
Conus regius-Derived Conotoxins: Novel Therapeutic Opportunities from a Marine Organism
 , ,
, ,
Abstract
:1. Introduction
2. Conus regius and Its Venom Composition
3. Conotoxins
4. α-Conotoxins and Their Targets
5. RgIA
5.1. Analgesic and Disease-Modifying Effects of RgIA
5.2. Anti-Colitis Effects of RgIA
5.3. Anticancer Effects of RgIA
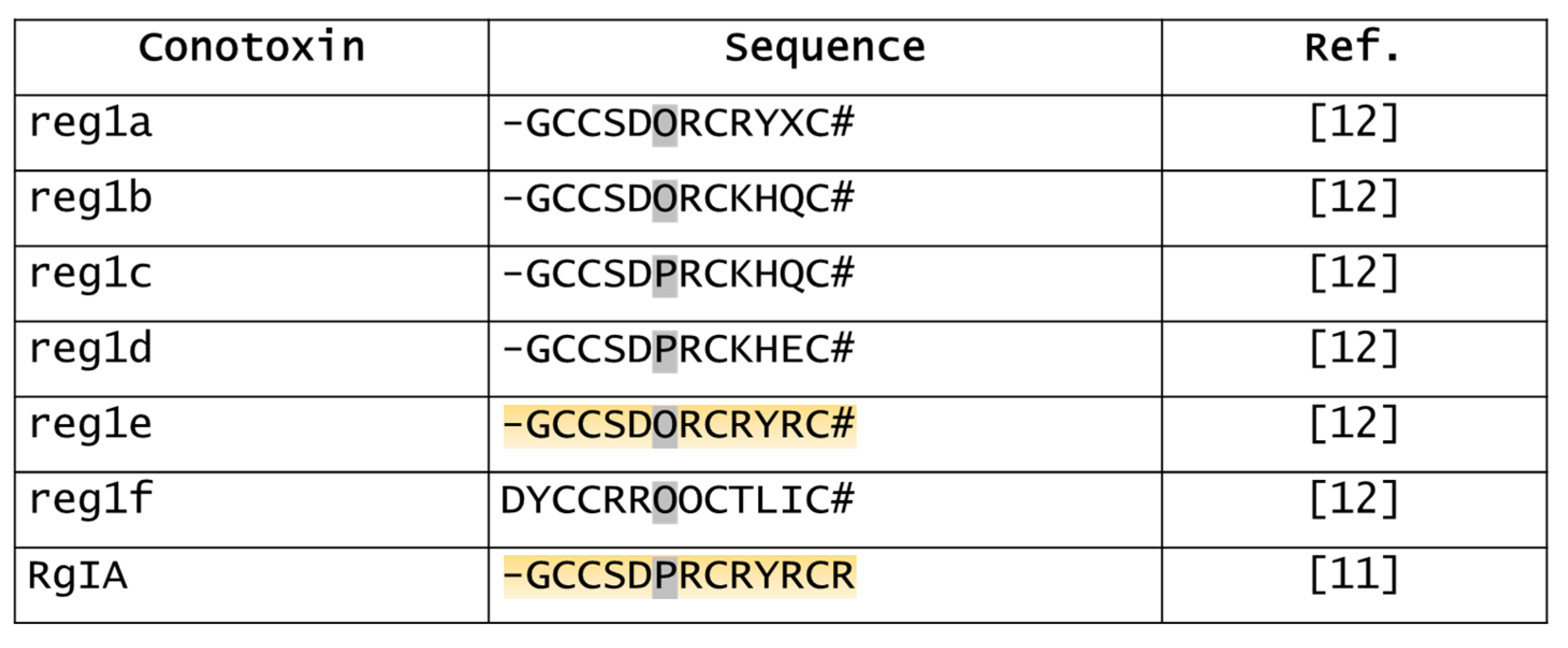
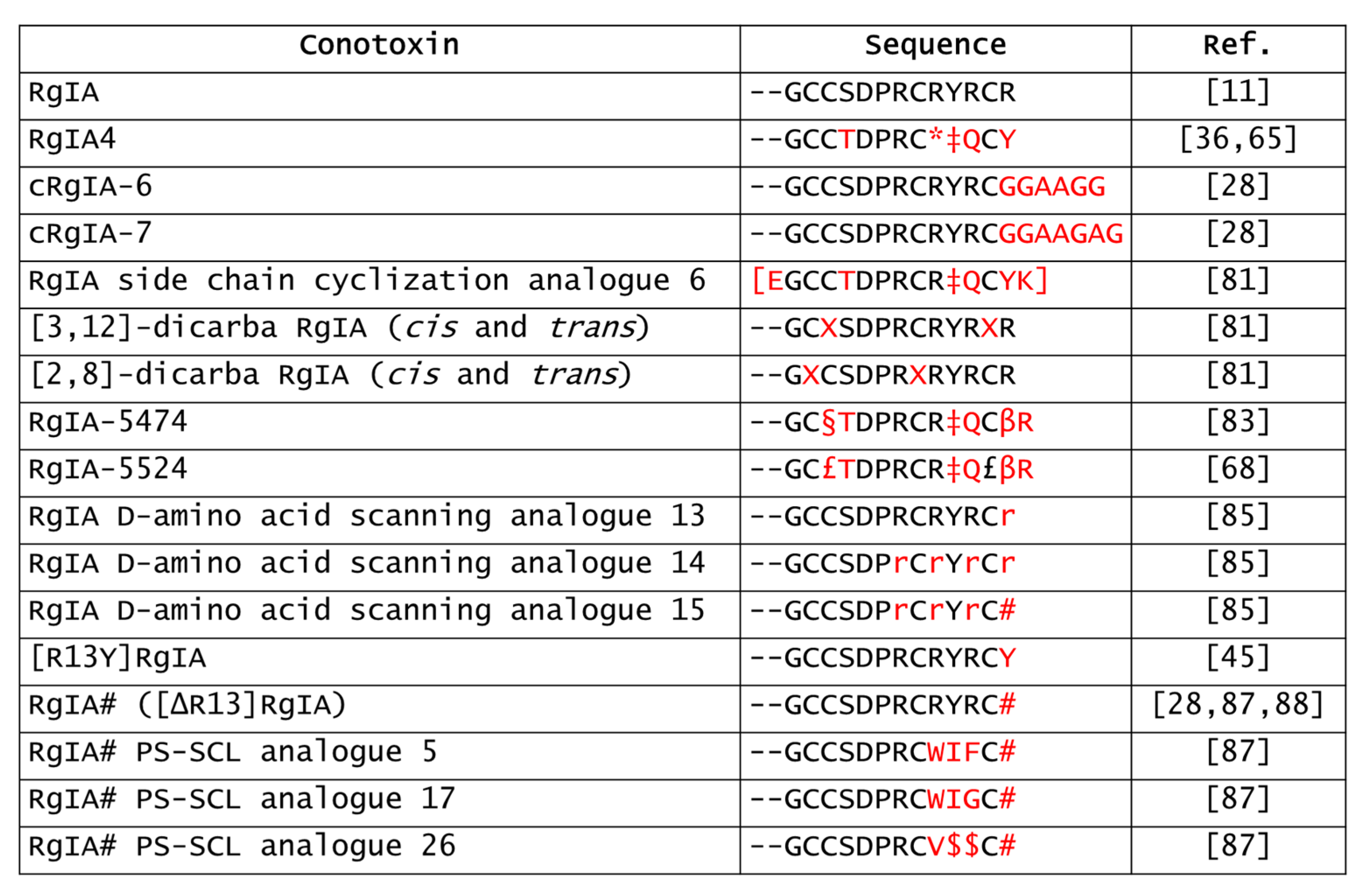
5.4. Derivatives of RgIA

6. RegIIA
6.1. Strategies to Improve RegIIA Selectivity towards Specific nAChRs Subtypes
6.2. Species-Specific Differences in RegIIA Sensitivity

7. RgIB
8. Reg3b and Mini-M Conotoxins
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Endean, R.; Gyr, P.; Surridge, J. The Pharmacological Actions on Guinea-Pig Ileum of Crude Venoms from the Marine Gastropods Conus striatus and Conus magus. Toxicon 1977, 15, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.J. Piscivorous Gastropods Of The Genus Conus. Proc. Natl. Acad. Sci. USA 1956, 42, 168–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, I.; Gillessen, D.; Gregson, R.P.; Quinn, R.J. Characterization of the Neurotoxic Constituents of Conus geographus (L.) Venom. Life Sci. 1977, 21, 1759–1769. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.-H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and Biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Olivera, B.M. EE Just Lecture, 1996. Mol. Biol. Cell 1997, 8, 2101–2109. [Google Scholar] [CrossRef] [Green Version]
- Olivera, B.M.; Rivier, J.; Clark, C.; Ramilo, C.A.; Corpuz, G.P.; Abogadie, F.C.; Mena, E.E.; Woodward, S.R.; Hillyard, D.R.; Cruz, L.J. Diversity of Conus Neuropeptides. Science 1990, 249, 257–263. [Google Scholar] [CrossRef]
- Vianna Braga, M.C.; Konno, K.; Portaro, F.C.V.; Carlos de Freitas, J.; Yamane, T.; Olivera, B.M.; Pimenta, D.C. Mass Spectrometric and High Performance Liquid Chromatography Profiling of the Venom of the Brazilian Vermivorous Mollusk Conus regius: Feeding Behavior and Identification of One Novel Conotoxin. Toxicon 2005, 45, 113–122. [Google Scholar] [CrossRef]
- Abraham, N.; Lewis, R. Neuronal Nicotinic Acetylcholine Receptor Modulators from Cone Snails. Mar. Drugs 2018, 16, 208. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Tae, H.-S.; Chu, Y.; Jiang, T.; Adams, D.J.; Yu, R. Medicinal Chemistry, Pharmacology, and Therapeutic Potential of α-Conotoxins Antagonizing the α9α10 Nicotinic Acetylcholine Receptor. Pharmacol. Ther. 2021, 222, 107792. [Google Scholar] [CrossRef]
- Walls, J.G. Cone Shells, a Synopsis of the Living Conidae. In Cone Shells, a Synopsis of the Living Conidae; T.F.H. Pulications: Neptune City, NJ, USA, 1979; pp. 831–833. [Google Scholar]
- Ellison, M.; Haberlandt, C.; Gomez-Casati, M.E.; Watkins, M.; Elgoyhen, A.B.; McIntosh, J.M.; Olivera, B.M. α-RgIA: A Novel Conotoxin That Specifically and Potently Blocks the α9α10 nAChR. Biochemistry 2006, 45, 1511–1517. [Google Scholar] [CrossRef]
- Franco, A.; Pisarewicz, K.; Moller, C.; Mora, D.; Fields, G.B.; Marí, F. Hyperhydroxylation: A New Strategy for Neuronal Targeting by Venomous Marine Molluscs. Prog. Mol. Subcell. Biol. 2006, 43, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Kompella, S.N.; Akondi, K.B.; Melaun, C.; Daly, N.L.; Luetje, C.W.; Alewood, P.F.; Craik, D.J.; Adams, D.J.; Marí, F. RegIIA: An α4/7-Conotoxin from the Venom of Conus regius That Potently Blocks α3β4 nAChRs. Biochem. Pharm. 2012, 83, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Braga, M.C.V.; Nery, A.A.; Ulrich, H.; Konno, K.; Sciani, J.M.; Pimenta, D.C. α-RgIB: A Novel Antagonist Peptide of Neuronal Acetylcholine Receptor Isolated from Conus regius Venom. Int. J. Pept. 2013, 2013, 543028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, A.; Dovell, S.; Möller, C.; Grandal, M.; Clark, E.; Marí, F. Structural Plasticity of Mini-M Conotoxins—Expression of All Mini-M Subtypes by Conus regius. FEBS J. 2018, 285, 887–902. [Google Scholar] [CrossRef] [Green Version]
- Halai, R.; Craik, D.J. Conotoxins: Natural Product Drug Leads. Nat. Prod. Rep. 2009, 26, 526. [Google Scholar] [CrossRef]
- Livett, B.G.; Gayler, K.R.; Khalil, Z. Drugs from the Sea: Conopeptides as Potential Therapeutics. Curr. Med. Chem. 2004, 11, 1715–1723. [Google Scholar] [CrossRef]
- Vetter, I.J.; Lewis, R. Therapeutic Potential of Cone Snail Venom Peptides (Conopeptides). Curr. Top. Med. Chem. 2012, 12, 1546–1552. [Google Scholar] [CrossRef]
- Kaas, Q.; Yu, R.; Jin, A.-H.; Dutertre, S.; Craik, D.J. ConoServer: Updated Content, Knowledge, and Discovery Tools in the Conopeptide Database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef]
- Góngora-Benítez, M.; Tulla-Puche, J.; Albericio, F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 2014, 114, 901–926. [Google Scholar] [CrossRef]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, Synthesis, and Structure–Activity Relationships of Conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.-C.; Halai, R.; Wang, C.K.L.; Craik, D.J. ConoServer, a Database for Conopeptide Sequences and Structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus Venom Peptide Pharmacology. Pharm. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide Characterization and Classifications: An Analysis Using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Lipovsek, M.; Marcovich, I.; Elgoyhen, A.B. The Hair Cell 9α10 Nicotinic Acetylcholine Receptor: Odd Cousin in an Old Family. Front. Cell. Neurosci. 2021, 15, 785265. [Google Scholar] [CrossRef]
- Hone, A.J.; McIntosh, J.M. Nicotinic Acetylcholine Receptors in Neuropathic and Inflammatory Pain. FEBS Lett. 2018, 592, 1045–1062. [Google Scholar] [CrossRef] [Green Version]
- Dutton, J.; Craik, D. Alpha Conotoxins Nicotinic Acetylcholine Receptor Antagonists as Pharmacological Tools and Potential Drug Leads. Curr. Med. Chem. 2001, 8, 327–344. [Google Scholar] [CrossRef]
- Halai, R.; Callaghan, B.; Daly, N.L.; Clark, R.J.; Adams, D.J.; Craik, D.J. Effects of Cyclization on Stability, Structure, and Activity of α-Conotoxin RgIA at the α9α10 Nicotinic Acetylcholine Receptor and GABAB Receptor. J. Med. Chem. 2011, 54, 6984–6992. [Google Scholar] [CrossRef]
- Safavi-Hemami, H.; Brogan, S.E.; Olivera, B.M. Pain Therapeutics from Cone Snail Venoms: From Ziconotide to Novel Non-Opioid Pathways. J. Proteom. 2019, 190, 12–20. [Google Scholar] [CrossRef]
- Clark, R.J.; Daly, N.L.; Halai, R.; Nevin, S.T.; Adams, D.J.; Craik, D.J. The Three-Dimensional Structure of the Analgesic α-Conotoxin, RgIA. FEBS Lett. 2008, 582, 597–602. [Google Scholar] [CrossRef] [Green Version]
- Ellison, M.; Feng, Z.-P.; Park, A.J.; Zhang, X.; Olivera, B.M.; McIntosh, J.M.; Norton, R.S. α-RgIA, a Novel Conotoxin That Blocks the α9α10 nAChR: Structure and Identification of Key Receptor-Binding Residues. J. Mol. Biol. 2008, 377, 1216–1227. [Google Scholar] [CrossRef]
- Del Bufalo, A.; Cesario, A.; Salinaro, G.; Fini, M.; Russo, P. Alpha9Alpha10 Nicotinic Acetylcholine Receptors as Target for the Treatment of Chronic Pain. Curr. Pharm. Des. 2014, 20, 6042–6047. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Cinci, L.; Micheli, L.; Zanardelli, M.; Pacini, A.; McIntosh, M.J.; Ghelardini, C. α-Conotoxin RgIA Protects against the Development of Nerve Injury-Induced Chronic Pain and Prevents Both Neuronal and Glial Derangement. Pain 2014, 155, 1986–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hone, A.J.; Servent, D.; McIntosh, J.M. α9-containing Nicotinic Acetylcholine Receptors and the Modulation of Pain. Br. J. Pharm. 2018, 175, 1915–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacini, A.; Micheli, L.; Maresca, M.; Branca, J.J.V.; McIntosh, J.M.; Ghelardini, C.; Di Cesare Mannelli, L. The α9α10 Nicotinic Receptor Antagonist α-Conotoxin RgIA Prevents Neuropathic Pain Induced by Oxaliplatin Treatment. Exp. Neurol. 2016, 282, 37–48. [Google Scholar] [CrossRef]
- Romero, H.K.; Christensen, S.B.; Di Cesare Mannelli, L.; Gajewiak, J.; Ramachandra, R.; Elmslie, K.S.; Vetter, D.E.; Ghelardini, C.; Iadonato, S.P.; Mercado, J.L.; et al. Inhibition of α9α10 Nicotinic Acetylcholine Receptors Prevents Chemotherapy-Induced Neuropathic Pain. Proc. Natl. Acad. Sci. USA 2017, 114, E1825–E1832. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Cheuk, I.W.Y.; Shin, V.Y.; Kwong, A. Acetylcholine Receptors: Key Players in Cancer Development. Surg. Oncol. 2019, 31, 46–53. [Google Scholar] [CrossRef]
- Dang, N.; Meng, X.; Song, H. Nicotinic Acetylcholine Receptors and Cancer. Biomed. Rep. 2016, 4, 515–518. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Richbart, S.D.; Merritt, J.C.; Brown, K.C.; Nolan, N.A.; Akers, A.T.; Lau, J.K.; Robateau, Z.R.; Miles, S.L.; Dasgupta, P. Acetylcholine Signaling System in Progression of Lung Cancers. Pharmacol. Ther. 2019, 194, 222–254. [Google Scholar] [CrossRef]
- AlSharari, S.D.; Toma, W.; Mahmood, H.M.; Michael McIntosh, J.; Imad Damaj, M. The α9α10 Nicotinic Acetylcholine Receptors Antagonist α-Conotoxin RgIA Reverses Colitis Signs in Murine Dextran Sodium Sulfate Model. Eur. J. Pharm. 2020, 883, 173320. [Google Scholar] [CrossRef]
- Richter, K.; Papke, R.L.; Stokes, C.; Roy, D.C.; Espinosa, E.S.; Wolf, P.M.K.; Hecker, A.; Liese, J.; Singh, V.K.; Padberg, W.; et al. Comparison of the Anti-Inflammatory Properties of Two Nicotinic Acetylcholine Receptor Ligands, Phosphocholine and PCF3-DiEPP. Front. Cell. Neurosci. 2022, 16, 779081. [Google Scholar] [CrossRef]
- Safronova, V.G.; Vulfius, C.A.; Astashev, M.E.; Tikhonova, I.V.; Serov, D.A.; Jirova, E.A.; Pershina, E.V.; Senko, D.A.; Zhmak, M.N.; Kasheverov, I.E.; et al. α9α10 Nicotinic Acetylcholine Receptors Regulate Murine Bone Marrow Granulocyte Functions. Immunobiology 2021, 226, 152047. [Google Scholar] [CrossRef] [PubMed]
- Terpinskaya, T.I.; Osipov, A.V.; Kuznetsova, T.E.; Ryzhkovskaya, E.L.; Ulaschik, V.S.; Ivanov, I.A.; Tsetlin, V.I.; Utkin, Y.N. α-Conotoxins Revealed Different Roles of Nicotinic Cholinergic Receptor Subtypes in Oncogenesis of Ehrlich Tumor and in the Associated Inflammation. Dokl. Biochem. Biophys. 2015, 463, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Tsetlin, V.; Haufe, Y.; Safronova, V.; Serov, D.; Shadamarshan, P.; Son, L.; Shelukhina, I.; Kudryavtsev, D.; Kryukova, E.; Kasheverov, I.; et al. Interaction of α9α10 Nicotinic Receptors With Peptides and Proteins From Animal Venoms. Front. Cell. Neurosci. 2021, 15, 765541. [Google Scholar] [CrossRef] [PubMed]
- Huynh, P.N.; Harvey, P.J.; Gajewiak, J.; Craik, D.J.; Michael McIntosh, J. Critical Residue Properties for Potency and Selectivity of α-Conotoxin RgIA towards α9α10 Nicotinic Acetylcholine Receptors. Biochem. Pharm. 2020, 181, 114124. [Google Scholar] [CrossRef]
- Vincler, M.; Wittenauer, S.; Parker, R.; Ellison, M.; Olivera, B.M.; McIntosh, J.M. Molecular Mechanism for Analgesia Involving Specific Antagonism of α9α10 Nicotinic Acetylcholine Receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 17880–17884. [Google Scholar] [CrossRef] [Green Version]
- Dyachenko, I.A.; Palikova, Y.A.; Palikov, V.A.; Korolkova, Y.V.; Kazakov, V.A.; Egorova, N.S.; Garifulina, A.I.; Utkin, Y.N.; Tsetlin, V.I.; Kryukova, E.V. α-Conotoxin RgIA and Oligoarginine R8 in the Mice Model Alleviate Long-Term Oxaliplatin Induced Neuropathy. Biochimie 2022, 194, 127–136. [Google Scholar] [CrossRef]
- Grau, V.; Richter, K.; Hone, A.J.; McIntosh, J.M. Conopeptides [V11L;V16D]ArIB and RgIA4: Powerful Tools for the Identification of Novel Nicotinic Acetylcholine Receptors in Monocytes. Front. Pharm. 2019, 9, 1499. [Google Scholar] [CrossRef] [Green Version]
- Lustig, L.R.; Peng, H.; Hiel, H.; Yamamoto, T.; Fuchs, P.A. Molecular Cloning and Mapping of the Human Nicotinic Acetylcholine Receptor alpha10 (CHRNA10). Genomics 2001, 73, 272–283. [Google Scholar] [CrossRef]
- Peng, H.; Ferris, R.L.; Matthews, T.; Hiel, H.; Lopez-Albaitero, A.; Lustig, L.R. Characterization of the Human Nicotinic Acetylcholine Receptor Subunit Alpha (α) 9 (CHRNA9) and Alpha (α) 10 (CHRNA10) in Lymphocytes. Life Sci. 2004, 76, 263–280. [Google Scholar] [CrossRef]
- Leskovar, A.; Moriarty, L.J.; Turek, J.J.; Schoenlein, I.A.; Borgens, R.B. The Macrophage in Acute Neural Injury: Changes in Cell Numbers over Time and Levels of Cytokine Production in Mammalian Central and Peripheral Nervous Systems. J. Exp. Biol. 2000, 203, 1783–1795. [Google Scholar] [CrossRef]
- Hendriks, J.J.A.; Teunissen, C.E.; de Vries, H.E.; Dijkstra, C.D. Macrophages and Neurodegeneration. Brain Res. Rev. 2005, 48, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Callaghan, B.; Berecki, G. Analgesic Conotoxins: Block and G Protein-Coupled Receptor Modulation of N-Type (CaV2.2) Calcium Channels. Br. J. Pharm. 2012, 166, 486–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaghan, B.; Adams, D.J. Analgesic α-Conotoxins Vc1.1 and Rg1A Inhibit N-Type Calcium Channels in Sensory Neurons of α9 Nicotinic Receptor Knockout Mice. Channels 2010, 4, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, B.; Haythornthwaite, A.; Berecki, G.; Clark, R.J.; Craik, D.J.; Adams, D.J. Analgesic Alpha-Conotoxins Vc1.1 and Rg1A Inhibit N-Type Calcium Channels in Rat Sensory Neurons via GABAB Receptor Activation. J. Neurosci. 2008, 28, 10943–10951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuny, H.; de Faoite, A.; Huynh, T.G.; Yasuda, T.; Berecki, G.; Adams, D.J. γ-Aminobutyric Acid Type B (GABAB) Receptor Expression Is Needed for Inhibition of N-Type (Cav2.2) Calcium Channels by Analgesic α-Conotoxins. J. Biol. Chem. 2012, 287, 23948–23957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, E.K.; Zamponi, G.W. The Calcium Channel Terminator: Hasta La Vista Pain. Trends Pharm. Sci. 2022, 43, 801–803. [Google Scholar] [CrossRef]
- Chalil, A.; Staudt, M.D.; Harland, T.A.; Leimer, E.M.; Bhullar, R.; Argoff, C.E. A Safety Review of Approved Intrathecal Analgesics for Chronic Pain Management. Expert Opin. Drug Saf. 2021, 20, 439–451. [Google Scholar] [CrossRef]
- Elmslie, K.S. Calcium Channel Blockers in the Treatment of Disease. J. Neurosci. Res. 2004, 75, 733–741. [Google Scholar] [CrossRef]
- McIntosh, J.M.; Absalom, N.; Chebib, M.; Elgoyhen, A.B.; Vincler, M. Alpha9 Nicotinic Acetylcholine Receptors and the Treatment of Pain. Biochem. Pharm. 2009, 78, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Blednov, Y.A.; Stoffel, M.; Alva, H.; Harris, R.A. A Pervasive Mechanism for Analgesia: Activation of GIRK2 Channels. Proc. Natl. Acad. Sci. USA 2003, 100, 277–282. [Google Scholar] [CrossRef]
- Bony, A.R.; McArthur, J.R.; Finol-Urdaneta, R.K.; Adams, D.J. Analgesic α-conotoxins Modulate Native and Recombinant GIRK1/2 Channels via Activation of GABAB Receptors and Reduce Neuroexcitability. Br. J. Pharm. 2022, 179, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.B.; Norimatsu, Y.; McIntosh, J.M.; Elmslie, K.S. Limited Efficacy of α-Conopeptides, Vc1.1 and RgIA, To Inhibit Sensory Neuron CaV Current. ENeuro 2015, 2, ENEURO.0057-14.2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napier, I.A.; Klimis, H.; Rycroft, B.K.; Jin, A.H.; Alewood, P.F.; Motin, L.; Adams, D.J.; Christie, M.J. Intrathecal α-Conotoxins Vc1.1, AuIB and MII Acting on Distinct Nicotinic Receptor Subtypes Reverse Signs of Neuropathic Pain. Neuropharmacology 2012, 62, 2202–2207. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.B.; Hone, A.J.; Roux, I.; Kniazeff, J.; Pin, J.-P.; Upert, G.; Servent, D.; Glowatzki, E.; McIntosh, J.M. RgIA4 Potently Blocks Mouse α9α10 nAChRs and Provides Long Lasting Protection against Oxaliplatin-Induced Cold Allodynia. Front. Cell. Neurosci. 2017, 11, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, P.N.; Giuvelis, D.; Christensen, S.; Tucker, K.L.; McIntosh, J.M. RgIA4 Accelerates Recovery from Paclitaxel-Induced Neuropathic Pain in Rats. Mar. Drugs 2019, 18, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, S.; Christie, M.J. α9-Nicotinic Acetylcholine Receptors Contribute to the Maintenance of Chronic Mechanical Hyperalgesia, but Not Thermal or Mechanical Allodynia. Mol. Pain 2014, 10, 1744–8069. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Christensen, S.B.; Dowell, C.; Purushottam, L.; Skalicky, J.J.; McIntosh, J.M.; Chou, D.H.-C. Discovery of Methylene Thioacetal-Incorporated α-RgIA Analogues as Potent and Stable Antagonists of the Human α9α10 Nicotinic Acetylcholine Receptor for the Treatment of Neuropathic Pain. J. Med. Chem. 2021, 64, 9513–9524. [Google Scholar] [CrossRef]
- Lee, C.-H.; Huang, C.-S.; Chen, C.-S.; Tu, S.-H.; Wang, Y.-J.; Chang, Y.-J.; Tam, K.-W.; Wei, P.-L.; Cheng, T.-C.; Chu, J.-S.; et al. Overexpression and Activation of the α9-Nicotinic Receptor During Tumorigenesis in Human Breast Epithelial Cells. J. Natl. Cancer Inst. 2010, 102, 1322–1335. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Qian, J.; Sun, Z.; Zhangsun, D.; Luo, S. Cervical Cancer Correlates with the Differential Expression of Nicotinic Acetylcholine Receptors and Reveals Therapeutic Targets. Mar. Drugs 2019, 17, 256. [Google Scholar] [CrossRef] [Green Version]
- Mucchietto, V.; Fasoli, F.; Pucci, S.; Moretti, M.; Benfante, R.; Maroli, A.; di Lascio, S.; Bolchi, C.; Pallavicini, M.; Dowell, C.; et al. α9- and α7-Containing Receptors Mediate the pro-Proliferative Effects of Nicotine in the A549 Adenocarcinoma Cell Line. Br. J. Pharm. 2018, 175, 1957–1972. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Gu, C.; Bao, W.; Bao, Y. Neuronal Acetylcholine Receptor Subunit Alpha-9 (CHRNA9) Polymorphisms Are Associated with NSCLC Risk in a Chinese Population. Med. Oncol. 2014, 31, 932. [Google Scholar] [CrossRef] [PubMed]
- Terpinskaya, T.I.; Osipov, A.V.; Balashevich, T.V.; Yanchanka, T.L.; Tamashionik, E.A.; Tsetlin, V.I.; Utkin, Y.N. Blockers of Nicotinic Acetylcholine Receptors Delay Tumor Growth and Increase Antitumor Activity of Mouse Splenocytes. Dokl. Biochem. Biophys. 2020, 491, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Osipov, A.V.; Terpinskaya, T.I.; Yanchanka, T.; Balashevich, T.; Zhmak, M.N.; Tsetlin, V.I.; Utkin, Y.N. α-Conotoxins Enhance Both the In Vivo Suppression of Ehrlich Carcinoma Growth and In Vitro Reduction in Cell Viability Elicited by Cyclooxygenase and Lipoxygenase Inhibitors. Mar. Drugs 2020, 18, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terpinskaya, T.I.; Osipov, A.V.; Kryukova, E.V.; Kudryavtsev, D.S.; Kopylova, N.V.; Yanchanka, T.L.; Palukoshka, A.F.; Gondarenko, E.A.; Zhmak, M.N.; Tsetlin, V.I.; et al. α-Conotoxins and α-Cobratoxin Promote, While Lipoxygenase and Cyclooxygenase Inhibitors Suppress the Proliferation of Glioma C6 Cells. Mar. Drugs 2021, 19, 118. [Google Scholar] [CrossRef] [PubMed]
- Zouridakis, M.; Papakyriakou, A.; Ivanov, I.A.; Kasheverov, I.E.; Tsetlin, V.; Tzartos, S.; Giastas, P. Crystal Structure of the Monomeric Extracellular Domain of α9 Nicotinic Receptor Subunit in Complex With α-Conotoxin RgIA: Molecular Dynamics Insights Into RgIA Binding to α9α10 Nicotinic Receptors. Front. Pharm. 2019, 10, 474. [Google Scholar] [CrossRef]
- Azam, L.; McIntosh, J.M. Molecular Basis for the Differential Sensitivity of Rat and Human α9α10 nAChRs to α-Conotoxin RgIA. J. Neurochem. 2012, 122, 1137–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azam, L.; Papakyriakou, A.; Zouridakis, M.; Giastas, P.; Tzartos, S.J.; McIntosh, J.M. Molecular Interaction of α -Conotoxin RgIA with the Rat α9α10 Nicotinic Acetylcholine Receptor. Mol. Pharm. 2015, 87, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Metab. Discontinues Clin. Drug Programme Neuropathic Pain Drug. ACV1. 2007. Available online: https://www.asx.com.au/asxpdf/20070814/pdf/313yjgpf7jl4lg.pdf (accessed on 6 November 2022).
- Zheng, N.; Christensen, S.B.; Blakely, A.; Dowell, C.; Purushottam, L.; McIntosh, J.M.; Chou, D.H.C. Development of Conformationally Constrained α-RgIA Analogues as Stable Peptide Antagonists of Human α9α10 Nicotinic Acetylcholine Receptors. J. Med. Chem. 2020, 63, 8380–8387. [Google Scholar] [CrossRef]
- Chhabra, S.; Belgi, A.; Bartels, P.; van Lierop, B.J.; Robinson, S.D.; Kompella, S.N.; Hung, A.; Callaghan, B.P.; Adams, D.J.; Robinson, A.J.; et al. Dicarba Analogues of α-Conotoxin RgIA. Structure, Stability, and Activity at Potential Pain Targets. J. Med. Chem. 2014, 57, 9933–9944. [Google Scholar] [CrossRef]
- van Lierop, B.J.; Robinson, S.D.; Kompella, S.N.; Belgi, A.; McArthur, J.R.; Hung, A.; MacRaild, C.A.; Adams, D.J.; Norton, R.S.; Robinson, A.J. Dicarba α-Conotoxin Vc1.1 Analogues with Differential Selectivity for Nicotinic Acetylcholine and GABAB Receptors. ACS Chem. Biol. 2013, 8, 1815–1821. [Google Scholar] [CrossRef]
- Gajewiak, J.; Christensen, S.B.; Dowell, C.; Hararah, F.; Fisher, F.; Huynh, P.N.; Olivera, B.M.; McIntosh, J.M. Selective Penicillamine Substitution Enables Development of a Potent Analgesic Peptide That Acts through a Non-Opioid-Based Mechanism. J. Med. Chem. 2021, 64, 9271–9278. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.; Zhang, Y.; Vincent, P.F.Y.; Gajewiak, J.; Gordon, T.J.; Glowatzki, E.; Fuchs, P.A.; McIntosh, J.M. Cy3-RgIA-5727 Labels and Inhibits α9-Containing nAChRs of Cochlear Hair Cells. Front. Cell. Neurosci. 2021, 15, 697560. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhu, X.; Xu, P.; Li, R.; Fu, Y.; Dong, S.; Zhangsun, D.; Wu, Y.; Luo, S. D-Amino Acid Substitution of α-Conotoxin RgIA Identifies Its Critical Residues and Improves the Enzymatic Stability. Mar. Drugs 2019, 17, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, E.L.; Craik, C.S. Positional Scanning Synthetic Combinatorial Libraries for Substrate Profiling. Methods Mol. Biol. 2009, 539, 59–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armishaw, C.J.; Banerjee, J.; Ganno, M.L.; Reilley, K.J.; Eans, S.O.; Mizrachi, E.; Gyanda, R.; Hoot, M.R.; Houghten, R.A.; McLaughlin, J.P. Discovery of Novel Antinociceptive α-Conotoxin Analogues from the Direct In Vivo Screening of a Synthetic Mixture-Based Combinatorial Library. ACS Comb. Sci. 2013, 15, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Tae, H.-S.; Xu, X.; Jiang, T.; Adams, D.J.; Yu, R. Dimerization of α-Conotoxins as a Strategy to Enhance the Inhibition of the Human α7 and α9α10 Nicotinic Acetylcholine Receptors. J. Med. Chem. 2020, 63, 2974–2985. [Google Scholar] [CrossRef]
- Jensen, A.A.; Frølund, B.; Liljefors, T.; Krogsgaard-Larsen, P. Neuronal Nicotinic Acetylcholine Receptors: Structural Revelations, Target Identifications, and Therapeutic Inspirations. J. Med. Chem. 2005, 48, 4705–4745. [Google Scholar] [CrossRef]
- Halldén, S.; Sjögren, M.; Hedblad, B.; Engström, G.; Hamrefors, V.; Manjer, J.; Melander, O. Gene Variance in the Nicotinic Receptor Cluster (CHRNA5-CHRNA3-CHRNB4) Predicts Death from Cardiopulmonary Disease and Cancer in Smokers. J. Intern. Med. 2016, 279, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Hurst, R.; Rollema, H.; Bertrand, D. Nicotinic Acetylcholine Receptors: From Basic Science to Therapeutics. Pharmacol. Ther. 2013, 137, 22–54. [Google Scholar] [CrossRef]
- Millar, N.S.; Gotti, C. Diversity of Vertebrate Nicotinic Acetylcholine Receptors. Neuropharmacology 2009, 56, 237–246. [Google Scholar] [CrossRef]
- Muldoon, P.P.; Jackson, K.J.; Perez, E.; Harenza, J.L.; Molas, S.; Rais, B.; Anwar, H.; Zaveri, N.T.; Maldonado, R.; Maskos, U.; et al. The α3β4* Nicotinic ACh Receptor Subtype Mediates Physical Dependence to Morphine: Mouse and Human Studies. Br. J. Pharm. 2014, 171, 3845–3857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuny, H.; Yu, R.; Tae, H.; Kompella, S.N.; Adams, D.J. α-Conotoxins Active at α3-containing Nicotinic Acetylcholine Receptors and Their Molecular Determinants for Selective Inhibition. Br. J. Pharm. 2018, 175, 1855–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Vijayaraghavan, S. Nicotinic Receptor Signaling in Nonexcitable Cells. J. Neurobiol. 2002, 53, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond Neurons: The Non-Neuronal Cholinergic System in Humans. Br. J. Pharm. 2008, 154, 1558–1571. [Google Scholar] [CrossRef] [Green Version]
- Kompella, S.N.; Hung, A.; Clark, R.J.; Marí, F.; Adams, D.J. Alanine Scan of α-Conotoxin RegIIA Reveals a Selective α3β4 Nicotinic Acetylcholine Receptor Antagonist. J. Biol. Chem. 2015, 290, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Cuny, H.; Kompella, S.N.; Tae, H.-S.; Yu, R.; Adams, D.J. Key Structural Determinants in the Agonist Binding Loops of Human β2 and β4 Nicotinic Acetylcholine Receptor Subunits Contribute to α3β4 Subtype Selectivity of α-Conotoxins. J. Biol. Chem. 2016, 291, 23779–23792. [Google Scholar] [CrossRef] [Green Version]
- Kompella, S.N.; Cuny, H.; Hung, A.; Adams, D.J. Molecular Basis for Differential Sensitivity of α-Conotoxin RegIIA at Rat and Human Neuronal Nicotinic Acetylcholine Receptors. Mol. Pharm. 2015, 88, 993–1001. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Tae, H.-S.; Xue, L.; Jiang, T.; Yu, R. Mechanism of Interactions between α-Conotoxin RegIIA and Carbohydrates at the Human α3β4 Nicotinic Acetylcholine Receptor. Mar. Life Sci. Technol. 2022, 4, 98–105. [Google Scholar] [CrossRef]
- Xu, Q.; Tae, H.-S.; Wang, Z.; Jiang, T.; Adams, D.J.; Yu, R. Rational Design of α-Conotoxin RegIIA Analogues Selectively Inhibiting the Human α3β2 Nicotinic Acetylcholine Receptor through Computational Scanning. ACS Chem. Neurosci. 2020, 11, 2804–2811. [Google Scholar] [CrossRef]
- Dinklo, T.; Shaban, H.; Thuring, J.W.; Lavreysen, H.; Stevens, K.E.; Zheng, L.; Mackie, C.; Grantham, C.; Vandenberk, I.; Meulders, G.; et al. Characterization of 2-[[4-Fluoro-3-(Trifluoromethyl)Phenyl]Amino]-4-(4-Pyridinyl)-5-Thiazolemethanol (JNJ-1930942), a Novel Positive Allosteric Modulator of the α 7 Nicotinic Acetylcholine Receptor. J. Pharmacol. Exp. Ther. 2011, 336, 560–574. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, X.; Zhang, L.; Kudryavtsev, D.; Kasheverov, I.; Lei, Y.; Zhangsun, D.; Tsetlin, V.; Luo, S. Species Specificity of Rat and Human α7 Nicotinic Acetylcholine Receptors towards Different Classes of Peptide and Protein Antagonists. Neuropharmacology 2018, 139, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.B.; McDougal, O.M. The M-Superfamily of Conotoxins: A Review. Cell. Mol. Life Sci. 2010, 67, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, S.; Norton, R. Conotoxin Gene Superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Sámano, A.C.; Falcón, A.; Zamudio, F.; Michel-Morfín, J.E.; Landa-Jaime, V.; López-Vera, E.; Jeziorski, M.C.; Aguilar, M.B. A Short Framework-III (Mini-M-2) Conotoxin from the Venom of a Vermivorous Species, Conus archon, Inhibits Human Neuronal Nicotinic Acetylcholine Receptors. Peptides 2022, 153, 170785. [Google Scholar] [CrossRef]
- Haque, N.; Parveen, S.; Tang, T.; Wei, J.; Huang, Z. Marine Natural Products in Clinical Use. Mar. Drugs 2022, 20, 528. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Framework | Cysteine Pattern | Disulfide Bonds |
|---|---|---|
| I | CC-C-C | 2 |
| II | CCC-C-C-C | 3 |
| III | CC-C-C-CC | 3 |
| IV | CC-C-C-C-C | 3 |
| V | CC-CC | 2 |
| VI | C-C-CC-C-C | 3 |
| VII | C-C-CC-C-C | 3 |
| VIII | C-C-C-C-C-C-C-C-C-C | 5 |
| IX | C-C-C-C-C-C | 3 |
| X | CC-CXOC 1 | 2 |
| XI | C-C-CC-CC-C-C | 4 |
| XII | C-C-C-C-CC-C-C | 4 |
| XIII | C-C-C-CC-C-C-C | 4 |
| XIV | C-C-C-C | 4 |
| XV | C-C-CC-C-C-C-C | 4 |
| XVI | C-C-CC | 4 |
| XVII | C-C-CC-C-CC-C | 4 |
| XVIII | C-C-CC-CC | 3 |
| XIX | C-C-C-CCC-C-C-C-C | 5 |
| XX | C-CC-C-CC-C-C-C-C | 5 |
| XXI | CC-C-C-C-CC-C-C-C | 5 |
| XXII | C-C-C-C-C-C-C-C | 4 |
| XXIII | C-C-C-CC-C | 3 |
| XXIV | C-CC-C | 2 |
| XXV | C-C-C-C-CC | 3 |
| XXVI | C-C-C-C-CC-CC | 4 |
| XXVII | C-C-C-CCC-C-C | 4 |
| XXVIII | C-C-C-CC-C-C-C-C-C | 5 |
| XXIX | CCC-C-CC-C-C | 4 |
| XXX | C-C-CCC-C-C-C-CC | 5 |
| XXXII | C-CC-C-C-C | 3 |
| XXXIII | C-C-C-C-C-C-C-C-C-C-C-C | N/D |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Margiotta, F.; Micheli, L.; Ciampi, C.; Ghelardini, C.; McIntosh, J.M.; Di Cesare Mannelli, L. Conus regius-Derived Conotoxins: Novel Therapeutic Opportunities from a Marine Organism. Mar. Drugs 2022, 20, 773. https://doi.org/10.3390/md20120773
Margiotta F, Micheli L, Ciampi C, Ghelardini C, McIntosh JM, Di Cesare Mannelli L. Conus regius-Derived Conotoxins: Novel Therapeutic Opportunities from a Marine Organism. Marine Drugs. 2022; 20(12):773. https://doi.org/10.3390/md20120773
Chicago/Turabian StyleMargiotta, Francesco, Laura Micheli, Clara Ciampi, Carla Ghelardini, J. Michael McIntosh, and Lorenzo Di Cesare Mannelli. 2022. "Conus regius-Derived Conotoxins: Novel Therapeutic Opportunities from a Marine Organism" Marine Drugs 20, no. 12: 773. https://doi.org/10.3390/md20120773