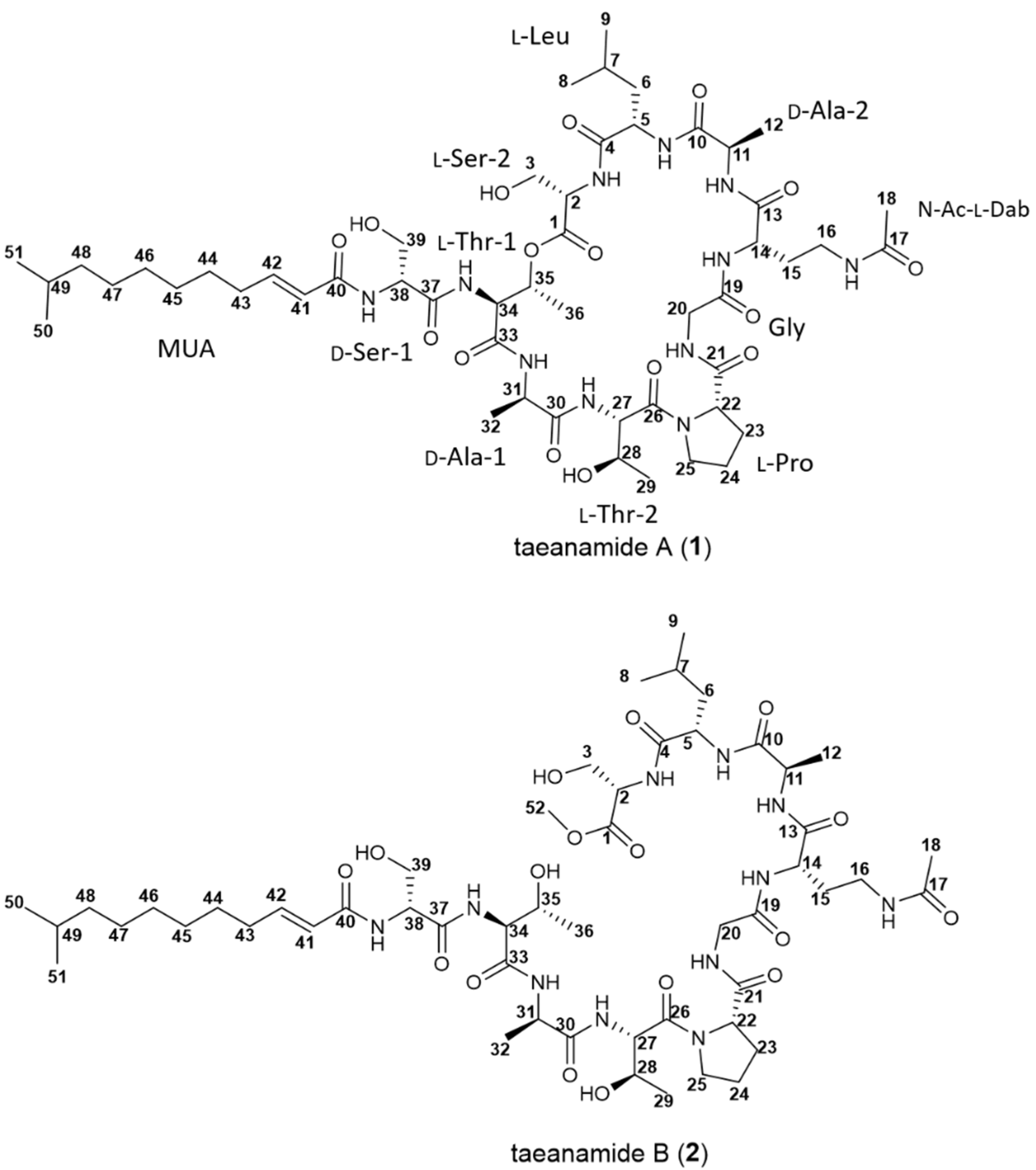
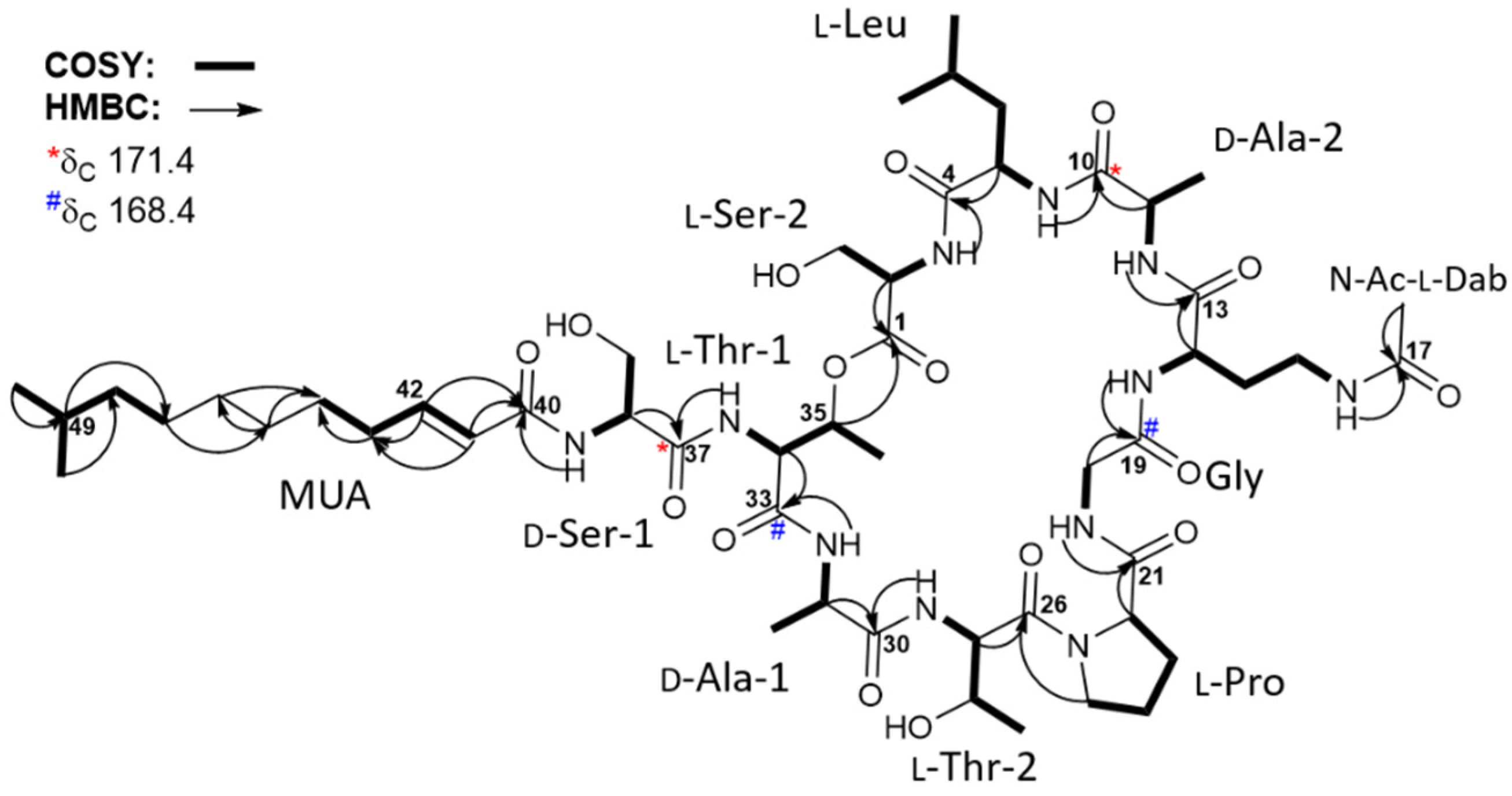
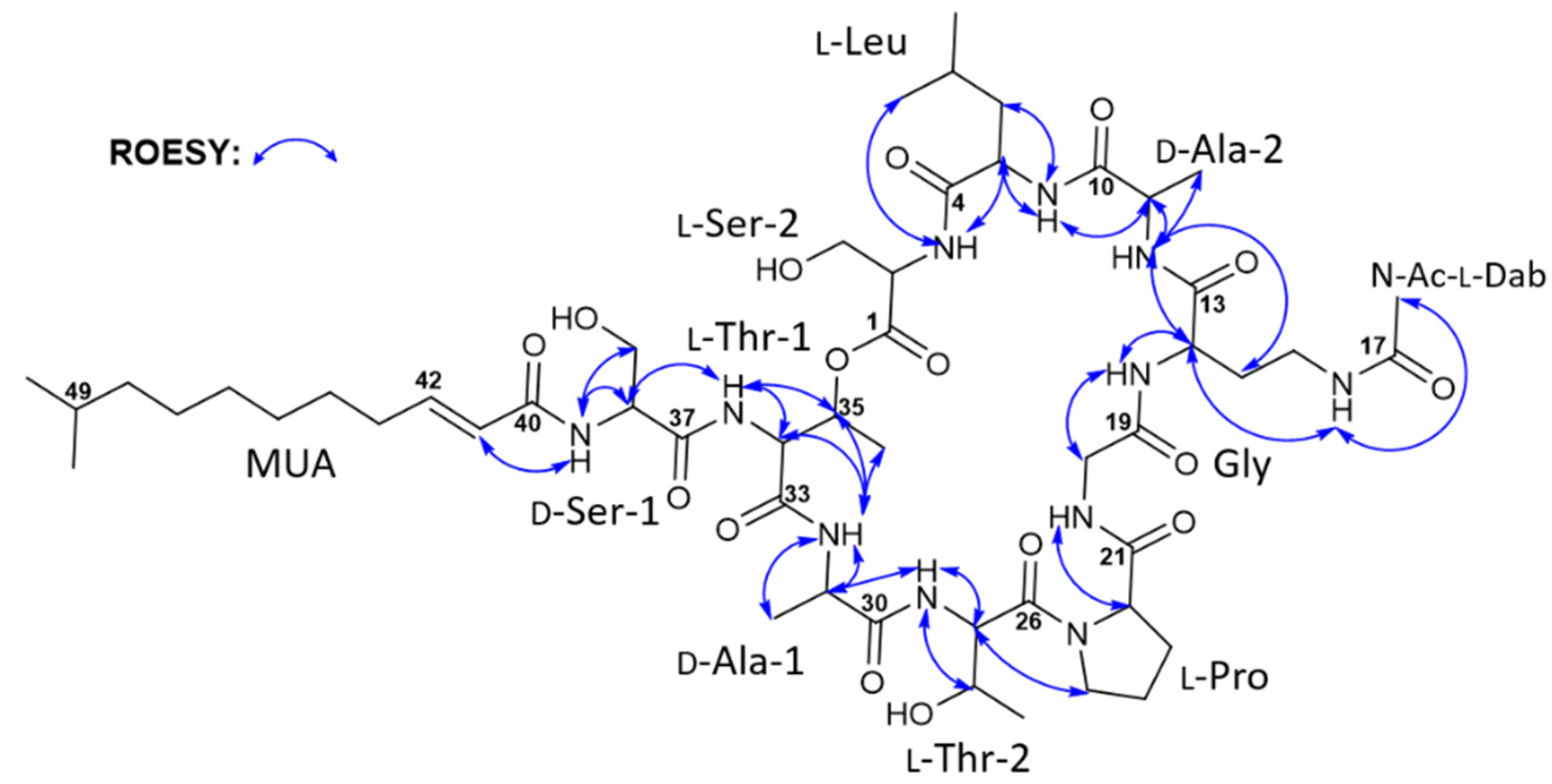
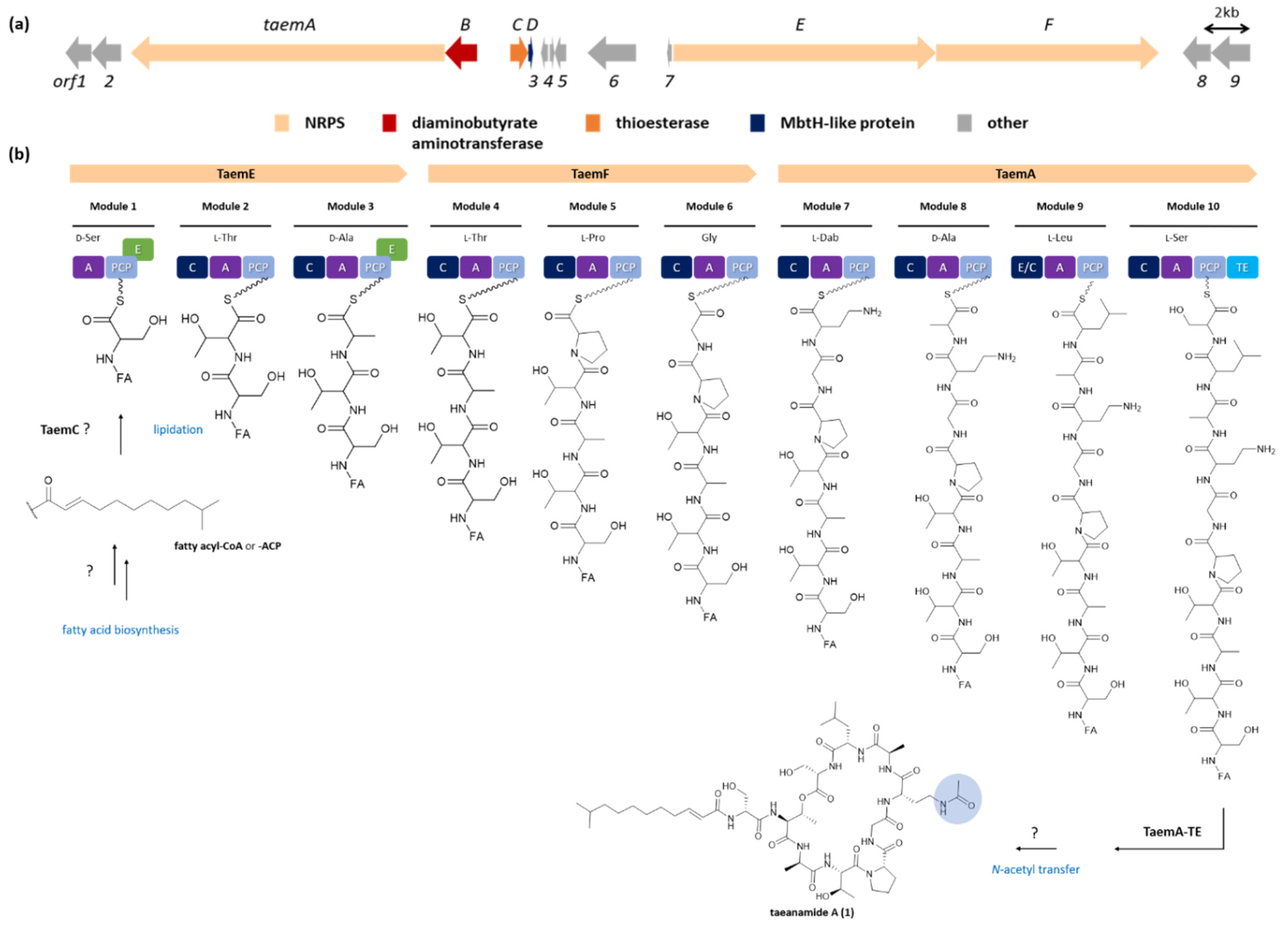
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Isolation and Identification of the Bacterial Strain Streptomyces sp. AMD43
3.3. Cultivation and Extraction
3.4. Isolation of Taeanamides A (1) and B (2)
3.5. qTOF-HR-MS/MS Analysis of Taeanamides A (1) and B (2)
3.6. The Advanced Marfey’s Method
3.7. Cytotoxicity Assay
3.8. Resazurin Microtiter Assay (REMA) Plate Testing of Mycobacterium Tuberculosis
3.9. Sequencing and Gene Annotation of Streptomyces sp. AMD43
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Süssmuth, R.D.; Mainz, A. Nonribosomal Peptide Synthesis—Principles and Prospects. Angew. Chem. Int. Ed. 2017, 56, 3770–3821. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Acharya, D.; Adholeya, A.; Barrow, C.J.; Deshmukh, S.K. Nonribosomal peptides from marine microbes and their antimicrobial and anticancer potential. Front. Pharmacol. 2017, 8, 828. [Google Scholar] [CrossRef] [PubMed]
- Felnagle, E.A.; Jackson, E.E.; Chan, Y.A.; Podevals, A.M.; Berti, A.D.; McMahon, M.D.; Thomas, M.G. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol. Pharm. 2008, 5, 191–211. [Google Scholar] [CrossRef] [PubMed]
- Behie, S.W.; Bonet, B.; Zacharia, V.M.; McClung, D.J.; Traxler, M.F. Molecules to ecosystems: Actinomycete natural products in situ. Front. Microbiol. 2017, 7, 2149. [Google Scholar] [CrossRef] [Green Version]
- Romero, F.; Espliego, F.; Baz, J.P.; Quesada, T.G.D.; Grávalos, D.; Calie, F.D.L.; Fernández-Puentes, J.L. Thiocoraline, a new depsipeptide with antitumor activity produced by a marine Micromonospora I. Taxonomy, fermentation, isolation, and biological activities. J. Antibiot. 1997, 50, 734–737. [Google Scholar] [CrossRef] [Green Version]
- Erba, E.; Bergamaschi, D.; Ronzoni, S.; Faretta, M.; Taverna, S.; Bonfanti, M.; Catapano, C.V.; Faircloth, G.; Jimeno, J.; D’incalci, M. Mode of action of thiocoraline, a natural marine compound with anti-tumour activity. Brit. J. Cancer 1999, 80, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Um, S.; Choi, T.J.; Kim, H.; Kim, B.Y.; Kim, S.H.; Lee, S.K.; Oh, K.B.; Shin, J.; Oh, D.C. Ohmyungsamycins A and B: Cytotoxic and antimicrobial cyclic peptides produced by Streptomyces sp. from a volcanic island. J. Org. Chem. 2013, 78, 12321–12329. [Google Scholar] [CrossRef]
- Bae, M.; Kim, H.; Moon, K.; Nam, S.-J.; Shin, J.; Oh, K.-B.; Oh, D.-C. Mohangamides A and B, new dilactone-tethered pseudo-dimeric peptides inhibiting Candida albicans isocitrate lyase. Org. Lett. 2015, 17, 712–715. [Google Scholar] [CrossRef]
- Kim, M.-S.; Bae, M.; Jung, Y.-E.; Kim, J.-M.; Hwang, S.; Song, M.C.; Ban, Y.H.; Bae, E.S.; Hong, S.; Lee, S.K.; et al. Unprecedented noncanonical features of the nonlinear nonribosomal peptide synthetase assembly line for WS9326A biosynthesis. Angew. Chem. Int. Ed. 2021, 60, 2–10. [Google Scholar]
- Shin, D.; Byun, W.S.; Moon, K.; Kwon, Y.; Bae, M.; Um, S.; Lee, S.K.; Oh, D.C. Coculture of marine Streptomyces sp. with Bacillus sp. produces a new piperazic acid-bearing cyclic peptide. Front. Chem. 2018, 6, 498. [Google Scholar] [CrossRef]
- Fujii, K.; Ikai, Y.; Oka, H.; Suzuki, M.; Harada, K. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: Combination of Marfey’s method with mass spectrometry and its practical application. Anal. Chem. 1997, 69, 5146–5151. [Google Scholar] [CrossRef]
- Du, Y.E.; Bae, E.S.; Lim, Y.; Cho, J.C.; Nam, S.J.; Shin, J.; Lee, S.K.; Nam, S.I.; Oh, D.C. Svalbamides A and B, pyrrolidinone-bearing lipodipeptides from Arctic Paenibacillus sp. Mar. Drugs 2021, 19, 229. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachelhaus, T.; Mootz, H.D.; Marahiel, M.A. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 1999, 6, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Rausch, C.; Hoof, I.; Weber, T.; Wohlleben, W.; Huson, D.H. Phylogenetic analysis of condensation domains in NRPS sheds light on their functional evolution. BMC Evol. Biol. 2007, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Linne, U.; Doekel, S.; Marahiel, M.A. Portability of epimerization domain and role of peptidyl carrier protein on epimerization activity in nonribosomal peptide synthetases. Biochemistry 2001, 40, 15824–15834. [Google Scholar] [CrossRef]
- Balibar, C.J.; Vaillancourt, F.H.; Walsh, C.T. Generation of D amino acid residues in assembly of arthrofactin by dual condensation/epimerization domains. Chem. Biol. 2005, 12, 1189–1200. [Google Scholar] [CrossRef] [Green Version]
- Kotowska, M.; Pawlik, K. Roles of type II thioesterases and their application for secondary metabolite yield improvement. Appl. Microbiol. Biotechnol. 2014, 98, 7735–7746. [Google Scholar] [CrossRef] [Green Version]
- Steller, S.; Sokoll, A.; Wilde, C.; Bernhard, F.; Franke, P.; Vater, J. Initiation of surfactin biosynthesis and the role of the SrfD-thioesterase protein. Biochemistry 2004, 43, 11331–11343. [Google Scholar] [CrossRef]
- Chooi, Y.-H.; Tang, Y. Adding the lipo to lipopeptides: Do more with less. Chem. Biol. 2010, 17, 791–793. [Google Scholar] [CrossRef] [Green Version]
- Vandenende, C.S.; Vlasschaert, M.; Seah, S.Y.K. Functional characterization of an aminotransferase required for pyoverdine siderophore biosynthesis in Pseudomonas aeruginosa PAO1. J. Bacteriol. 2004, 186, 5596–5602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshetnikov, A.S.; Khmelenina, V.N.; Trotsenko, Y.A. Characterization of the ectoine biosynthesis genes of haloalkalotolerant obligate methanotroph “Methylomicrobium alcaliphilum 20Z”. Arch. Microbiol. 2006, 184, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yu, J.; Jeong, L.S.; Lee, S.K. A novel cytarabine analog evokes synthetic lethality by targeting MK2 in p53-deficient cancer cells. Cancer Lett. 2021, 497, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Peypoux, F.; Guinand, M.; Michel, G.; Delcambe, L.; Das, B.C.; Lederer, E. Structure of iturine A, a peptidolipid antibiotic from Bacillus subtilis. Biochemistry 1978, 17, 3992–3996. [Google Scholar] [CrossRef]
- Peypoux, F.; Besson, F.; Michel, G.; Delcambe, L. Structure of bacillomycin D, a new antibiotic of the iturin group. Eur. J. Biochem. 1981, 118, 323–327. [Google Scholar] [CrossRef]
- Peypoux, F.; Michel, G.; Delcambe, L. Structure de la mycosubtiline, antibiotique isolé de Bacillus subtilis. Eur. J. Biochem. 1976, 63, 391–398. [Google Scholar] [CrossRef]
- Arima, K.; Kakinuma, A.; Tamura, G. Surfactin, a crystalline peptidelipid surfactant produced by Bacillus subtilis: Isolation, characterization and its inhibition of fibrin clot formation. Biochem. Biophys. Res. Commun. 1968, 31, 488–494. [Google Scholar] [CrossRef]
- Iwasaki, A.; Tadenuma, T.; Sumimoto, S.; Shiota, I.; Matsubara, T.; Saito-Nakano, Y.; Nozaki, T.; Sato, T.; Suenaga, K. Hoshinoamides A and B, acyclic lipopeptides from the marine cyanobacterium Caldora penicillate. J. Nat. Prod. 2018, 81, 2545–2552. [Google Scholar] [CrossRef]
- Bornancin, L.; Alonso, E.; Alvariño, R.; Inguimbert, N.; Bonnard, I.; Botana, L.M.; Banaigs, B. Structure and biological evaluation of new cyclic and acyclic laxaphycin-A type peptides. Bioorganic Med. Chem. 2019, 27, 1966–1980. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Murai, A.; Takahara, Y.; Kainosho, K. The structure of permetin A, a new polypeptin type antibiotic produced by Bacillus circulans. J. Antibiot. 1979, 32, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Shen, B.; Korshalla, J.; Carter, G.T. Circulocins, new antibacterial lipopeptides from Bacillus circulans J2154. Tetrahedron 2001, 57, 1189–1195. [Google Scholar] [CrossRef]
- Hirota-Takahara, Y.; Kozuma, S.; Nahoki, K.; Fukuda, D.; Nakajima, M.; Ando, O. Pedopeptins, novel inhibitors of LPS: Taxonomy of producing organism, fermentation, isolation, physicochemical properties and structure elucidation. J. Antibiot. 2014, 67, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Herath, K.B.; Zhang, C.; Jayasuriya, H.; Ondeyka, J.G.; Zink, D.L.; Burgess, B.; Wang, J.; Singh, S.B. Structure and semisynthesis of platensimide A, produced by Streptomyces platensis. Org. Lett. 2009, 10, 1699–1702. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taeanamide A (1) | Taeanamide B (2) | Taeanamide A (1) | Taeanamide B (2) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C/H | δC, Type | δH, Mult (J in Hz) | C/H | δC, Type | δH, Mult (J in Hz) | C/H | δC, Type | δH, Mult (J in Hz) | C/H | δC, Type | δH, Mult (J in Hz) |
| 1 | 169.3, C | 1 | 172.5, C | 26 | 169.8, C | 26 | 171.7, C | ||||
| 2 | 54.2, CH | 4.50, m | 2 | 56.7, CH | 5.11, m | 27 | 55.3, CH | 4.53, m | 27 | 57.8, CH | 5.22, (dd, 8.5, 4.5) |
| 2-NH | 8.18, m | 2-NH | 9.35, (d, 6.5) | 27-NH | 8.08, (d, 8.5) | 27-NH | 8.55, (d, 8.5) | ||||
| 3a | 61.4, CH2 | 3.89, (dd, 11.0, 4.5) | 3a | 63.0, CH2 | 4.36. m | 28 | 66.5, CH | 3.92, m | 28 | 68.5, CH | 4.58, m |
| 3b | 3.62, m | 3b | 4.22, m | 28-OH | - | 28-OH | 6.49, (d, 5.0) | ||||
| 3-OH | - | 3-OH | 6.84, (t, 5.5) | 29 | 19.3, CH3 | 1.02, (d, 6.5) | 29 | 20.8, CH3 | 1.58, (d, 6.5) | ||
| 4 | 171.8, C | 4 | 174.3, C | 30 | 172.1, C | 30 | 174.1, C | ||||
| 5 | 50.3, CH | 4.51, m | 5 | 52.6, CH | 5.25, m | 31 | 48.8, CH | 4.28, m | 31 | 51.0, CH | 5.03, m |
| 5-NH | 7.67, (d, 8.0) | 5-NH | 8.84, (d, 9.5) | 31-NH | 7.76, (d, 6.5) | 31-NH | 9.07, (d, 6.5) | ||||
| 6 | 41.7, CH2 | 1.41, m | 6 | 41.9, CH2 | 2.02, m | 32 | 17.9, CH3 | 1.23, (d, 7.0) | 32 | 18.7, CH3 | 1.70, (d, 7.0) |
| 7 | 23.8, CH | 1.53, m | 7 | 25.4, CH | 1.94, m | 33 | 168.4, C | 33 | 172.5, C | ||
| 8 | 21.3, CH3 | 0.82, (d, 6.5) | 8 | 23.9, CH3 | 0.90, (d, 6.5) | 34 | 55.7, CH | 4.43, (dd, 9.0, 2.5) | 34 | 60.6, CH | 5.06, m |
| 9 | 23.3, CH3 | 0.80, (d, 6.5) | 9 | 19.8, CH3 | 0.83, (d, 6.5) | 34-NH | 8.26, (d, 9.0) | 34-NH | 9.05, (d, 8.0) | ||
| 10 | 171.4, C | 10 | 174.0, C | 35 | 70.2, CH | 5.34, m | 35 | 67.8, CH | 4.92, m | ||
| 11 | 48.6, CH | 4.22, m | 11 | 50.7, CH | 5.04, m | 35-OH | - | 35-OH | 6.53, (d, 3.5) | ||
| 11-NH | 8.33, (d, 7.0) | 11-NH | 9.41, (d, 6.5) | 36 | 16.1, CH3 | 1.08, (d, 6.5) | 36 | 20.8, CH3 | 1.48, (d, 6.5) | ||
| 12 | 18.0, CH3 | 1.22, (d, 7.0) | 12 | 18.7, CH3 | 1.70, (d, 7.0) | 37 | 171.4, C | 37 | 173.1, C | ||
| 13 | 171.0, C | 13 | 173.6, C | 38 | 55.7, CH | 4.50, m | 38 | 58.2, CH | 5.32, (dd, 12.0, 6.0) | ||
| 14 | 51.1, CH | 4.19, m | 14 | 52.9, CH | 4.97, m | 38-NH | 8.17, m | 38-NH | 9.22, (d, 6.0) | ||
| 14-NH | 8.15, (d, 6.5) | 14-NH | 8.67, (d, 7.0) | 39 | 61.4, CH2 | 3.63, m | 39a | 63.3, CH2 | 4.46, m | ||
| 15a | 31.2, CH2 | 1.77, m | 15a | 32.8, CH2 | 2.59, (td, 13.5, 6.5) | 39b | 4.36, m | ||||
| 15b | 1.64, (td, 14.0, 7.5) | 15b | 2.35, (td, 13.5, 7.0) | 39-OH | - | 39-OH | 7.00, (t, 5.5) | ||||
| 16 | 35.5, CH2 | 3.03, m | 16a | 37.3, CH2 | 3.85, m | 40 | 165.4, C | 40 | 167.6, C | ||
| 16b | 3.55, m | 41 | 123.8, CH | 6.06, (d, 15.0) | 41 | 124.9, CH | 6.35, (d, 15.5) | ||||
| 16-NH | 7.81, (t, 5.5) | 16-NH | 8.58, (t, 6.0) | 42 | 143.5, CH | 6.65, (dt, 15.0, 7.0) | 42 | 145.4, CH | 7.15, m | ||
| 17 | 169.2, C | 17 | 171.5, C | 43 | 31.2, CH2 | 2.14, (dd, 14.0, 7.0) | 43 | 32.7, CH2 | 2.09, m | ||
| 18 | 22.6, CH3 | 1.79, m | 18 | 23.6, CH3 | 2.01, s | 44 | 27.8, CH2 | 1.38, m | 44 | 29.1, CH2 | 1.30, m |
| 19 | 168.4, C | 19 | 170.8, C | 45 | 28.6, CH2 | 1.27, m | 45 | 30.0, CH2 | 1.19, m | ||
| 20a | 41.9, CH2 | 3.85, (dd, 17.0, 6.0) | 20a | 44.3, CH2 | 4.40, m | 46 | 29.1, CH2 | 1.25, m | 46 | 30.5, CH2 | 1.15, m |
| 20b | 3.55, (dd, 17.0, 3.5) | 20b | 4.04, (dd, 17.0, 5.0) | 47 | 26.7, CH2 | 1.24, m | 47a | 37.3, CH2 | 1.02, m | ||
| 20-NH | 7.67, (d, 8.0) | 20-NH | 9.25, (t, 6.0) | 47b | 1.22, m | ||||||
| 21 | 171.5, C | 21 | 174.0, C | 48 | 38.5, CH2 | 1.13, m | 48 | 30.2, CH2 | 1.30, m | ||
| 22 | 60.3, CH | 4.26, m | 22 | 62.3, CH | 4.69, (t, 7.5) | 49 | 27.4, CH | 1.50, m | 49 | 35.1, CH | 1.24, m |
| 23a | 29.0, CH2 | 1.98, m | 23 | 30.2, CH2 | 2.15, m | 50 | 22.5, CH3 | 0.84, (d, 6.5) | 50 | 12.1, CH3 | 0.86, (d, 7.5) |
| 23b | 1.78, m | 51 | 22.5, CH3 | 0.84, (d, 6.5) | 51 | 22.1, CH3 | 0.83, (d, 7.5) | ||||
| 24a | 24.3, CH2 | 1.79, m | 24 | 26.1, CH2 | 1.94, m | 52 | 52.5, CH3 | 3.63, s | |||
| 24b | 1.73, m | ||||||||||
| 25a | 46.8, CH2 | 3.43, m | 25 | 48.8, CH2 | 3.79, m | ||||||
| 25b | 3.59, m | ||||||||||
| IC50 (μM) | A549 | HCT116 | MCF-7 | MDA-MB-231 | SK-Hep-1 | SNU638 |
|---|---|---|---|---|---|---|
| 1 | >20 | >20 | >20 | >20 | >20 | >20 |
| 2 | 0.60 ± 0.29 | 0.26 ± 0.07 | 1.13 ± 0.74 | 0.8 ± 0.69 | 0.67 ± 0.66 | 0.33 ± 0.02 |
| Etoposide | 0.15 ± 0.09 | 0.73 ± 0.15 | 1.43 ± 1.00 | 3.42 ± 3.18 | 2.26 ± 3.76 | 0.28 ± 0.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Kim, E.; Moon, D.H.; Kim, T.H.; Kang, I.; Lim, Y.; Shin, D.; Hwang, S.; Du, Y.E.; Song, M.C.; et al. Taeanamides A and B, Nonribosomal Lipo-Decapeptides Isolated from an Intertidal-Mudflat-Derived Streptomyces sp. Mar. Drugs 2022, 20, 400. https://doi.org/10.3390/md20060400
Cui J, Kim E, Moon DH, Kim TH, Kang I, Lim Y, Shin D, Hwang S, Du YE, Song MC, et al. Taeanamides A and B, Nonribosomal Lipo-Decapeptides Isolated from an Intertidal-Mudflat-Derived Streptomyces sp. Marine Drugs. 2022; 20(6):400. https://doi.org/10.3390/md20060400
Chicago/Turabian StyleCui, Jinsheng, Eunji Kim, Dong Hyun Moon, Tae Ho Kim, Ilnam Kang, Yeonjung Lim, Daniel Shin, Sunghoon Hwang, Young Eun Du, Myoung Chong Song, and et al. 2022. "Taeanamides A and B, Nonribosomal Lipo-Decapeptides Isolated from an Intertidal-Mudflat-Derived Streptomyces sp." Marine Drugs 20, no. 6: 400. https://doi.org/10.3390/md20060400