
Acute Toxicity by Oral Co-Exposure to Palytoxin and Okadaic Acid in Mice

Abstract
:1. Introduction
2. Results
2.1. Mortality
2.2. Signs of Toxicity
2.3. Body Weight
2.4. Food Consumption
2.5. Gross Pathology and Relative Organs Weight
2.6. Blood Chemistry
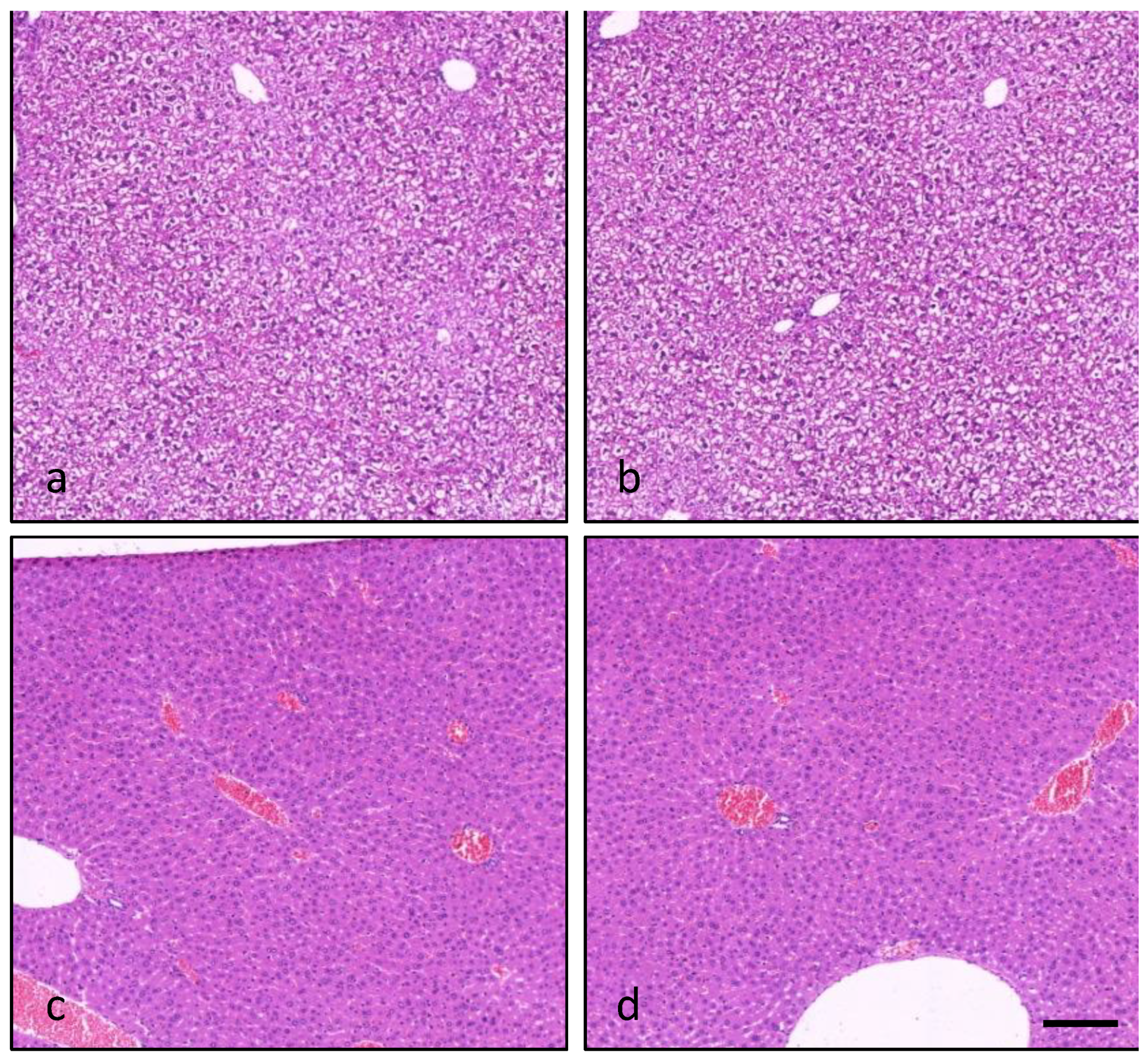
2.7. Light Microscopy
3. Discussion
4. Materials and Methods
4.1. Toxins and Chemicals
4.2. Animals and Experimental Conditions
4.3. Dose Selection and Experimental Design
4.4. Blood Chemistry Analyses
4.5. Histological Analysis
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Farabegoli, F.; Blanco, L.; Rodríguez, L.P.; Vieites, J.M.; García Cabado, A. Phycotoxins in marine shellfish: Origin, occurrence and effects on humans. Mar. Drugs 2018, 16, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdalet, E.; Fleming, L.; Gowen, R.; Davidson, K.; Hess, P.; Backer, L.C.; Moore, S.K.; Hoagland, P.; Enevoldsen, H. Marine harmful algal blooms, human health and wellbeing: Challenges and opportunities in the 21st century. J. Mar. Biol. Assoc. UK 2016, 96, 61–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcan, J.; Biré, R.; Le Hégarat, L.; Fessard, V. Mixtures of lipophilic phycotoxins: Exposure data and toxicological assessment. Mar. Drugs 2018, 16, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EFSA (European Food Safety Authority). Scientific Opinion of the Panel on Contaminants in the Food Chain (CONTAM) on a Request from the European Commission on Marine Biotoxins in Shellfish—Summary on regulated marine biotoxins. EFSA J. 2009, 7, 1306. [Google Scholar]
- Ciminiello, P.; Dell’Aversano, C.; Fattorusso, E.; Forino, M.; Magno, S.; Santelia, F.; Tsoukatou, M. Investigation of the toxin profile of Greek mussels Mytilus galloprovincialis by liquid chromatography-mass spectrometry. Toxicon 2006, 47, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Kacem, I.; Hajjem, B.; Bouaïcha, N. First evidence of okadaic acid in Mytilus galloprovincialis mussels, collected in a Mediterranean Lagoon, Tunisia. Bull. Environ. Contam. Toxicol. 2009, 82, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Nincevic Gladan, Z.; Ujevic, I.; Milandri, A.; Marasovic, I.; Ceredi, A.; Pigozzi, S.; Arapov, J.; Skejic, S. Lipophilic toxin profile in Mytilus galloprovincialis during episodes of diarrhetic shellfish poisoning (DSP) in the N.E. Adriatic Sea in 2006. Molecules 2011, 16, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Valdiglesias, V.; Prego-Faraldo, M.V.; Pásaro, E.; Méndez, J.; Laffon, B. Okadaic acid: More than a diarrheic toxin. Mar. Drugs 2013, 11, 4328–4349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacchiocchi, S.; Siracusa, M.; Ruzzi, A.; Gorbi, S.; Ercolessi, M.; Cosentino, M.A.; Ammazzalorso, P.; Orletti, R. Two-year study of lipophilic marine toxin profile in mussels of the North-central Adriatic Sea: First report of azaspiracids in Mediterranean seafood. Toxicon 2015, 108, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, A.M.; Mudadu, A.G.; Lorenzoni, G.; Soro, B.; Bardino, N.; Arras, I.; Sanna, G.; Vodret, B.; Bazzardi, R.; Marongiu, E.; et al. Detection of Dinophysis species and associated okadaic acid in farmed shellfish: A two-year study from the Western Mediterranean Area. J. Vet. Res. 2018, 62, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, R.; Mamán, L.; Jaén, D.; Fernández Fuentes, L.; Ocaña, M.A.; Gordillo, M.M. Dinophysis species and diarrhetic shellfish toxins: 20 years of monitoring program in Andalusia, south of Spain. Toxins 2019, 11, 189. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, T.; Murata, M. Marine toxins. Chem. Rev. 1993, 93, 1897–1909. [Google Scholar] [CrossRef]
- Bialojan, C.; Takai, A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J. 1988, 256, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.F.; Holmes, C.F. Molecular mechanisms underlying inhibition of protein phosphatases by marine toxins. Front. Biosci. 1999, 4, D646–D658. [Google Scholar] [CrossRef] [Green Version]
- Haystead, T.; Sim, A.; Carling, D.; Honnor, R.C.; Tsukitani, Y.; Cohen, P.; Hardie, D.G. Effects of the tumour promoter okadaic acid on intracellular protein phosphorylation and metabolism. Nature 1989, 337, 78–81. [Google Scholar] [CrossRef]
- Cohen, P.; Holmes, C.F.; Tsukitani, Y. Okadaic acid: A new probe for the study of cellular regulation. Trends Biochem. Sci. 1990, 15, 98–102. [Google Scholar] [CrossRef]
- Tripuraneni, J.; Koutsouris, A.; Pestic, L.; De Lanerolle, P.; Hecht, G. The toxin of diarrheic shellfish poisoning, okadaic acid, increases intestinal epithelial paracellular permeability. Gastroenterology 1997, 112, 100–108. [Google Scholar] [CrossRef]
- Louzao, M.C.; Vieytes, M.R.; Botana, L.M. Effect of okadaic acid on glucose regulation. Mini Rev. Med. Chem. 2005, 5, 207–215. [Google Scholar] [CrossRef]
- Munday, R. Is protein phosphatase inhibition responsible for the toxic effects of okadaic acid in animals? Toxins 2013, 5, 267–285. [Google Scholar] [CrossRef] [Green Version]
- Louzao, M.C.; Costas, C.; Abal, P.; Suzuki, T.; Watanabe, R.; Vilariño, N.; Carrera, C.; Boente-Juncal, A.; Vale, C.; Vieytes, M.R.; et al. Serotonin involvement in okadaic acid-induced diarrhoea in vivo. Arch. Toxicol. 2021, 95, 2797–2813. [Google Scholar] [CrossRef]
- Tubaro, A.; Sosa, S.; Carbonatto, M.; Altinier, G.; Vita, F.; Melato, M.; Satake, M.; Yasumoto, T. Oral and intraperitoneal acute toxicity studies of yessotoxin and homoyessotoxins in mice. Toxicon 2003, 41, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Aune, T.; Espenes, A.; Aasen, J.A.; Quilliam, M.A.; Hess, P.; Larsen, S. Study of possible combined toxic effects of azaspiracid-1 and okadaic acid in mice via the oral route. Toxicon 2012, 60, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Abal, P.; Louzao, M.C.; Suzuki, T.; Watanabe, R.; Vilariño, N.; Carrera, C.; Botana, A.M.; Vieytes, M.R.; Botana, L.M. Toxic action reevaluation of okadaic acid, dinophysistoxin-1 and dinophysistoxin-2: Toxicity equivalency factors based on the oral toxicity study. Cell Physiol. Biochem. 2018, 49, 743–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terao, K.; Ito, E.; Ohkusu, M.; Yasumoto, T. A comparative study of the effects of DSP-toxins on mice and rats. In Toxic Phytoplankton Blooms in the Sea; Smayda, T.J., Shimizu, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 1993; pp. 581–586. [Google Scholar]
- Ito, E.; Terao, K. Injury and recovery process of intestine caused by okadaic acid and related compounds. Nat. Toxins 1994, 2, 371–377. [Google Scholar] [PubMed]
- Yuasa, H.; Yoshida, K.; Iwata, H.; Nakanishi, H.; Suganuma, M.; Tatematsu, M. Increase of labeling indices in gastrointestinal mucosae of mice and rats by compounds of the okadaic acid type. J. Cancer Res. Clin. Oncol. 1994, 120, 208–212. [Google Scholar] [CrossRef]
- Aune, T.; Stabell, O.B.; Nordstoga, K.; Tjøtta, K. Oral toxicity in mice of algal toxins from the diarrheic shellfish toxin (DST) complex and associated toxins. J. Nat. Toxins 1998, 7, 141–158. [Google Scholar]
- Ito, E.; Satake, M.; Ofuji, K.; Kurita, N.; McMahon, T.; James, K.; Yasumoto, T. Multiple organ damage caused by a new toxin azaspiracid, isolated from mussels produced in Ireland. Toxicon 2000, 38, 917–930. [Google Scholar] [CrossRef]
- Berven, G.; Sætre, F.; Halvorsen, K.; Seglen, P.O. Effects of the diarrhetic shellfish toxin, okadaic acid, on cytoskeletal elements, viability and functionality of rat liver and intestinal cells. Toxicon 2001, 39, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Ito, E.; Yasumoto, T.; Akira, T.; Imanishi, S.; Harada, K. Investigation of the distribution and excretion of okadaic acid in mice using immunostaining method. Toxicon 2002, 40, 159–165. [Google Scholar] [CrossRef]
- Le Hégarat, L.; Jacquin, A.-G.; Bazin, E.; Fessard, V. Genotoxicity of the marine toxin okadaic acid, in human Caco-2 cells and in mice gut cells. Environ. Toxicol. 2006, 21, 55–64. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.-Y.; Lin, L.; Gao, Y.; Hong, H.-S.; Wang, D.-Z. Quantitative proteomic analysis of okadaic acid treated mouse small intestines reveals differentially expressed proteins involved in diarrhetic shellfish poisoning. J. Proteom. 2012, 75, 2038–2052. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.C.; Rubiolo, J.A.; López-Alonso, H.; Cifuentes, J.M.; Alfonso, A.; Bermúdez, R.; Otero, P.; Vieytes, M.R.; Vega, F.V.; Botana, L.M. Oral toxicity of okadaic acid in mice: Study of lethality, organ damage, distribution and effects on detoxifying gene expression. Toxins 2013, 5, 2093–2108. [Google Scholar] [CrossRef] [PubMed]
- Tubaro, A.; Sosa, S.; Altinier, G.; Soranzo, M.R.; Satake, M.; Della Loggia, R.; Yasumoto, T. Short-term oral toxicity of homoyessotoxins, yessotoxin and okadaic acid in mice. Toxicon 2004, 43, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Sosa, S.; Ardizzone, M.; Beltramo, D.; Vita, F.; Dell’Ovo, V.; Barreras, A.; Yasumoto, T.; Tubaro, A. Repeated oral co-exposure to yessotoxin and okadaic acid: A short term toxicity study in mice. Toxicon 2013, 76, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Ciminiello, P.; Dell’Aversano, C.; Fattorusso, E.; Forino, M.; Tartaglione, L.; Grillo, C.; Melchiorre, N. Putative palytoxin and its new analogue, ovatoxin-a, in Ostreopsis ovata collected along the Ligurian coasts during the 2006 toxic outbreak. J. Am. Soc. Mass Spectrom. 2008, 19, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciminiello, P.; Dell’Aversano, C.; Dello Iacovo, E.; Fattorusso, E.; Forino, M.; Tartaglione, L.; Benedettini, G.; Onorari, M.; Serena, F.; Battocchi, C.; et al. First finding of Ostreopsis cf. ovata toxins in marine aerosols. Environ. Sci. Technol. 2014, 48, 3532–3540. [Google Scholar] [CrossRef]
- Honsell, G.; De Bortoli, M.; Boscolo, S.; Dell’Aversano, C.; Battocchi, C.; Fontanive, G.; Penna, A.; Berti, F.; Sosa, S.; Yasumoto, T.; et al. Harmful dinoflagellate Ostreopsis cf. ovata Fukuyo: Detection of ovatoxins in field samples and cell immunolocalization using antipalytoxin antibodies. Environ. Sci. Technol. 2011, 45, 7051–7059. [Google Scholar] [CrossRef]
- Mangialajo, L.; Ganzin, N.; Accoroni, S.; Asnaghi, V.; Blanfuné, A.; Cabrini, M.; Cattaneo-Vietti, R.; Chavanon, F.; Chiantore, M.; Cohu, S.; et al. Trends in Ostreopsis proliferation along the Northern Mediterranean coasts. Toxicon 2011, 57, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Pfannkuchen, M.; Godrijan, J.; Marić Pfannkuchen, D.; Ivesa, L.; Kruzić, P.; Ciminiello, P.; Dell’Aversano, C.; Dello Iacovo, E.; Fattorusso, E.; Forino, M.; et al. Toxin-producing Ostreopsis cf. ovata are likely to bloom undetected along coastal areas. Environ. Sci. Technol. 2012, 46, 5574–5582. [Google Scholar] [CrossRef]
- Accoroni, S.; Totti, C. The toxic benthic dinoflagellates of the genus Ostreopsis in temperate areas: A review. Adv. Oceanogr. Limnol. 2016, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Tartaglione, L.; Dello Iacovo, E.; Mazzeo, A.; Casabianca, S.; Ciminiello, P.; Penna, A.; Dell’Aversano, C. Variability in toxin profiles of the Mediterranean Ostreopsis cf. ovata and in structural features of the produced ovatoxins. Environ. Sci. Technol. 2017, 51, 13920–13928. [Google Scholar] [CrossRef] [PubMed]
- Ninčević Gladan, Ž.; Arapov, J.; Casabianca, S.; Penna, A.; Honsell, G.; Brovedani, V.; Pelin, M.; Tartaglione, L.; Sosa, S.; Dell’Aversano, C.; et al. Massive occurrence of the harmful benthic dinoflagellate Ostreopsis cf. ovata in the Eastern Adriatic Sea. Toxins 2019, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Marampouti, C.; Buma, A.G.J.; de Boer, M.K. Mediterranean alien harmful algal blooms: Origins and impacts. Environ. Sci. Pollut. Res. 2021, 28, 3837–3851. [Google Scholar] [CrossRef] [PubMed]
- Durando, P.; Ansaldi, F.; Oreste, P.; Moscatelli, P.; Marensi, L.; Grillo, C.; Gasparini, R.; Icardi, G. Ostreopsis ovata and human health: Epidemiological and clinical features of respiratory syndrome outbreaks from a two-year syndromic surveillance, 2005–2006, in north-west Italy. Eurosurveillance 2007, 12, E070607.1. [Google Scholar]
- Tichadou, L.; Glaizal, M.; Armengaud, A.; Grossel, H.; Lemée, R.; Kantin, R.; Lasalle, J.L.; Drouet, G.; Rambaud, L.; Malfait, P.; et al. Health impact of unicellular algae of the Ostreopsis genus blooms in the Mediterranean Sea: Experience of the French Mediterranean coast surveillance network from 2006 to 2009. Clin. Toxicol. 2010, 48, 839–844. [Google Scholar] [CrossRef] [Green Version]
- Del Favero, G.; Sosa, S.; Pelin, M.; D’Orlando, E.; Florio, C.; Lorenzon, P.; Poli, M.; Tubaro, A. Sanitary problems related to the presence of Ostreopsis spp. in the Mediterranean Sea: A multidisciplinary scientific approach. Ann. Dell’istituto Super. Sanità 2012, 48, 407–414. [Google Scholar] [CrossRef]
- Tubaro, A.; Durando, P.; Del Favero, G.; Ansaldi, F.; Icardi, G.; Deeds, J.R.; Sosa, S. Case definitions for human poisonings postulated to palytoxins exposure. Toxicon 2011, 57, 478–495. [Google Scholar] [CrossRef]
- Alcala, A.C.; Alcala, L.C.; Garth, J.S.; Yasumura, D.; Yasumoto, T. Human fatality due to ingestion of the crab Demania reynaudii that contained a palytoxin-like toxin. Toxicon 1988, 26, 105–107. [Google Scholar] [CrossRef]
- Noguchi, T.; Hwang, D.F.; Arakawa, O.; Daigo, K.; Sato, S.; Ozaki, H.; Kawai, N.; Ito, M.; Hashimoto, K. Palytoxin as the causative agent in the parrotfish poisoning. In Progress in Venom and Toxin Research, Proceedings of the First Asia–Pacific Congress on Animal, Plant and Microbial Toxins, Singapore, 24–27 June 1987; Gopalakrishnakone, P., Tan, C.K., Eds.; Faculty of Medicine, National University of Singapore: Singapore, 1987; pp. 325–335. [Google Scholar]
- Onuma, Y.; Satake, M.; Ukena, T.; Roux, J.; Chanteau, S.; Rasolofonirina, N.; Ratsimaloto, M.; Naoki, H.; Yasumoto, T. Identification of putative palytoxin as the cause of clupeotoxism. Toxicon 1999, 37, 55–65. [Google Scholar] [CrossRef]
- Taniyama, S.; Mahmud, Y.; Terada, M.; Takatani, T.; Arakawa, O.; Noguchi, T. Occurrence of a food poisoning incident by palytoxin from a serranid Epinephelus sp. in Japan. J. Nat. Toxins 2002, 11, 277–282. [Google Scholar]
- Wu, M.L.; Yang, C.C.; Deng, J.F.; Wang, K.Y. Hyperkalemia, hyperphosphatemia, acute kidney injury, and fatal dysrhythmias after consumption of palytoxin-contaminated goldspot herring. Ann. Emerg. Med. 2014, 64, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Habermann, E. Palytoxin acts through Na+, K+-ATPase. Toxicon 1989, 27, 1171–1187. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Marx, K.A.; Wu, C.H. Involvement of the Na,K-ATPase in the induction of ion channels by palytoxin. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1995, 351, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Rossini, G.P.; Bigiani, A. Palytoxin action on the Na+,K+-ATPase and the disruption of ion equilibria in biological systems. Toxicon 2011, 57, 429–439. [Google Scholar] [CrossRef]
- Wu, C.H. Palytoxin: Membrane mechanisms of action. Toxicon 2009, 54, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Munday, R. Occurrence and toxicology of palytoxin. In Seafood and Freshwater Toxins. Pharmacology, Physiology and Detection; Botana, L.M., Ed.; CRC Press: Boca Raton, FL, USA, 2008; pp. 693–713. [Google Scholar]
- Sosa, S.; Del Favero, G.; De Bortoli, M.; Vita, F.; Soranzo, M.R.; Beltramo, D.; Ardizzone, M.; Tubaro, A. Palytoxin toxicity after acute oral administration in mice. Toxicol. Lett. 2009, 191, 253–259. [Google Scholar] [CrossRef]
- Boente-Juncal, A.; Vale, C.; Camiña, M.; Cifuentes, J.M.; Vieytes, M.R.; Botana, L.M. Reevaluation of the acute toxicity of palytoxin in mice: Determination of lethal dose 50 (LD50) and No-observed-adverse-effect level (NOAEL). Toxicon 2020, 177, 16–24. [Google Scholar] [CrossRef]
- Del Favero, G.; Beltramo, D.; Sciancalepore, M.; Lorenzon, P.; Coslovich, T.; Poli, M.; Testai, E.; Sosa, S.; Tubaro, A. Toxicity of palytoxin after repeated oral exposure in mice and in vitro effects on cardiomyocytes. Toxicon 2013, 75, 3–15. [Google Scholar] [CrossRef]
- Boente-Juncal, A.; Raposo-García, S.; Vale, C.; Louzao, M.C.; Otero, P.; Botana, L.M. In vivo evaluation of the chronic oral toxicity of the marine toxin palytoxin. Toxins 2020, 12, 489. [Google Scholar] [CrossRef]
- European Commission. Regulation (EC) No 853/2004 of the European Parliament and of the Council of 29 April 2004 Laying down Specific Hygiene Rules for Food of Animal Origin; 25.6.2004: L 226/22; Publications Office of the European Union: Luxembourg, 2004. [Google Scholar]
- EFSA (European Food Safety Authority) Panel on Contaminants in the Food Chain (CONTAM). Scientific opinion on marine biotoxins in shellfish—Palytoxin group. EFSA J. 2009, 7, 1393. [Google Scholar] [CrossRef]
- Aligizaki, K.; Katikou, P.; Nikolaidis, G.; Panou, A. First episode of shellfish contamination by palytoxin-like compounds from Ostreopsis species (Aegean Sea, Greece). Toxicon 2008, 51, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Aligizaki, K.; Katikou, P.; Milandri, A.; Diogène, J. Occurrence of palytoxin-group toxins in seafood and future strategies to complement the present state of the art. Toxicon 2011, 57, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Amzil, Z.; Sibat, M.; Chomerat, N.; Grossel, H.; Marco-Miralles, F.; Lemee, R.; Nezan, E.; Sechet, V. Ovatoxin-a and palytoxin accumulation in seafood in relation to Ostreopsis cf. ovata blooms on the French Mediterranean coast. Mar. Drugs 2012, 10, 477–496. [Google Scholar] [CrossRef] [Green Version]
- Biré, R.; Trotereau, S.; Lemée, R.; Delpont, C.; Chabot, B.; Aumond, Y.; Krys, S. Occurrence of palytoxins in marine organisms from different trophic levels of the French Mediterranean coast harvested in 2009. Harmful Algae 2013, 28, 10–12. [Google Scholar] [CrossRef]
- Brissard, C.; Herrenknecht, C.; Séchet, V.; Hervé, F.; Pisapia, F.; Harcouet, J.; Lémée, R.; Chomérat, N.; Hess, P.; Amzil, Z. Complex toxin profile of French Mediterranean Ostreopsis cf. ovata strains, seafood accumulation and ovatoxins prepurification. Mar. Drugs 2014, 12, 2851–2876. [Google Scholar] [CrossRef] [Green Version]
- Ciminiello, P.; Dell’Aversano, C.; Dello Iacovo, E.; Forino, M.; Tartaglione, L. Liquid chromatography-high-resolution mass spectrometry for palytoxins in mussels. Anal. Bioanal. Chem. 2015, 407, 1463–1473. [Google Scholar] [CrossRef]
- Pelin, M.; Stocco, G.; Florio, C.; Sosa, S.; Tubaro, A. In vitro cell sensitivity to palytoxin correlates with high gene expression of the Na+/K+-ATPase β2 subunit isoform. Int. J. Mol. Sci. 2020, 21, 5833. [Google Scholar] [CrossRef]
- Tubaro, A.; Del Favero, G.; Beltramo, D.; Ardizzone, M.; Forino, M.; De Bortoli, M.; Pelin, M.; Poli, M.; Bignami, G.; Ciminiello, P.; et al. Acute oral toxicity in mice of a new palytoxin analog: 42-hydroxy-palytoxin. Toxicon 2011, 57, 755–763. [Google Scholar] [CrossRef]
- Ito, E.; Yasumoto, T. Toxicological studies on palytoxin and ostreocin-D administered to mice by three different routes. Toxicon 2009, 54, 244–251. [Google Scholar] [CrossRef]
- Serfilippi, L.M.; Stackhouse Pallman, D.R.; Russell, B. Serum clinical chemistry and hematology reference values in outbred stocks of albino mice from three commonly used vendors and two inbred strains of albino mice. J. Am. Assoc. Lab. Anim. Sci. 2003, 42, 46–52. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLTX Dose (μg/kg) | OA Dose (μg/kg) | Lethality at 24 h 1 (Survival Time, h:min) | Lethality during 14-Day Recovery 1 (D: Day of Death) |
|---|---|---|---|
| 0 | 0 | 0/16 (--) | 0/6 (--) |
| 0 | 370 | 0/16 (--) | 0/6 (--) |
| 30 | 0 | 0/8 (--) | 0/3 (--) |
| 90 | 0 | 2/8 (04:52–06:50) | 1/3 (D8) |
| 270 | 0 | 2/8 (01:48–04:47) | 0/3 (--) |
| 30 | 370 | 0/8 (--) | 0/3 (--) |
| 90 | 370 | 2/8 (00:39–02:30) | 0/3 (--) |
| 270 | 370 | 2/8 (03:46–06:49) | 1/3 (D5) |
| Sign | Controls | OA 370 1 | PLTX 30 1 | PLTX 90 1 | PLTX 270 1 | PLTX+OA 30+370 1 | PLTX+OA 90+370 1 | PLTX+OA 270+370 1 |
|---|---|---|---|---|---|---|---|---|
| Diarrhea | 0/16 (--) | 4/16 (01:12–05:25) | 0/8 (--) | 0/8 (--) | 0/8 (--) | 2/8 (01:06–05:30) | 3/8 (01:00–06:10) | 3/8 (00:55–06:20) |
| Scratching | 0/16 (--) | 0/16 (--) | 0/8 (--) | 1/8 (00:30–02:06) | 1/8 (00:24–02:18) | 3/8 (00:30–02:18) | 1/8 (0:24–02:.25) | 1/8 (00:20–02:35) |
| Piloerection | 0/16 (--) | 0/16 (--) | 0/8 (--) | 2/8 (01:30–05.48) | 2/8 (00.35–03:12) | 1/8 (01:48–24:00) | 2/8 (00:20–01:35) | 3/8 (00:18–11:30) |
| Righting reflex loss | 0/16 (--) | 0/16 (--) | 0/8 (--) | 0/8 (--) | 0/8 (--) | 0/8 (--) | 2/8 (00:25–01:35) | 2/8 (00:30–05:15) |
| Sedation | 0/16 (--) | 0/16 (--) | 0/8 (--) | 2/8 (03:00–05:48) | 2/8 (01:00–03:18) | 0/8 (--) | 2/8 (00:15–01:36) | 2/8 (00:30–05:15) |
| Tremors | 0/16 (--) | 0/16 (--) | 0/8 (--) | 0/8 (--) | 2/8 (01:30–03:12) | 0/8 (--) | 0/8 (--) | 2/8 (01:18–05:15) |
| Jumping | 0/16 (--) | 0/16 (--) | 0/8 (--) | 0/8 (--) | 2/8 (01:35–03:00) | 0/8 (--) | 0/8 (--) | 0/8 (--) |
| Paralysis | 0/16 (--) | 0/16 (--) | 0/8 (--) | 0/8 (--) | 3/8 (01:42–10:12) | 0/8 (--) | 2/8 (00:42–01:35) | 4/8 (00:48–11:30) |
| Dyspnea | 0/16 (--) | 0/16 (--) | 0/8 (--) | 0/8 (--) | 2/8 (01:42–03:12) | 0/8 (--) | 2/8 (01:12–01:36) | 2/8 (01:18–05:15) |
| Muscular spasms | 0/16 (--) | 0/16 (--) | 0/8 (--) | 2/8 (04:48–05:48) | 2/8 (01:30–03:12) | 0/8 (--) | 2/8 (00:24–01:35) | 2/8 (00:24–05.12) |
| Abdomen dilation | 0/16 (--) | 0/16 (--) | 0/8 (--) | 1/8 (03:00–06:48) | 1/8 (01:30–04:42) | 0/8 (--) | 1/8 (02:30–24:00) | 2/8 (01:24–15:25) |
| Alteration | Controls | OA 370 1 | PLTX 30 1 | PLTX 90 1 | PLTX 270 1 | PLTX+OA 30+370 1 | PLTX+OA 90+370 1 | PLTX+OA 270+370 1 |
|---|---|---|---|---|---|---|---|---|
| Intestinal redness | 0/10 | 0/10 | 0/5 | 1/5 | 1/5 | 3/5 * | 5/5 *,§ | 5/5 *,§ |
| Intestinal fluid | 0/10 | 6/10 * | 0/5 | 0/5 | 0/5 | 4/5 *,§ | 5/5 *,§ | 5/5 *,§ |
| Gastric redness | 0/10 | 0/10 | 0/5 | 0/5 | 0/5 | 0/5 | 5/5 *,§ | 5/5 *,§ |
| Group | PLTX Dose (μg/kg) | OA Dose (μg/kg) | N° of Treated Animals | N° of Sacrificed Animals (24 h) | N° of Sacrificed Animals (14 Days) |
|---|---|---|---|---|---|
| Experiment 1 | |||||
| 1 | 0 | 0 | 8 | 5 | 3 |
| 2 | 0 | 370 | 8 | 5 | 3 |
| 3 | 30 | 0 | 8 | 5 | 3 |
| 4 | 30 | 370 | 8 | 5 | 3 |
| Experiment 2 | |||||
| 1 | 0 | 0 | 8 | 5 | 3 |
| 2 | 0 | 370 | 8 | 5 | 3 |
| 3 | 90 | 0 | 8 | 5 | 3 |
| 4 | 270 | 0 | 8 | 5 | 3 |
| 5 | 90 | 370 | 8 | 5 | 3 |
| 6 | 270 | 370 | 8 | 5 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sosa, S.; Pelin, M.; Ponti, C.; Carlin, M.; Tubaro, A. Acute Toxicity by Oral Co-Exposure to Palytoxin and Okadaic Acid in Mice. Mar. Drugs 2022, 20, 735. https://doi.org/10.3390/md20120735
Sosa S, Pelin M, Ponti C, Carlin M, Tubaro A. Acute Toxicity by Oral Co-Exposure to Palytoxin and Okadaic Acid in Mice. Marine Drugs. 2022; 20(12):735. https://doi.org/10.3390/md20120735
Chicago/Turabian StyleSosa, Silvio, Marco Pelin, Cristina Ponti, Michela Carlin, and Aurelia Tubaro. 2022. "Acute Toxicity by Oral Co-Exposure to Palytoxin and Okadaic Acid in Mice" Marine Drugs 20, no. 12: 735. https://doi.org/10.3390/md20120735