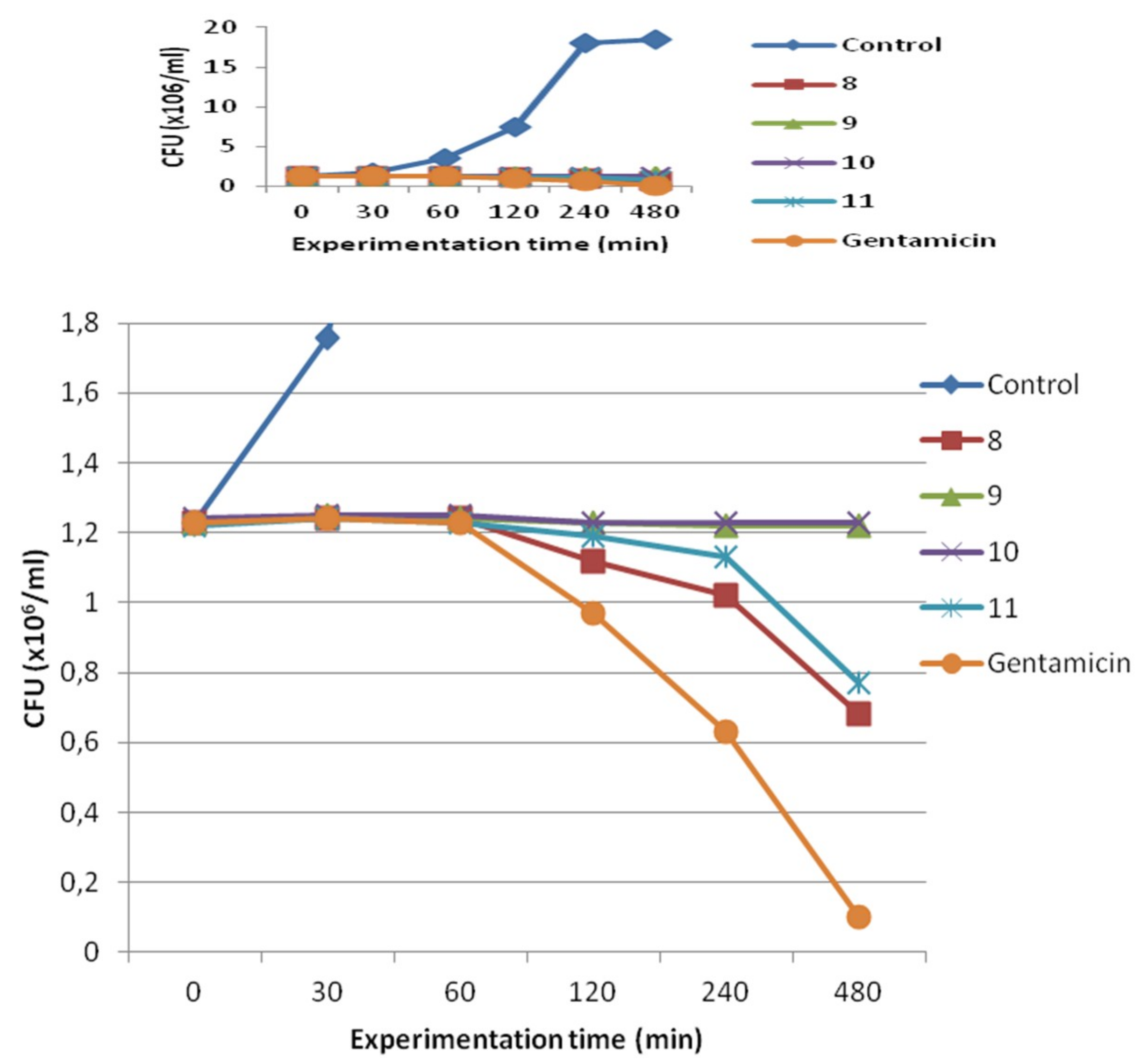
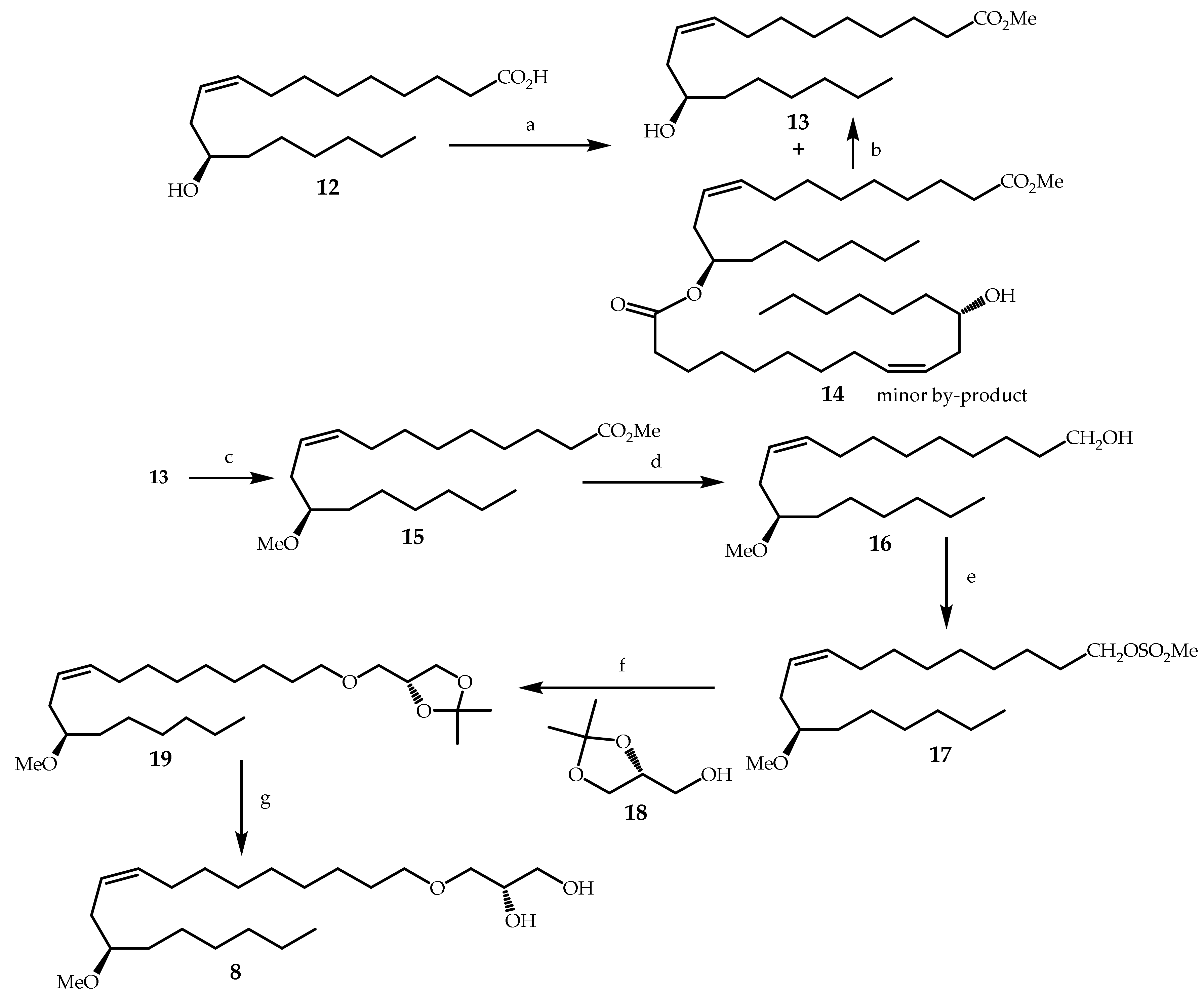
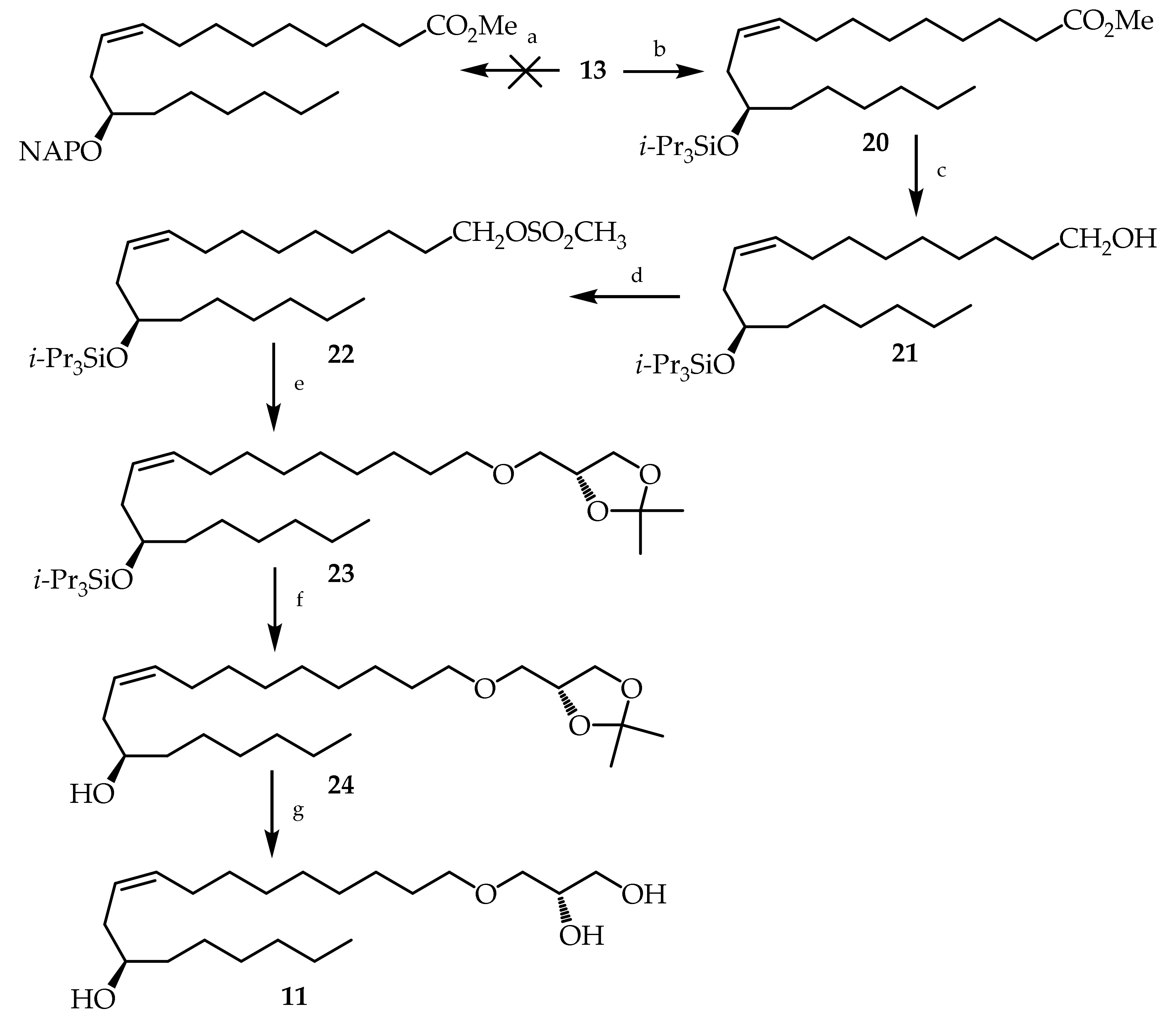
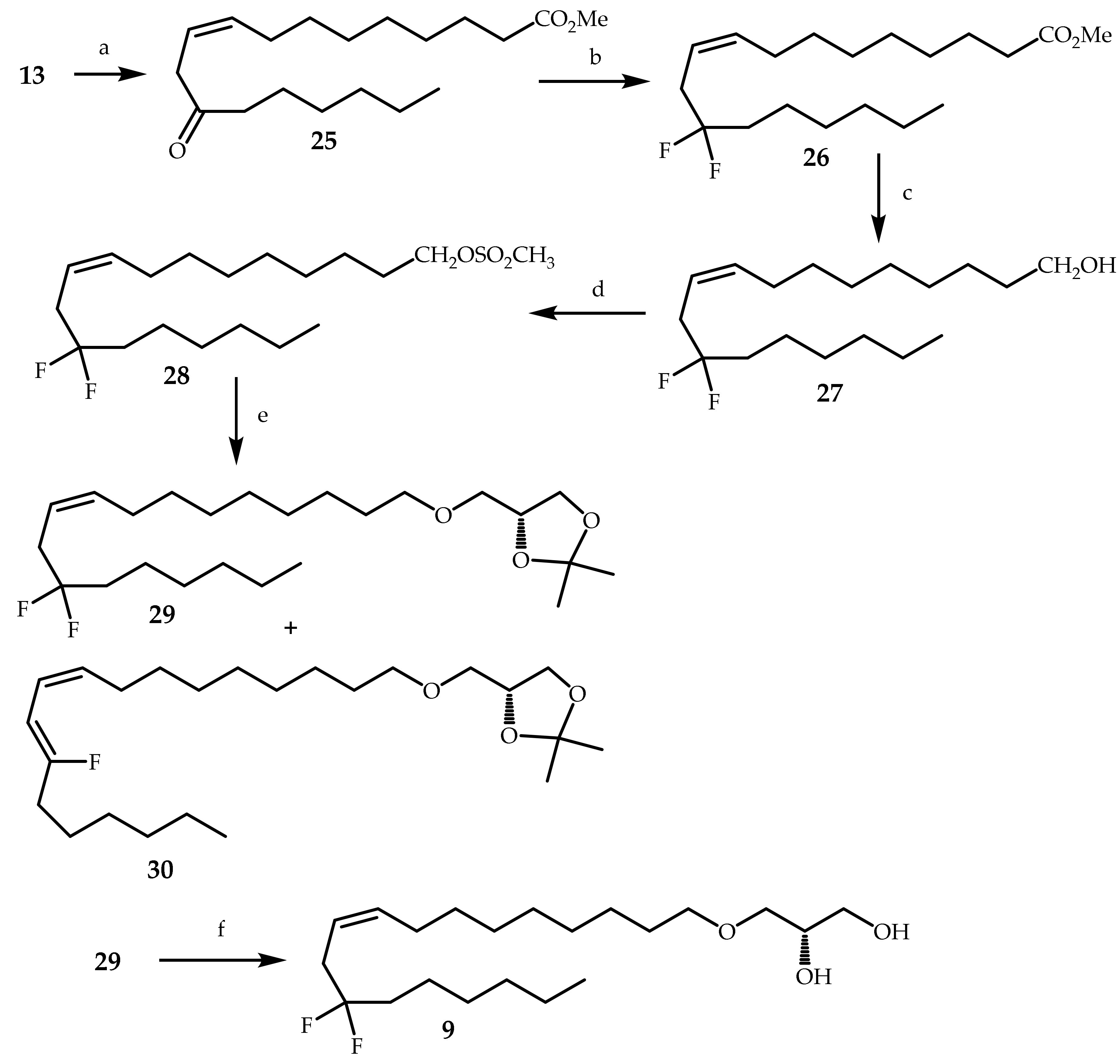
3.2. Synthesis of Compounds 8–11,13–17,19–29 and 31–36
3.2.1. (R,Z)-Methyl 12-hydroxyoctadec-9-enoate (13) (Methyl ricinoleate)
To a solution of ricinoleic acid (12) (21 g, technical, ~80% pure, ca. 56 mmol) in methanol (140 mL) with stirring, was added BF3.2MeOH (3.85 mL, 35.5 mmol, 0.63 equiv). Stirring was continued overnight at 50 °C. TLC monitoring showed completion of the reaction mixture after 16 h. Methanol was removed in vacuo, and the resulting oily residue was transferred into a separating funnel with ethyl acetate (100 mL). After washing with brine (3 × 30 mL) and drying over Na2SO4, ethyl acetate was removed under reduced pressure, and the crude product was purified by column chromatography on silica gel (86 g, 0–4% acetone in petroleum ether) to afford methyl ricinoleate 13 as a colorless oil (16.59 g, 75%) along with a small amount (4%) of a slightly less polar by-product 14. It eluted after a minor amount of a less polar mixture of methyl oleate and methyl linoleate, Rf ca. 0.75 (petroleum ether/acetone 80:20), which was the result of the esterification of the ~15% oleic + linoleic acids, which were contained in technical ricinoleic acid. When potassium carbonate (600 mg) was added to a solution of 13 containing 14 (3.17 g) in methanol (30 mL) with stirring for 42 h (followed by quenching with a solution of citric acid (834 mg) in water (7.5 mL)), the by-product 14 disappeared to afford compound 13 alone, Rf = 0.4 (petroleum ether/acetone 80:20).
IR (KBr) ν 3445 (broad, O–H), 3007, 2928, 2855, 1741 (C=O), 1461, 1436, 1246, 1198, 1173, 725 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.56 (dtt, 1H, J = 10.9, 7.3, 1.5 Hz, CH=CH–CH2–CHOH), 5.40 (dtt, 1H, J = 10.9, 7.5, 1.5 Hz, CH=CH–CH2–CHOH), 3.67 (s, 3H, CO2Me), 3.66–3.56 (m, 1H, CHOH), 2.30 (dd, 2H, J = 7.7, 7.4 Hz, CH2CO2Me), 2.24–2.18 (m, 2H, CHOH–CH2–CH=CH), 2.05 (br qd, 2H, J = 7.2, 1.2 Hz, CH=CH–CH2), 1.62 (br tt, 2H, J = 7.5, 7.3 Hz, CH2CH2CO2Me), 1.58 (br d, 1H, J = 0.9 Hz, OH, could be s and overlapped at another δ), 1.51–1.41 (m, 3H, CH2CHOH and 1H of CH2CH2CHOH), 1.39–1.23 (m, 15H, 1H of CH2CH2CHOH, CH3(CH2)3 and (CH2)4CH2CH2CO2Me), 0.88 (t (approximately t because not first order due to coupling to a rather close CH2 at ~1.25 ppm), 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 174.35 (Cquat, CO), 133.39 (CH=CH), 125.24 (CH=CH), 71.52 (CHOH), 51.46 (CO2CH3), 36.87 (CH2), 35.37 (CH2), 34.10 (CH2CO2Me), 31.85 (CH2CH2CH3), 29.58 (CH2), 29.37 (CH2), 29.13 (CH2), 29.10 (2 CH2), 27.38 (CH2), 25.73 (CH2), 24.93 (CH2CH2CO2Me), 22.63 (CH2CH3), 14.10 (CH3).
[α]D22: +4.2; [α]57822: +4.3; [α]54622: +4.7; [α]43622: +7.0; [α]36522: +8.7 (c 6.00, CHCl3),
[α]D22: +5.7; [α]57822: +5.9; [α]54622: +6.6; [α]43622: +10.7; [α]36522: +15.4 (c 6.00, acetone).
3.2.2. Physical data for (R,Z)-(R,Z)-18-methoxy-18-oxooctadec-9-en-7-yl 12-hydroxyoctadec-9-enoate (14)
Rf = 0.61 (petroleum ether/acetone 80:20).
IR (KBr) ν 3465 (O–H), 3010, 2928, 2855, 1737 (C=O), 1466, 1456, 1436, 1245, 1194, 1175, 725 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.56 (dtt, 1H, J = 10.9, 7.3, 1.5 Hz, CH=CH–CH2–CHOH), 5.46 (dtt, 1H, J = 10.9, 7.2, 1.5 Hz, CH=CH–CH2–CHOCO), 5.40 (dtt, 1H, J = 10.9, 7.5, 1.5 Hz, CH=CH–CH2–CHOH), 5.32 (dtt, 1H, J = 10.9, 7.3, 1.5 Hz, CH=CH–CH2–CHOCO), 4.88 (tt, 1H, J = 6.3, 6.3 Hz, CHOCO), 3.67 (s, 3H, CO2Me), 3.61 (br tt, 1H, J = 6.1, 5.7 Hz, CHOH), 2.33–2.24 (m, 6H), 2.23–2.18 (m, 2H), 2.08–1.98 (m, 4H, 2 CH=CH–CH2), 1.66–1.43 (m, ~12H, 2 CH2CH2CO2, CH2CHOCO, CH2CHOH,OH, H2O), 1.38–1.21 (m, 32H, 2 CH3(CH2)4 and 2 (CH2)4CH2CH2CO2), 0.884 (t, 3H, J = 6.9 Hz, CH3), 0.876 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 174.33 (Cquat, CO2Me), 173.59 (CO2CH), 133.39 (CH=CH), 132.53 (CH=CH), 125.22 (CH=CH), 124.35 (CH=CH), 73.70 (CHOCO), 71.51 (CHOH), 51.46 (CO2CH3), 36.87 (CH2), 35.38 (CH2), 34.68 (CH2), 34.10 (CH2CO2Me), 33.65 (CH2CO2CH), 32.00 (CH2), 31.85 (CH2CH2CH3), 31.76 (CH2), 29.62 (CH2), 29.53 (CH2), 29.36 (CH2), 29.19 (CH2), 29.18 (CH2), 29.16 (CH2), 29.14 (CH2), 29.13 (CH2), 29.12 (2 CH2), 27.40 (CH2), 27.34 (CH2), 25.73 (CH2), 25.37 (CH2), 25.10 (CH2), 24.95 (CH2CH2CO2Me), 22.63 (CH2), 22.59 (CH2), 14.10 (CH3), 14.08 (CH3).
[α]D21: +17.4; [α]57821: +18.1; [α]54621: +20.7; [α]43621: +34.9; [α]36521: +54.8 (c 2.56, CHCl3).
HRMS (ESI, m/z) calculated for C37H68O5Na [M + Na]+: 615.4964, found: 615.4971.
3.2.3. (R,Z)-Methyl 12-methoxyoctadec-9-enoate (15)
In a flask containing a solution of 13 (625 mg, 2.0 mmol), tetra-n-butylammonium bromide (709.2 mg, 2.2 mmol, 1.2 equiv) in DMSO (2.0 mL) with stirring, was added finely crushed (with a mortar and pestle) sodium hydroxide (250 mg, 6 mmol, 3 equiv) and methyl iodide (0.63 mL, 10 mmol, 5 equiv). The reaction flask was flushed under nitrogen, tightly stoppered and protected from light by wrapping with an aluminum foil. After stirring overnight for 18 h, TLC monitoring showed that the reaction was mostly done. An aqueous solution of 10% citric acid (10 mL) was added, and extraction was done with petroleum ether/EtOAc (80:20). Organic layers were dried over Na2SO4, and solvent was removed under reduced pressure. Then, the residue was purified by column chromatography on basic alumina (5 g, 0%–0.2% acetone in petroleum ether) to afford 15 as a colorless oil (471 mg, 72%). Rf = 0.6 (petroleum ether/acetone 90:10).
IR (KBr) ν 3465 (small, harmonic of C=O), 3009, 2929, 2855, 1742 (C=O), 1463, 1456, 1436, 1360, 1245, 1195, 1173, 1099 (C–O of OMe), 725 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.50–5.34 (m, 2H: CH=CH partly distorted due to strong coupling at 5.45 and 5.38 ppm (dtt, J = 10.9, 6.9, 1.4 Hz)), 3.67 (s, 3H, CO2Me), 3.34 (s, 3H, CHOCH3), 3.17 (tt, 1H, J = 6.2, 5.5 Hz, CHOMe), 2.30 (dd, 2H, J = 7.7, 7.4 Hz, CH2CO2Me), 2.30–2.17 (m, 2H, CHOMe–CH2–CH=CH), 2.03 (br q, 2H, J = 6.7 Hz, CH=CH–CH2), 1.67–1.57 (m, 2H, CH2CH2CO2Me), 1.49–1.41 (m, 2H, CH2CHOMe), 1.40–1.21 (m, 16H, CH3(CH2)4 and (CH2)4CH2CH2CO2Me), 0.88 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 174.33 (Cquat, CO), 131.73 (CH=CH), 125.42 (CH=CH), 80.99 (CHOMe), 56.58 (CHOCH3), 51.46 (CO2CH3), 34.11 (CH2CO2Me), 33.57 (CH2), 31.88 (CH2CH2CH3), 31.05 (CH2), 29.56 (CH2), 29.50 (CH2), 29.18 (CH2), 29.15 (CH2), 29.13 (CH2), 27.41 (CH2), 25.36 (CH2), 24.95 (CH2CH2CO2Me), 22.65 (CH2CH3), 14.10 (CH3).
[α]D17.5: +13.6; [α]57817.5: +14.1; [α]54617.5: +16.1; [α]43617.5: +27.5; [α]36517.5: +43.2 (c 5.00, CHCl3)
[α]D17.5: +16.2; [α]57817.5: +16.9; [α]54617.5: +19.2; [α]43617.5: +32.6 (neat liquid).
3.2.4. (R,Z)-12-Methoxyoctadec-9-en-1-ol (16)
Red-Al (0.52 mL, ~3 M in toluene, 1.56 mmol, 1.2 equiv) was added dropwise to a cooled solution of 15 (422 mg, 1.29 mmol) in anhydrous Et2O (4 mL) at 0 °C with stirring and under nitrogen. After the addition, the stirring was continued overnight for 18 h at 0 °C (use of a Dewar with ice-cooling). TLC monitoring confirmed disappearance of the starting material. A solution of citric acid (400 mg) in distilled water (5 mL) was added to the reaction mixture, which was allowed to stir again for 30 min. Extraction was done with petroleum ether/EtOAc (80:20). Organic layers were dried over Na2SO4, and solvent was evaporated under reduced pressure. The crude product was then purified by column chromatography on basic alumina (5 g, 0%–3% acetone in petroleum ether) to afford 16 as a colorless oil (318 mg, 82%). Rf = 0.32 (petroleum ether/acetone 85:15).
IR (KBr) ν 3372 (broad, O–H), 3009, 2927, 2855, 1464, 1456, 1377, 1357, 1099 (C–O of OMe), 1058, 724 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.50–5.34 (m, 2H: CH=CH partly distorted due to strong coupling at 5.46 and 5.38 ppm (dtt, J = 10.9, 7.0, 1.4 Hz)), 3.64 (t, 2H, J = 6.6 Hz, CH2OH), 3.34 (s, 3H, CHOCH3), 3.17 (tt, 1H, J = 6.2, 5.5 Hz, CHOMe), 2.31–2.17 (m, 2H, CHOMe–CH2–CH=CH), 2.03 (br q, 2H, J = 6.7 Hz, CH=CH–CH2), 1.66 (br s, 1H, OH), 1.56 (br tt, 2H, J = 7.5, 6.6 Hz, CH2CH2OH), 1.49–1.41 (m, 2H, CH2CHOMe), 1.41–1.23 (m, 18H, CH3(CH2)4 and (CH2)5CH2CH2OH), 0.88 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 131.81 (CH=CH), 125.38 (CH=CH), 81.02 (CHOMe), 63.06 (CH2OH), 56.58 (CHOCH3), 33.57 (CH2), 32.80 (CH2CH2OH), 31.88 (CH2), 31.07 (CH2), 29.61 (CH2), 29.50 (2 CH2), 29.40 (CH2), 29.27 (CH2), 27.43 (CH2), 25.74 (CH2), 25.36 (CH2), 22.65 (CH2), 14.12 (CH3).
[α]D23.5: +12.8; [α]57823.5: +13.4; [α]54623.5: +15.2; [α]43623.5: +25.9; [α]36523.5: +40,6 (c 5.02, CHCl3).
[α]D23.5: +17.0; [α]57823.5: +17.4; [α]54623.5: +19.8; [α]43623.5: +33.6; [α]36523.5: +53.2 (c 5.02, acetone).
[α]D23: +15.7; [α]57823: +16.4; [α]54623: +18.6; [α]43623: +31.7 (neat liquid).
HRMS (ESI, m/z) calculated for C19H38O2Na [M + Na]+: 321.4892, found: 321.4886.
3.2.5. (R,Z)-12-Methoxyoctadec-9-en-1-yl methanesulfonate (17)
To a stirred solution of 16 (6.64 g, 22.2 mmol), Et3N (4.7 mL, 33.4 mmol, 1.5 equiv) in DCM (67 mL) under nitrogen at −30 °C, mesyl chloride (2.2 mL, 28 mmol, 1.25 equiv) in DCM (9 mL) was added dropwise. The addition of mesyl chloride was completed by rinsing with DCM (3 × 0.3 mL). The corresponding mixture was then stirred for 15 h at −30 °C. TLC monitoring (elution with DCM, mesylate showed far bigger Rf than starting alcohol with this eluent) showed completion of the reaction, and distilled water (75 mL) was added to quench the reaction. Extraction was done with DCM. Organic layers were washed with brine and dried over Na2SO4. Solvent was removed under reduced pressure to provide the crude product as a light yellow oil. The crude product was then purified by column chromatography on silica gel (10 g, 0%–2% acetone in petroleum ether) to provide 17 as a colorless oil (5.40 g, 74%). Rf = 0.45 (petroleum ether/acetone 80:20).
IR (KBr) ν 3011, 2928, 2855, 1464, 1358, 1177 (S=O), 1098 (C–O of OMe), 974, 945, 831, 724, 529 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.50–5.34 (m, 2H: CH=CH partly distorted due to strong coupling at 5.46 and 5.38 ppm (dtt, J = 10.9, 6.9, 1.3 Hz)), 4.22 (t, 2H, J = 6.6 Hz, CH2OMs), 3.34 (s, 3H, CHOCH3), 3.17 (tt, 1H, J = 6.1, 5.5 Hz, CHOMe), 3.00 (s, 3H, SO2CH3), 2.31–2.17 (m, 2H, CHOMe–CH2–CH=CH), 2.03 (br q, 2H, J = 6.7 Hz, CH=CH–CH2), 1.75 (br tt, 2H, J = 7.5, 6.6 Hz, CH2CH2OMs), 1.50–1.22 (m, 20H, CH3(CH2)5 and (CH2)5CH2CH2OMs), 0.88 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 131.70 (CH=CH), 125.47 (CH=CH), 80.98 (CHOMe), 70.16 (CH2OMs), 56.59 (CHOCH3), 37.38 (SO2CH3), 33.57 (CH2), 31.88 (CH2), 31.08 (CH2), 29.57 (CH2), 29.49 (CH2), 29.34 (CH2), 29.21 (CH2), 29.13 (CH2), 29.02 (CH2), 27.41 (CH2), 25.43 (CH2), 25.36 (CH2), 22.64 (CH2), 14.10 (CH3).
[α]D23.5: +13.7; [α]57823.5: +14.1; [α]54623.5: +16.0; [α]43623.5: +27.5; [α]36523.5: +43.4 (c 5.01, acetone).
[α]D23.5: +10.3; [α]57823.5: +10.5; [α]54623.5: +11.9; [α]43623.5: +20.3; [α]36523.5: +31.5 (c 2.60, CHCl3).
[α]D23.5: +13.7; [α]57823.5: +14.3; [α]54623.5: +16.3; [α]43623.5: +27.2 (neat liquid).
3.2.6. (R)-4-((((R,Z)-12-Methoxyoctadec-9-en-1-yl)oxy)methyl)-2,2-dimethyl-1,3-dioxolane (19)
In a flask containing a solution of 17 (4.97 g, 13.2 mmol), n-Bu4NBr (1.06 g, 3.3 mmol, 0.25 equiv), and finely crushed (with a mortar and pestle) potassium hydroxide (3.48 g, 52.75 mmol, ~85% KOH, 4 equiv) in DMSO (26.4 mL) with stirring and under nitrogen at rt for 10 min, was added (R)-solketal (18) (2.07 g, ≥95% pure, 14.9 mmol, 1.13 equiv), and the corresponding mixture was stirred overnight for 14 h at 35 °C. TLC monitoring showed completion of the reaction and distilled water (50 mL) was added to the reaction mixture. Extraction was done with petroleum ether/EtOAc (80:20). Organic layers were washed with brine and dried over Na2SO4. Solvent was removed under reduced pressure. The crude product was then purified by column chromatography on basic alumina (25 g, 0%–1% acetone in petroleum ether) to afford 19 as a colorless oil (4.65 g, 93%). Rf = 0.45 (petroleum ether/acetone 95:5).
IR (KBr) ν 2985, 2929, 2856, 2821, 1466, 1456, 1379, 1369, 1256, 1237, 1214, 1118, 1100 (C–O of OMe), 1056, 847, 724, 514 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.50–5.34 (m, 2H: CH=CH partly distorted due to strong coupling at 5.46 and 5.38 ppm (dtt, J = 10.9, 6.9, 1.3 Hz)), 4.27 (dddd (apparent tt), 1H, J = 6.4, 6.4, 5.7, 5.6 Hz, CH–O in dioxolane), 4.06 (dd, 1H, J = 8.2, 6.4 Hz, CH2O in dioxolane), 3.73 (dd, 1H, J = 8.2, 6.4 Hz, CH2O in dioxolane), 3.54–3.39 (m, 4H, CH2OCH2O with 1H dd at 3.52 ppm, J = 9.9, 5.7 Hz and 1H dd at 3.42 ppm, J = 9.9, 5.6 Hz), 3.34 (s, 3H, CHOCH3), 3.17 (br tt, 1H, J = 6.2, 5.4 Hz, CHOMe), 2.31–2.17 (m, 2H, CHOMe–CH2–CH=CH), 2.03 (br q, 2H, J = 6.7 Hz, CH=CH–CH2), 1.62–1.52 (m, 2H, CH2CH2O), 1.50–1.41 (m, 2H, CH2CHOMe), 1.42 (q, 3H, J = 0.7 Hz (w coupling), CH3), 1.36 (q, 3H, J = 0.7 Hz (w coupling), CH3), 1.39–1.23 (m, 18H, CH3(CH2)4 and (CH2)3CH2CH2O), 0.88 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 131.82 (CH=CH), 125.35 (CH=CH), 109.37 (CMe2), 80.99 (CHOMe), 74.76 (CH–O), 71.88 and 71.83 (CH2OCH2(CH2)7), 66.93 (CH2OCMe2), 56.58 (CHOCH3), 33.57 (CH2), 31.88 (CH2), 31.04 (CH2), 29.63 (CH2), 29.56 (CH2), 29.51 (CH2), 29.50 (CH2), 29.45 (CH2), 29.30 (CH2), 27.44 (CH2), 26.78 (C–CH3), 26.06 (CH2), 25.43 (C–CH3), 25.36 (CH2), 22.64 (CH2), 14.11 (CH3).
[α]D18: +4.4; [α]57818: +4.1; [α]54618: +4.7; [α]43618: +7.3; [α]36518: +10.7 (c 3.01, acetone).
[α]D18: +2.7; [α]57818: +2.8; [α]54618: +3.0; [α]43618: +4.8; [α]36518: +6.3 (c 2.42, CHCl3).
HRMS (ESI, m/z) calculated for C25H48O4Na [M + Na]+: 435.3450, found: 435.3452.
Elementary analysis calculated for C25H48O4: C, 72.77; H, 11.72, found: C, 73.23; H, 11.94.
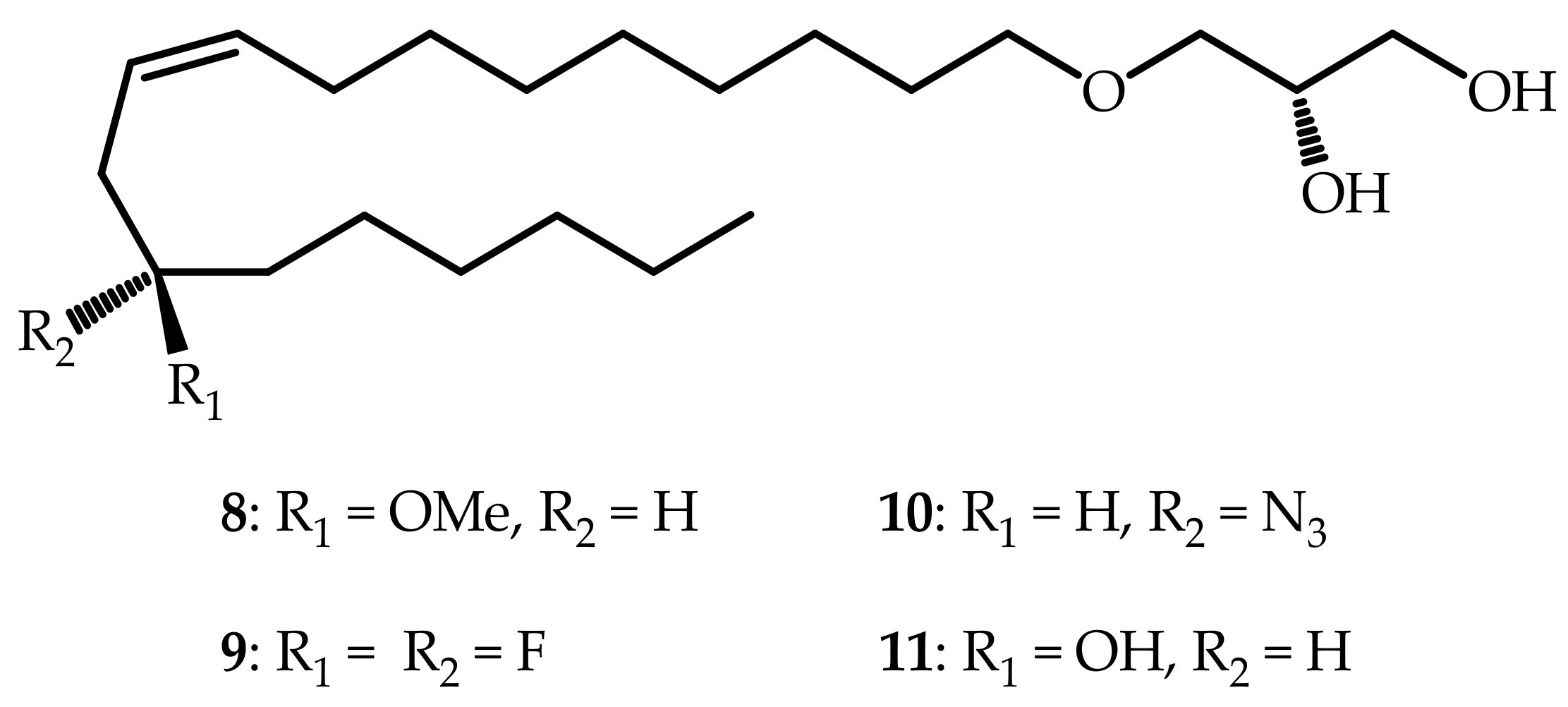
3.2.7. (S)-3-(((R,Z)-12-Methoxyoctadec-9-en-1-yl)oxy)propane-1,2-diol (8)
To a solution of acetonide 19 (4.65 g, 11.2 mmol) in methanol (45.1 mL), was added p-toluenesulfonic acid monohydrate (107.3 mg, 0.55 mmol, 0.05 equiv) and distilled water (4.5 mL). The flask was then purged with nitrogen, stoppered and dipped in a preheated bath at 60 °C. Stirring was maintained for 5 h at 60 °C. TLC monitoring showed completion of the reaction, and sodium bicarbonate (52.1 mg, 0.62 mmol, 0.055 equiv) was added. Stirring was continued for 1 h at 60 °C. Methanol was then removed under reduced pressure, and the crude product was purified by column chromatography on silica gel (20 g, 0%–5% acetone in petroleum ether and then petroleum ether + 5% acetone + 12% methanol) to afford 8 as a colorless oil (4.16 g, 99%). Rf = 0.03 (petroleum ether/acetone 90:10).
1H NMR (400 MHz, CDCl3): δ 5.50–5.34 (m, 2H: CH=CH partly distorted due to strong coupling at 5.46 and 5.38 ppm (dtt, J = 10.9, 7.0, 1.4 Hz)), 3.90–3.83 (m, 1H, CHOH), 3.72 (broad ddd, 1H, J = 11.3, 6.7 (with OH), 3.9 Hz, CH2OH), 3.65 (broad ddd, 1H, J = 11.3, 5.1 (with OH), 4.9 Hz, CH2OH), 3.56–3.42 (m, 4H, CH2OCH2 with 1H dd at 3.54 ppm, J = 9.7, 4.0 Hz), 3.34 (s, 3H, CHOCH3), 3.17 (tt, 1H, J = 6.2, 5.4 Hz, CHOMe), 2.67 (d, 1H, J = 5.0 Hz, CHOH), 2.28 (broad dd, 1H, J = 6.7, 5.1 Hz, resolution ω = 1.3 Hz, CH2OH), 2.30–2.17 (m, 2H, CHOMe–CH2–CH=CH), 2.03 (br q, 2H, J = 6.7 Hz, CH=CH–CH2), 1.57 (broad tt, 2H, J = 7.1, 6.7 Hz, CH2CH2O), 1.50–1.41 (m, 2H, CH2CHOMe), 1.41–1.23 (m, 18H, CH3(CH2)4 and (CH2)5CH2CH2O), 0.88 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 131.81 (CH=CH), 125.36 (CH=CH), 81.00 (CHOMe), 72.51 and 71.82 (CH2OCH2(CH2)7), 70.41 (CHOH), 64.29 (CH2OH), 56.59 (CHOCH3), 33.54 (CH2), 31.88 (CH2), 31.04 (CH2), 29.60 (CH2), 29.57 (CH2), 29.49 (CH2), 29.46 (CH2), 29.40 (CH2), 29.26 (CH2), 27.42 (CH2), 26.06 (CH2), 25.35 (CH2), 22.64 (CH2), 14.11 (CH3).
[α]D22.5: +9.8; [α]57822.5: +9.6; [α]54622.5: +11.1; [α]43622.5: +19.6; [α]36522.5: +31.5 (c 1.10, acetone).
HRMS (ESI, m/z) calculated for C22H44O4Na [M + Na]+: 395.3137, found: 395.3132.
3.2.8. (R,Z)-Methyl 12-((triisopropylsilyl)oxy)octadec-9-enoate (20)
To a vigorously stirred solution of 13 (625 mg, 2.0 mmol) and imidazole (334 mg, 4.9 mmol, 2.45 equiv) in DMF (1.6 mL), which was cooled under nitrogen at 0 °C, was added dropwise triisopropylsilyl chloride (0.53 mL, 97% pure, 2.4 mmol, 1.2 equiv). The corresponding mixture was allowed to stir for 48 h at rt. TLC monitoring showed completion of the reaction. Petroleum ether/EtOAc (80:20) was added. After washing with brine and drying over MgSO4, solvent was removed under reduced pressure, and the residue was purified by column chromatography on silica gel (5 g, 0%–0.2% acetone in petroleum ether) to provide 20 as a colorless oil (574.7 mg, 61%). Rf = 0.73 (petroleum ether/acetone 95:5).
IR (KBr) ν 3006, 2929, 2865, 1744, 1464, 1436, 1366, 1096, 883, 678 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.46–5.36 (m, 2H), 3.83 (tt, 1H, J = 5.8, 5.6 Hz), 3.67 (s, 3H), 2.30 (dd, 2H, J = 7.8, 7.4 Hz), 2.27–2.22 (m, 2H), 2.06–1.97 (m, 2H), 1.67–1.57 (m, 2H), 1.54–1.38 (m, 2H), 1.38–1.20 (m, 16H), 1.06 (s, 21H, Si(CH(CH3)2)3, due to the shielding effect on neighboring CH, CH and CH3 of isopropyl groups being very close, so coupling was not seen and these signals were superimposed, HSQC showed CH at 1.061 ppm and CH3 at 1.059 ppm), 0.88 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 174.33 (CO), 131.31 (CH=CH), 125.72 (CH=CH), 72.26 (CH), 51.46 (CH3), 36.56 (CH2), 34.68 (CH2), 34.11 (CH2), 31.92 (CH2), 29.64 (2 CH2), 29.20 (CH2), 29.19 (CH2), 29.15 (CH2), 27.51 (CH2), 24.96 (CH2), 24.81 (CH2), 22.64 (CH2), 18.21 (6 CH3), 14.11 (CH3), 12.63 (3 CH).
[α]D18: +12.3; [α]57818: +12.5; [α]54618: +14.2; [α]43618: +24.3; [α]36518: +38.6 (c 4.00, acetone).
[α]D18: +10.6; [α]57818: +11.0; [α]54618: +12.6; [α]43618: +21.5; [α]36518: +33.7 (c 4.00, CHCl3).
HRMS (ESI, m/z) calculated for C26H51O3Si [M–C2H5]+: 439.3607, found: 439.3607, calculated for C25H49O3Si [M–iPr]+: 425.3451, found: 425.3455, calculated for C24H45O2Si [M–iPr-MeOH]+: 393.3189, found: 393.3177.
3.2.9. (R,Z)-12-((Triisopropylsilyl)oxy)octadec-9-en-1-ol (21)
In a flame-dried two-necked flask, a solution of 20 (574.7 mg, 1.22 mmol) in anhydrous Et2O (3.7 mL) was cooled at 0 °C under nitrogen. A solution of Red-Al (65% in toluene, ~3 M, 0.5 mL, 1.5 mmol, 1.23 equiv) was added dropwise under stirring. Stirring was continued for 5 h at 0 °C. TLC monitoring confirmed completion of the reaction. Citric acid (400 mg) and distilled water (5 mL) were added to the mixture and stirring was continued for 30 min. Extraction was then done with petroleum ether/EtOAc (80:20), and organic layers were dried over Na2SO4. Solvent was evaporated under reduced pressure, and the crude product was purified by column chromatography on silica gel (2.5 g, 0%–0.5% acetone in petroleum ether) to afford 21 as a colorless oil (509.6 mg, 94%). Rf = 0.39 (petroleum ether/acetone 85:15).
1H NMR (400 MHz, CDCl3): δ 5.47–5.36 (m, 2H), 3.83 (tt, 1H, J = 6.0, 5.6 Hz), 3.64 (t, 2H, J = 6.6 Hz, CH2OH), 2.27–2.22 (m, 2H), 2.02 (br q, 2H, J = 6.5 Hz), 1.61–1.52 (m, 3H, CH2 and OH), 1.52–1.40 (m, 2H), 1.40–1.19 (m, 18H), 1.06 (s, 21H), 0.88 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 131.38 (CH=CH), 125.68 (CH=CH), 72.27 (CH), 63.10 (CH2), 36.55 (CH2), 34.68 (CH2), 32.81 (CH2), 31.92 (CH2), 29.69 (CH2), 29.64 (CH2), 29.54 (CH2), 29.42 (CH2), 29.32 (CH2), 27.54 (CH2), 25.74 (CH2), 24.81 (CH2), 22.64 (CH2), 18.21 (6 CH3), 14.11 (CH3), 12.63 (3 CH).
3.2.10. (R,Z)-12-((Triisopropylsilyl)oxy)octadec-9-en-1-yl methanesulfonate (22)
To a solution of 21 (509.6 mg, 1.15 mmol) and Et3N (0.245 mL, 1.75 mmol, 1.5 equiv) in DCM (4.6 mL) with stirring and under nitrogen cooled at −50 °C, was added dropwise mesyl chloride (0.112 mL, 1.45 mmol, 1.25 equiv) in DCM (0.6 mL). Transfer of mesyl chloride was completed by rinsing twice with a few drops of DCM. The reaction mixture was then allowed to warm up slowly in 2 h up to −5 °C. TLC monitoring (elution with DCM) showed completion of the reaction, and distilled water (5.8 mL) was added to quench the reaction. Extraction was done with DCM. Organic layers were washed with brine and dried over Na2SO4. DCM was removed under reduced pressure, and the crude product was purified by column chromatography on silica gel (4 g, 0%–1% acetone in petroleum ether) to afford 22 as a colorless oil (453 mg, 76%). Rf = 0.41 (petroleum ether/acetone 80:20).
1H NMR (400 MHz, CDCl3): δ 5.47–5.37 (m, 2H), 4.22 (t, 2H, J = 6.6 Hz, CH2OMs), 3.83 (tt, 1H, J = 6.0, 5.6 Hz), 3.00 (s, 3H), 2.29–2.19 (m, 2H), 2.08–1.96 (m, 2H), 1.75 (dq, 2H, J = 8.2, 6.6 Hz), 1.54–1.20 (m, 20H), 1.06 (s, 21H), 0.88 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 131.28 (CH=CH), 125.75 (CH=CH), 72.25 (CH), 70.16 (CH2), 37.37 (CH3), 36.56 (CH2), 34.69 (CH2), 31.91 (CH2), 29.65 (CH2), 29.63 (CH2), 29.37 (CH2), 29.25 (CH2), 29.14 (CH2), 29.04 (CH2), 27.51 (CH2), 25.44 (CH2), 24.81 (CH2), 22.64 (CH2), 18.22 (6 CH3), 14.11 (CH3), 12.64 (3 CH).
HRMS (ESI, m/z) calculated for C28H58O4NaSiS [M + Na]+: 541.3723, found: 541.3724.
3.2.11. (((R,Z)-18-(((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)methoxy)octadec-9-en-7-yl)oxy)triisopropylsilane (23)
A 60% dispersion of sodium hydride in mineral oil (29.5 mg, 0.7 mmol, 2.5 equiv) was washed three times with petroleum ether under nitrogen and with stirring. Anhydrous DMF (0.2 mL) was then added and the mixture was cooled at 0 °C. Following this, a solution of 2,3 isopropylidene-sn-glycerol 18 (50.2 mg, ≥95% pure, 0.36 mmol, 1.25 equiv) in DMF (0.2 mL) was added dropwise to the mixture, and the flask containing 18 was rinsed with DMF (2 × 0.1 mL). The corresponding mixture was allowed to stir for 10 min at 0 °C, and a solution of 22 (153 mg, 0.29 mmol) in DMF (0.2 mL) was added to the resulting white suspension. Transfer of 22 was completed by rinsing with DMF (2 × 0.1 mL). The mixture was then vigorously stirred overnight for 16 h at rt. TLC monitoring showed completion of the reaction, and 10% aqueous ammonium acetate was added as a buffer. Extraction was done with petroleum ether. Organic layers were dried over Na2SO4. Solvent was removed under reduced pressure, and the crude product was purified by column chromatography on silica gel (1.5 g, 0%–0.5% acetone in petroleum ether) to provide 23 as a colorless oil (110.8 mg, 68%). Rf = 0.71 (petroleum ether/acetone 95:5).
IR (KBr) ν 2930, 2865, 1464, 1379, 1369, 1255, 1097, 883, 849, 678 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.47–5.36 (symmetrical m, 2H), 4.27 (tt, 1H, J = 6.4, 5.6 Hz), 4.06 (dd, 1H, J = 8.2, 6.4 Hz), 3.83 (broad tt, 1H, J = 5.9, 5.5 Hz), 3.73 (dd, 1H, J = 8.2, 6.4 Hz), 3.54–3.39 (m, 4H with 1H dd at 3.52 ppm, J = 9.9, 5.6 Hz and 1H dd at 3.42 ppm, J = 9.9, 5.6 Hz), 2.29–2.20 (m, 2H), 2.07–1.96 (m, 2H), 1.62–1.53 (m, 2H plus signal of water as a singlet at 1.59 ppm), 1.53–1.39 (m, 5H including CH3, q, 3H at 1.42 ppm, J = 0.6 Hz), 1.37 (q, 3H, J = 0.6 Hz, CH3), 1.36–1.20 (m, 18H), 1.06 (s, 21H), 0.88 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 131.40 (CH=CH), 125.66 (CH=CH), 109.37 (CMe2), 74.76 (CH), 72.27 (CH), 71.89 (CH2), 71.83 (CH2), 66.94 (CH2), 36.55 (CH2), 34.68 (CH2), 31.92 (CH2), 29.71 (CH2), 29.64 (CH2), 29.57 (CH2), 29.53 (CH2), 29.47 (CH2), 29.34 (CH2), 27.55 (CH2), 26.78 (CH3), 26.07 (CH2), 25.43 (CH3), 24.81 (CH2), 22.64 (CH2), 18.22 (6 CH3), 14.11 (CH3), 12.64 (3 CH).
[α]D19: +4.1; [α]57819: +4.2; [α]54619: +4.9; [α]43619: +8.2; [α]36519: +12.4 (c 3.62, CHCl3).
[α]D19: +4.0; [α]57819: +3.9; [α]54619: +4.5; [α]43619: +7.9; [α]36519: +12.2 (c 2.51, acetone).
HRMS (ESI, m/z) calculated for C33H66O4NaSi [M + Na]+: 577.4628, found: 577.4627.
Elementary analysis calculated for C33H66O4Si: C, 71.42; H, 11.99, found: C, 71.88; H, 12.09.
3.2.12. (R,Z)-18-(((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)methoxy)octadec-9-en-7-ol (24)
To a stirred solution at of 23 (278 mg, 0.5 mmol) in anhydrous THF (1.5 mL) under nitrogen, which was cooled at -20 °C, was added a 1 M solution of TBAF in THF (0.67 mL, 0.67 mmol, 1.3 equiv). The reaction mixture was then left under stirring for 20 h at rt. TLC monitoring showed completion of the reaction, and distilled water (1.5 mL) was added to the mixture. Extraction was done with EtOAc and organic layers were washed with brine and dried over Na2SO4. Solvent was removed under reduced pressure, and the crude product was purified by column chromatography on silica gel (2.5 g, 0%–4% acetone in petroleum ether) to provide 24 as a colorless oil (184.2 mg, 92%). Rf = 0.18 (petroleum ether/acetone 95:5).
IR (KBr) ν 3457, 2928, 2856, 1466, 1370, 1256, 1214, 1120, 846, 724 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.56 (dtt, 1H, J = 10.9, 7.3, 1.5 Hz), 5.40 (dtt, 1H, J = 10.9, 7.3, 1.5 Hz), 4.27 (dddd, 1H, J = 6.4, 6.4, 5.7, 5.7 Hz), 4.06 (dd, 1H, J = 8.2, 6.4 Hz, ddd with improving the resolution, J = 8.2, 6.4, 0.2 Hz), 3.73 (dd, 1H, J = 8.2, 6.4 Hz), 3.66–3.56 (m, 1H), 3.54–3.39 (m, 4H with 1H dd at 3.52 ppm, J = 9.9, 5.7 Hz (ddd with improving the resolution, J = 9.9, 5.7, 0.3 Hz) and 1H dd at 3.42 ppm, J = 9.9, 5.6 Hz), 2.24–2.18 (m, 2H), 2.05 (br dtd (apparent qd), 2H, J = 7.2, 7.2, 1 Hz), 1.61–1.52 (m, 3H, CH2 and OH), 1.51–1.43 (m, 2H), 1.43 (q, 3H, J = 0.6 Hz, CH3), 1.37 (q, 3H, J = 0.6 Hz, CH3), 1.37–1.23 (m, 18H), 0.88 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 133.49 (CH=CH), 125.16 (CH=CH), 109.37 (CMe2), 74.76 (CH), 71.87 (CH2), 71.83 (CH2), 71.51 (CH), 66.94 (CH2), 36.86 (CH2), 35.37 (CH2), 31.85 (CH2), 29.66 (CH2), 29.55 (CH2), 29.46 (CH2), 29.41 (CH2), 29.36 (CH2), 29.25 (CH2), 27.42 (CH2), 26.78 (CH3), 26.04 (CH2), 25.73 (CH2), 25.43 (CH3), 22.63 (CH2), 14.10 (CH3).
[α]D20: -5.0; [α]57820: −5.4; [α]54620: −12.5; [α]43620: −11.9; [α]36520: −21.2 (c 6.00, CHCl3).
Elementary analysis calculated for C24H46O4: C, 72.31; H, 11.63, found: C, 71.79; H, 11.66.
3.2.13. (S)-3-(((R,Z)-12-Hydroxyoctadec-9-en-1-yl)oxy)propane-1,2-diol (11)
To a solution of 24 (1.26 g, 3.15 mmol) in methanol (12.6 mL), was added p-toluenesulfonic acid monohydrate (30 mg, 0.16 mmol, 0.05 eq) and distilled water (1.26 mL). The flask was then purged with nitrogen, stoppered and dipped in a preheated bath at 60 °C. Stirring was maintained for 4 h at 60 °C. TLC monitoring showed completion of the reaction, and sodium bicarbonate (14.5 mg, 0.173 mmol, 0.055 equiv) was added and stirring was continued for 1 h at 60 °C. Methanol was removed under reduced pressure, and the crude product was purified by column chromatography on silica gel eluting with 0%–5% acetone in petroleum ether to remove non polar impurities and then with petroleum ether + 5% acetone + 12% methanol to give 11 as a colorless oil (945.6 mg, 84%). Rf = 0.02 (petroleum ether/acetone 80:20).
IR (KBr) ν 3383, 2926, 2855, 1654, 1466, 1124, 1045, 858, 724 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.56 (dtt, 1H, J = 10.8, 7.3, 1.4 Hz), 5.40 (dtt, 1H, J = 10.7, 7.4, 1.5 Hz), 3.91–3.81 (m, 1H), 3.72 (very broad dd, 1H, J = 10, 4 Hz (br dd after exchange with D2O, J = 11.3, 3.5 Hz), 1H of CH2OH), 3.69–3.58 (m, 2H, 1H of CH2OH and H12), 3.57–3.41 (m, 4H, CH2OCH2), 2.80–2.50 (envelope, 1H, CHOH), 2.42–2.12 (envelope, 1H, CH2OH), 2.21 (broad ddd, 2H, J = 7.4, 6.2, 1.1 Hz), 2.05 (br dt (apparent q), 2H J = 7.0, 6.9 Hz), 1.90–1.51 (m, 6H with an envelope centered at 1.63 ppm), 1.51–1.41 (m, 3H), 1.40–1.19 (m, 18H), 0.88 (pseudo t, 3H, J = 6.8 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 133.50 (CH=CH), 125.18 (CH=CH), 72.53 (CH2), 71.79 (CH2), 71.52 (CH), 70.40 (CH), 64.31 (CH2), 36.83 (CH2), 35.36 (CH2), 31.85 (CH2), 29.61 (CH2), 29.53 (CH2), 29.38 (CH2), 29.36 (CH2), 29.32 (CH2), 29.17 (CH2), 27.39 (CH2), 26.02 (CH2), 25.73 (CH2), 22.63 (CH2), 14.10 (CH3).
[α]D22: +1.2; [α]57822: +0.5; [α]54622: +0.6; [α]43622: +1.3; [α]36522: +1.6 (c 1.115, acetone).
HRMS (ESI, m/z) calculated for C21H42O4Na [M + Na]+: 381.2981, found: 381.2982.
3.2.14. Methyl (Z)-12-oxooctadec-9-enoate (25)
Pyridium chlorochromate (18.06 g, 83.8 mmol, 2.6 equiv) was suspended in DCM (111.7 mL) with stirring for 5 min. Following this, a solution of methyl ricinoleate 13 (10 g, 32 mmol) in DCM (15 mL) was added rapidly to the mixture and the flask containing 13 was rinsed with DCM (3 x 2 mL). Stirring was pursued for 1 h at rt under nitrogen. TLC monitoring showed completion of the reaction, and petroleum ether/EtOAc (90:10) (111.7 mL) was added. The resulting mixture was filtered over a short plug of silica gel with rinsing of silica gel by petroleum ether/EtOAc (90:10). Evaporation of the filtrate under reduced pressure followed by the purification of the crude by column chromatography on silica gel (55 g, 0%–1% acetone in petroleum ether) afforded 25 as a colorless oil (6.73 g, 68%). Rf = 0.36 (petroleum ether/acetone 90:10).
1H NMR (400 MHz, CDCl3): δ 5.62–5.50 (m, 2H), 3.67 (s, 3H), 3.15 (br d, 2H, J = 6.0 Hz), 2.43 (dd, 2H, J = 7.5, 7.4, Hz), 2.30 (dd, 2H, J = 7.7, 7.4 Hz), 2.02 (br dt (apparent q), 2H, J = 7.5, 7.0 Hz), 1.67–1.51 (m, 4H), 1.40–1.20 (m, 14H), 0.88 (t, 3H, J = 6.8 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 209.36 (CO), 174.31(CO), 133.53 (CH=CH), 120.99 (CH=CH), 51.45 (CH3), 42.37 (CH2), 41.64 (CH2), 34.04 (CH2), 31.59 (CH2), 29.25 (CH2), 29.11 (CH2), 29.06 (CH2), 29.05 (CH2), 28.88 (CH2), 27.46 (CH2), 24.90 (CH2), 23.77 (CH2), 22.49 (CH2), 14.04 (CH3).
3.2.15. Methyl (Z)-12,12-difluorooctadec-9-enoate (26)
To a solution of 25 (6 g 19.3 mmol) in DCM (22.5 mL) with stirring and under nitrogen at rt, was added dropwise DAST (6.22 mL, 47 mmol, 2.4 equiv). The corresponding mixture was stirred for 21 days at rt. TLC monitoring showed that the reaction was mostly done and saturated aqueous sodium bicarbonate (32 mL) plus water (20 mL) were added to quench unreacted DAST. After partitioning and extraction of the aqueous layer with DCM, combined organic layers were washed with brine and dried over Na2SO4. DCM was removed under reduced pressure, and the crude product was purified by column chromatography on silica gel (20 g). First, elution with 0%–0.5% acetone in petroleum ether) afforded 26 as a colorless oil (3.48 g, 54%). Rf = 0.39 (petroleum ether/acetone 95:5). Then unreacted 25 (2.00 g, 33%) was eluted with 1% acetone in petroleum ether.
IR (KBr) ν 3465, 3021, 2953, 2930, 2856, 1742, 1467, 1436, 1198, 1170, 876, 726 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.64–5.55 (m, 1H), 5.39 (dtt, 1H, J = 10.9, 7.3, 1.5 Hz), 3.67 (s, 3H), 2.65–2.52 (m, which could be analyzed as a td (JHF = 15.9 Hz, JHH = 7.3 Hz) with further very small couplings, 2H), 2.30 (dd, 2H, J = 7.7, 7.4 Hz), 2.03 (br dt (apparent q), 2H, J = 7, 7 Hz), 1.87–1.72 (m, 2H), 1.67–1.57 (m, 2H), 1.51–1.41 (m, 2H), 1.40–1.22 (m, 14H), 0.89 (t, 3H, J = 6.8 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 174.32 (CO), 134.52 (CH=CH), 124.87 (C12, t, J = 241.2 Hz), 120.30 (CH=CH, t, J = 5.8 Hz), 51.47 (CH3), 35.98 (CH2, t, J = 25.0 Hz), 34.62 (CH2, t, J = 26.4 Hz), 34.09 (CH2), 31.60 (CH2), 29.30 (CH2), 29.14 (CH2), 29.09 (CH2), 29.08 (CH2), 29.06 (CH2), 27.40 (CH2), 24.93 (CH2), 22.51 (CH2), 22.16 (CH2, t, J =4.6 Hz), 14.04 (CH3).
19F NMR (376 MHz, CDCl3): δ −96.88 (pentuplet, J = 16.3 Hz on 19F-undecoupled spectrum).
3.2.16. (Z)-12,12-Difluorooctadec-9-en-1-ol (27)
To a stirred solution of 26 (199.5 mg, 0.6 mmol) in anhydrous Et2O (2 mL) under nitrogen, which was cooled at 0 °C, was added dropwise a solution of Red-Al (65% in toluene, ~3 M, 0.3 mL, 0.9 mmol, 1.5 equiv); then stirring was continued for 5 h at 0 °C. TLC monitoring confirmed completion of the starting material. A solution of citric acid (400 mg) in water (5 mL) was added, and stirring was still continued for 30 min. Extraction was done with petroleum ether/EtOAc (80:20), and organic layers were dried over Na2SO4. Solvent was evaporated under reduced pressure, and the crude product was purified by column chromatography on silica gel (4 g, 0%–2% acetone in petroleum ether) to afford 27 as a colorless oil (141.3 mg, 77%). Rf = 0.21 (petroleum ether/acetone 85:15). This reduction was subsequently performed on a larger scale (15 ×), and the crude alcohol, which was thus obtained, was used as such for the next step.
1H NMR (400 MHz, CDCl3): δ 5.65–5.56 (m, 1H), 5.39 (dtt, 1H, J = 10.9, 7.4, 1.6 Hz), 3.64 (t, 2H, J = 6.6 Hz), 2.65–2.53 (m, which could be analyzed as a tdd with further very small couplings at 2.59 ppm, 2H, JHF = 15.9 Hz, JHH = 7.3, 1.4 Hz), 2.04 (br dt (apparent q), 2H, J = 7, 7 Hz), 1.87–1.72 (m, 2H), 1.61–1.52 (m, 2H), 1.50–1.41 (m, 2H), 1.40–1.23 (m, 16H), 0.89 t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 134.59 (CH=CH), 124.89 (CF2, t, J = 241.2 Hz), 120.25 (t, CH=CH, J = 5.8 Hz), 63.07 (CH2), 35.97 (t, CH2, J = 25.0 Hz), 34.62 (t, CH2, J = 26.4 Hz), 32.78 (CH2), 31.60 (CH2), 29.47 (CH2), 29.38 (CH2), 29.36 (CH2), 29.22 (CH2), 29.06 (CH2), 27.42 (CH2), 25.73 (CH2), 22.51 (CH2), 22.16 (t, CH2, J = 4.6 Hz), 14.04 (CH3).
19F NMR (376 MHz, CDCl3): δ −96.85 (pentuplet, J = 16.3 Hz on 19F-undecoupled spectrum).
HRMS (ESI, m/z) calculated for C18H34OF2Na [M + Na]+: 327.2475 found: 327.2478, calculated for C18H33OFNa [M – HF + Na]+: 307.2413, found: 307.2415.
3.2.17. (Z)-12,12-Difluorooctadec-9-en-1-yl methanesulfonate (28)
To a stirred solution of crude 27 (made from 3.09 g of 26, 9.3 mmol) and Et3N (1.95 mL, 14.0 mmol, 1.5 equiv) in DCM (28 mL) under nitrogen, which was cooled at −35 °C, was added dropwise mesyl chloride (0.9 mL, 11.6 mmol, 1.25 equiv) in DCM (3.7 mL). Transfer of mesyl chloride was completed by rinsing with DCM (3 × 0.2 mL). The reaction mixture was then allowed to warm up slowly to −5 °C (in 4–5 h) and TLC monitoring (elution with DCM) showed completion of the reaction. Distilled water (96 mL) was added to quench the reaction, and extraction of the aqueous layer was done with DCM. Combined organic layers were washed with brine and dried over Na2SO4. DCM was removed under reduced pressure, and the crude product was purified by column chromatography on silica gel (10 g, 0%–1% acetone in petroleum ether) to afford 28 as a colorless oil (2.44 g, 69% from 26). Rf = 0.61 (petroleum ether/acetone 80:20).
1H NMR (400 MHz, CDCl3): δ 5.65–5.55 (m, 1H), 5.39 (dtt, 1H, J = 10.9, 7.3, 1.6 Hz), 4.22 (t, 2H, J = 6.6 Hz), 3.00 (s, 3H), 2.65–2.52 (m, which could be analyzed as a tdd with further very small couplings at 2.59 ppm, 2H, JHF = 16.0 Hz, JHH = 7.3, 1.3 Hz), 2.04 (br dt (apparent q), 2H, J = 7, 7 Hz), 1.87–1.70 (m, 4H), 1.58–1.21 (m, 18H), 0.89 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 134.51 (CH=CH), 124.87 (C12, t, J = 241.2 Hz), 120.30 (t, CH=CH, J = 5.8 Hz), 70.16 (CH2), 37.36 (CH3), 35.97 (t, CH2, J = 25.0 Hz), 34.61 (t, CH2, J = 26.4 Hz), 31.60 (CH2), 29.31 (CH2), 29.29 (CH2), 29.13 (CH2), 29.12 (CH2), 29.05 (CH2), 28.98, (CH2), 27.39 (CH2), 25.40 (CH2), 22.50 (CH2), 22.16 (t, CH2, J = 4.6 Hz), 14.05 (CH3).
19F NMR (376 MHz, CDCl3): δ −96.86 (pentuplet, J = 16.3 Hz on 19F-undecoupled spectrum).
3.2.18. (R,Z)-4-(((12, 12-Difluorooctadec-9-en-1-yl)oxy)methyl)-2,2-dimethyl-1,3-dioxolane (29)
To a stirred mixture of 28 (115 mg, 0.3 mmol), n-Bu4NBr (24.2 mg, 0.075 mmol, 0.25 equiv), DMSO (0.6 mL) and 50% aqueous NaOH (63 µL, 1.2 mmol of NaOH, 4 equiv), was added (R)-solketal 18 (49 mg, ≥95% pure, 0.35 mmol, 1.17 equiv). The corresponding mixture was stirred for 5 h at 60 °C. TLC monitoring showed completion of the reaction, and distilled water was added. Extraction was done with petroleum ether/EtOAc (80:20). Organic layers were washed again with brine and dried over Na2SO4. Solvent was removed under reduced pressure, and the crude product was purified by column chromatography on silica gel (5 g, 0%–0.5% acetone in petroleum ether) to provide 29 as a colorless oil (79.3 mg, 63%). Rf = 0.44 (petroleum ether/acetone 95:5).
1H NMR (400 MHz, CDCl3): δ 5.64–5.56 (m, 1H), 5.38 (dtt, 1H, J = 10.9, 7.3, 1.6 Hz), 4.27 (dddd (apparent tt), 1H, J = 6.4, 6.4, 5.7, 5.7 Hz), 4.06 (dd, 1H, J = 8.2, 6.4 Hz), 3.73 (dd, 1H, J = 8.2, 6.4 Hz), 3.54–3.39 (m, 4H), 2.65–2.53 (m, which could be analyzed as a tdd with further very small couplings at 2.59 ppm, 2H, JHF = 15.9 Hz, JHH = 7.4, 1.3 Hz), 2.03 (br dt (apparent q), 2H, J = 7, 7 Hz), 1.87–1.72 (m, 2H), 1.62–1.51 (m, 2H), 1.43 (q, 3H, J = 0.6 Hz), 1.37 (q, 3H, J = 0.6 Hz), 1.40–1.21 (m, 18H), 0.89 (t, 3H, J = 6.8 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 134.59 (CH=CH), 124.88 (CF2, t, J = 241.2 Hz), 120.23 (t, CH=CH, J = 5.8 Hz), 109.37 (CMe2), 74.76 (CH), 71.87 (CH2), 71.84 (CH2), 66.93 (CH2), 35.96 (t, CH2, J = 25.0 Hz), 34.62 (t, CH2, J = 26.4 Hz), 31.60 (CH2), 29.55 (CH2), 29.45 (CH2), 29.41 (CH2), 29.37 (CH2), 29.23 (CH2), 29.06 (CH2), 27.43 (CH2), 26.78 (CH3), 26.05 (CH2), 25.43 (CH3), 22.50 (CH2), 22.15 (t, CH2, J = 4.6 Hz), 14.04 (CH3).
19F NMR (376 MHz, CDCl3): δ −96.85 (pentuplet, J = 16.3 Hz on 19F-undecoupled spectrum).
HRMS (ESI, m/z) calculated for C24H44O3F2Na [M + Na]+: 441.3156, found: 441.3149, calculated for C24H43O3FNa [M – HF + Na]+: 421.3094, found: 421.3100, calculated for C24H42O3Na [M – 2HF + Na]+: 401.3032, found: 401.3044.
3.2.19. (S,Z)-3-((12,12-Difluorooctadec-9-en-1-yl)oxy)propane-1,2-diol (9)
To a solution of acetonide 29 (971.9 mg, 2.32 mmol) in methanol (9.3 mL) and distilled water (0.93 mL) was added p-toluenesulfonic acid monohydrate (22.1 mg, 0.116 mmol, 0.05 equiv). The flask was then purged with nitrogen, stoppered and dipped in a preheated bath at 60 °C. Stirring was maintained for 5 h at 60 °C. TLC monitoring showed completion of the reaction, and sodium bicarbonate (10.9 mg, 0.13 mmol, 0.056 equiv) was added to the mixture and the stirring was continued for 1 h at 60 °C. Methanol was removed under reduced pressure, and then the crude product was purified by column chromatography on silica gel (6 g, 0%–5% acetone in petroleum ether and then petroleum ether + 5% acetone + 12% methanol) to provide 9 as a green oil (811.2 mg, 92%). Rf = 0.04 (petroleum ether/acetone 90:10).
1H NMR (400 MHz, CDCl3): δ 5.65–5.55 (m, 1H), 5.39 (dtt, 1H, J = 10.9, 7.3, 1.6 Hz), 3.91–3.82 (m (ddt after exchange with D2O, J = 6.0, 5.2, 3.9 Hz), 1H), 3.72 (ddd, 1H, J = 11.4, 6.9, 3.8 Hz (dd after exchange with D2O, J = 11.4, 3.9 Hz)), 3.65 (ddd, 1H, J = 11.4, 5.1, 5.0 Hz (dd after exchange with D2O, J = 11.4, 5.2 Hz)), 3.56–3.42 (m with 1H dd at 3.54 ppm, J = 9.7, 3.9 Hz, 4H), 2.67 (d, which was suppressed after exchange with D2O, 1H, J = 5.1, Hz, OH), 2.65–2.53 (m, which could be analyzed as a tdd with further very small couplings at 2.59 ppm, 2H, JHF = 15.9 Hz, JHH = 7.3, 1.2 Hz, 2H), 2.26 (br dd, 1H, J = 6.6, 5.6 Hz), 2.04 (br dt (apparent q), 2H, J = 7, 7 Hz), 1.87–1.73 (m, 2H), 1.58 (br tt, 2H, J = 7.2, 6.7 Hz), 1.51–1.41 (m, 2H), 1.40–1.22 (m, 16H), 0.89 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 134.58 (CH=CH), 124.89 (CF2, t, J = 241.2 Hz), 120.25 (t, CH=CH, J = 5.8 Hz), 72.53 (CH2), 71.82 (CH2), 70.41 (CH), 64.30 (CH2), 35.96 (t, CH2, J= 25.0 Hz), 34.62 (t, CH2, J = 26.4 Hz), 31.60 (CH2), 29.57 (CH2), 29.44 (CH2), 29.40 (CH2), 29.36 (CH2), 29.22 (CH2), 29.05 (CH2), 27.42 (CH2), 26.07 (CH2), 22.50 (CH2), 22.15 (t, CH2, J= 4.6 Hz), 14.05 (CH3).
19F NMR (376 MHz, CDCl3): δ −96.84 (pentuplet, J= 16.3 Hz on 19F-undecoupled spectrum).
[α]D24: −4.6; [α]57824: −5.8; [α]54624: −6.4; [α]43624: −9.9; [α]36524: −14.3 (c 0.94, acetone).
HRMS (ESI, m/z) calculated for C21H40O3F2Na [M + Na]+: 401.2843, found: 401.2840, calculated for C21H39O3FNa [M – HF + Na]+: 381.2781, found: 381.2791, calculated for C21H38O3Na [M – 2HF + Na]+: 361.2719, found: 361.2726.
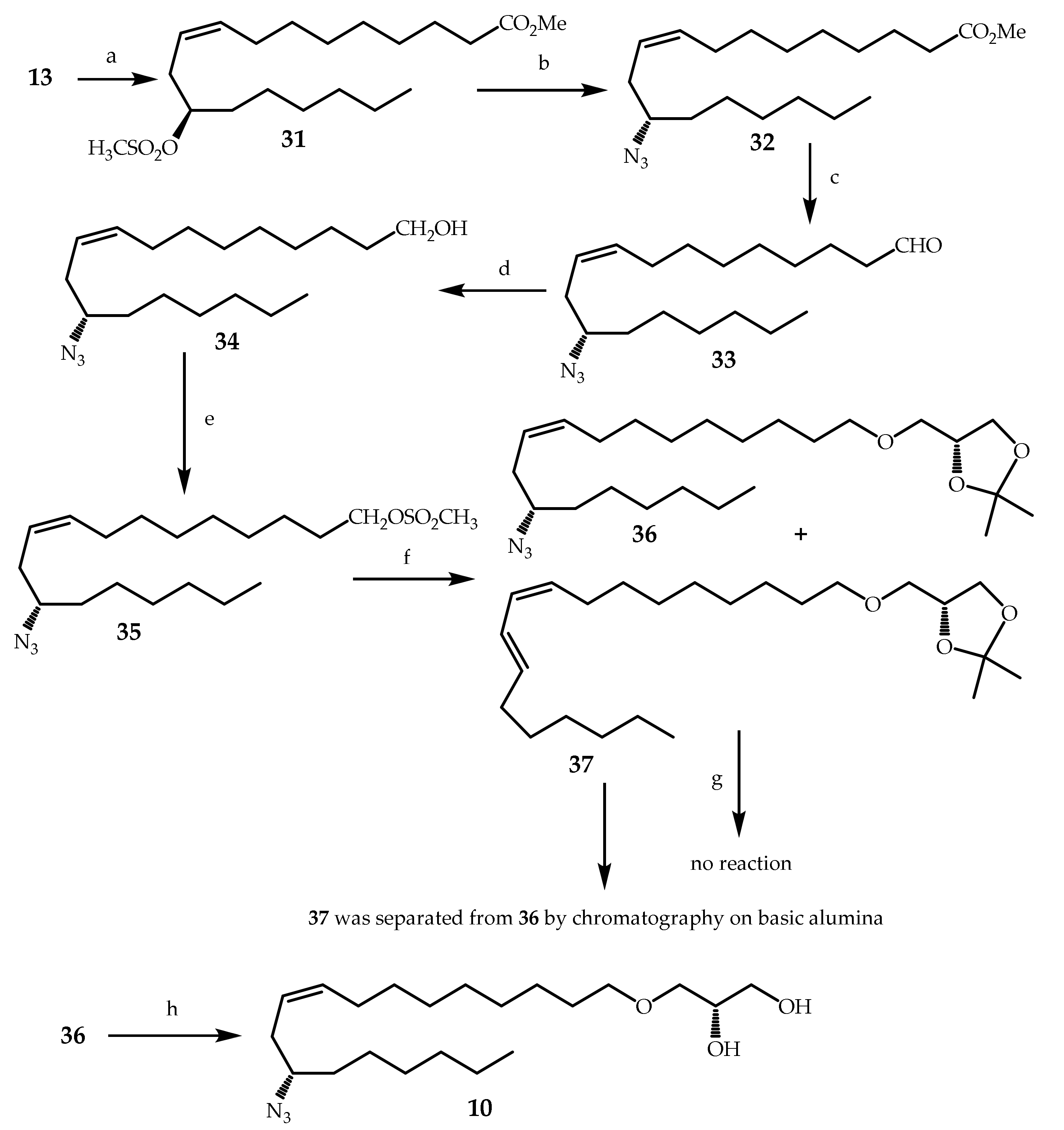
3.2.20. Methyl (R,Z)-12-((methylsulfonyl)oxy)octadec-9-enoate (31)
To a stirred solution of 13 (10.0 g, 32.0 mmol) and Et3N (9.15 mL, 65.5 mmol, 2.05 equiv) in DCM (80 mL), which was cooled under nitrogen cooled at −40 °C, mesyl chloride (5.0 mL, 64.0 mmol, 2.0 equiv) in DCM (10 mL) was added dropwise. Transfer of mesyl chloride was completed by rinsing with DCM (2 × 1 mL). The reaction mixture was allowed to warm up slowly to −10 °C and then further stirred for 4 h at −10 °C. TLC monitoring showed completion of the reaction, and distilled water (120 mL) was added. Extraction of the aqueous layer was done with DCM. Combined organic layers were washed with brine and dried over Na2SO4. DCM was removed under reduced pressure, and the crude product was purified by column chromatography on silica gel (36 g, 0%–1% acetone in petroleum ether) to afford 31 as a colorless oil (8.06 g, 65%). Rf = 0.26 (petroleum ether/acetone 80:20).
1H NMR (400 MHz, CDCl3): δ 5.55 (dtt, 1H, J = 10.9, 7.3, 1.5 Hz), 5.37 (dtt, 1H, J = 10.9, 7.2, 1.5 Hz), 4.69 (tt, 1H, J = 6.2, 6.1 Hz), 3.67 (s, 3H), 2.99 (s, 3H), 2.55–2.37 (m, 2H), 2.30 (t, 2H, J = 7.5 Hz), 2.03 (br dt (apparent q), 2H, J = 7, 6.5 Hz), 1.72–1.57 (m, 4H), 1.49–1.18 (m, 16H), 0.88 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 174.32 (Cquat, CO), 133.81 (CH=CH), 123.03 (CH=CH), 83.66 (CH), 51.47 (CH3), 38.68 (CH3), 34.25 (CH2), 34.08 (CH2), 32.54 (CH2), 31.64 (CH2), 29.39 (CH2), 29.14 (CH2), 29.10 (CH2), 29.08 (CH2), 29.00 (CH2), 27.42 (CH2), 25.05 (CH2), 24.92 (CH2), 22.56 (CH2), 14.05 (CH3).
[α]D18: +12.3; [α]57818: +12.5; [α]54618: +14.2; [α]43618: +24.3; [α]36518: +38.6 (c 4.00, acetone).
[α]D18: +10.6; [α]57818: +11.0; [α]54618: +12.6; [α]43618: +21.5; [α]36518: +33.7 (c 4.00, CHCl3).
3.2.21. Methyl (S,Z)-12-azidooctadec-9-enoate (32)
To a solution of 31 (9.1 g, 23.3 mmol) in DMSO (34.9 mL), was added NaN3 (2.6 g, 40.0 mmol, 1.7 equiv). The flask was then purged with nitrogen, stoppered and dipped in a preheated bath at 80 °C. Stirring was maintained for 16 h at the same temperature. TLC monitoring showed completion of the reaction, and it was brought back to rt, then quenched with an aqueous solution of NH4Cl. Extraction was done with petroleum ether/EtOAc (80:20, 185 mL), and organic layers were washed with brine and dried over Na2SO4. Solvent was removed under reduced pressure to obtain the crude product as a light-yellow oil. The crude product was then purified by column chromatography on silica gel (30 g, 0%–0.5% acetone in petroleum ether) to provide 32 as an oil (6.26 g, 80%). Rf = 0.58 (petroleum ether/acetone 80:20).
IR (KBr) ν 3011, 2929, 2855, 2100, 1742, 1456, 1250, 1197, 726 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.52 (dtt, 1H, J = 10.9, 7.3, 1.5 Hz), 5.38 (dtt, 1H, J = 10.9, 7.2, 1.5 Hz), 3.67 (s, 3H), 3.33–3.25 (m, which could be analyzed as a dddd at 3.29 ppm with J= 7.6, 6.5, 6.5, 5.3 Hz, 1H, CHN3), 2.33–2.25 (m, 4H with a dd at 2.34 ppm integrating for 2 protons, J = 7.7, 7.4 Hz), 2.04 (br td (apparent q), 2H, J = 7.0, 6.8 Hz), 1.68–1.57 (m, 2H), 1.56–1.40 (m, 3H), 1.40–1.22 (m, 15H), 0.89 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 174.33 (Cquat, CO), 133.10 (CH=CH), 124.58 (CH=CH), 62.95 (CH), 51.46 (CH3), 34.10 (CH2), 33.99 (CH2), 32.28 (CH2), 31.72 (CH2), 29.46 (CH2), 29.15 (CH2), 29.10 (2 CH2), 29.09 (CH2), 27.40 (CH2), 26.13 (CH2), 24.93 (CH2), 22.59 (CH2), 14.07 (CH3).
3.2.22. (S,Z)-12-Azidooctadec-9-enal (33)
To a vigorously stirred solution of 32 (5.063 g, 15 mmol) in Et2O (60 mL), which was cooled at −80 °C under nitrogen, was added dropwise a solution of Dibal (25% in hexane, 28 mL, 34.5 mmol, 2.3 equiv). The resulting mixture was allowed to stir for 2 h at −80 °C. TLC monitoring showed completion of the reaction. A saturated aqueous solution of potassium sodium tartrate (25 mL) was added, and stirring was continued for 10 min. Extraction was then done with petroleum ether/EtOAc (80:20). Organic layers were washed with brine and dried over Na2SO4. Solvent was evaporated under reduced pressure, and the crude product was used as such for the next step. For analytical purposes, the crude reaction product, which was initially obtained from 10 times less product, was purified by column chromatography on silica gel (5.2 g, 0%–1% acetone in petroleum ether) to afford 33 as an ochre oil (313.1 mg from 506.3 mg of 32, 68%). Rf = 0.4 (petroleum ether/acetone 85:15).
IR (KBr) ν 34,340, 2929, 2856, 2716, 2100, 1727, 1466, 1273, 725 cm−1.
1H NMR (400 MHz, CDCl3): δ 9.77 (t, 1H, J = 1.9 Hz, CHO), 5.52 (dtt, 1H, J = 10.9, 7.2, 1.5 Hz, CH=CH–CH2–CHN3), 5.39 (dtt, 1H, J = 10.9, 7.2, 1.5 Hz, CH=CH–CH2–CHN3), 3.33–3.25 (m, which could be analyzed as a dddd at 3.29 ppm with J = 7.6, 6.6, 6.5, 5.2 Hz, 1H, CHN3), 2.42 (td, 2H, J = 7.4, 1.9 Hz, CH2CHO), 2.35–2.20 (m, 2H, CHCH2CH=CH), 2.05 (br td (apparent q), 2H, J = 7.0, 6.8 Hz, CH=CHCH2), 1.68–1.58 (m, 2H, CH2CH2CHO), 1.57–1.47 (m, 2H, CH2CHN3), 1.47–1.41 (m, 1H, of CH2CH2CHN3), 1.41–1.24 (m, 15H, 1H of CH2CH2CHN3, CH3(CH2)3 and (CH2)4CH2CH2CHO), 0.89 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 202.92 (CHO), 133.05 (CH=CH), 124.62 (CH=CH), 62.94 (CH–N3), 43.91 (CH2), 33.99 (CH2), 32.29 (CH2), 31.72 (CH2CH2CH3), 29.44 (CH2), 29.25 (CH2), 29.11 (CH2), 29.08 (CH2), 29.07 (CH2), 27.39 (CH2), 26.13 (CH2), 22.58 (CH2CH3), 22.05 (CH2), 14.07 (CH3).
[α]D18.5: −31.5; [α]57818.5: −33.4; [α]54618.5: −38.2; [α]43618.5: −67.5; [α]36518.5: −111.0 (c 2.00, acetone).
[α]D18.5: −24.8; [α]57818.5: −25.9; [α]54618.5: −29.7; [α]43618.5: −52.0; [α]36518.5: −85.4 (c 2.60, CHCl3).
Elementary analysis calculated for C18H33N3O: C, 70.31; H, 10.82; N, 13.67 found: C, 70.39; H, 11.01; N, 13.38.
3.2.23. (S,Z)-12-Azidooctadec-9-en-1-ol (34)
To a vigorous stirred solution of 33 (crude made from 5.063 g, 15 mmol of 32) in ethanol (15 mL), which was cooled at 0 °C under nitrogen, was added NaBH4 (435 mg, 11.25 mmol, 0.75 equiv). The resulting mixture was allowed to stir for 1 h at 0 °C. TLC monitoring showed completion of the reaction. About 10–20 drops of acetone were added, and ethanol was removed under reduced pressure. After addition of water, extraction with petroleum ether/EtOAc (80:20), organic layers were washed with brine and dried over Na2SO4. Solvent was removed under reduced pressure, and the crude product was purified by two successive column chromatographies on silica gel (15 g, 0%–2% acetone in petroleum ether) to afford 34 as an ochre oil (3.44 g, 74% from 32). Rf = 0.36 (petroleum ether/acetone 85:15).
1H NMR (400 MHz, CDCl3): δ 5.52 (dtt, 1H, J = 10.9, 7.3, 1.5 Hz), 5.38 (dtt, 1H, J = 10.9, 7.3, 1.5 Hz), 3.64 (t, 2H, J = 6.6 Hz), 3.33–3.25 (m, which could be analyzed as a dddd at 3.29 ppm with J = 7.8, 6.8, 6.4, 5.2 Hz, 1H, CHN3), 2.35–2.20 (m, 2H), 2.04 (br td (apparent q), 2H, J = 7.0, 7.0 Hz), 1.61–1.41 (m, 5H), 1.41–1.23 (m, 17H), 0.89 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 133.17 (CH=CH), 124.53 (CH=CH), 63.07 (CH2), 62.96 (CH), 33.98 (CH2), 32.79 (CH2), 32.28 (CH2), 31.72 (CH2), 29.51 (CH2), 29.49 (CH2), 29.39 (CH2), 29.24 (CH2), 29.09 (CH2), 27.43 (CH2), 26.13 (CH2) 25.73 (CH2), 22.59 (CH2), 14.07 (CH3).
3.2.24. (S,Z)-12-Azidooctadec-9-en-1-yl methanesulfonate (35)
To a stirred solution of 34 (3.93 g, 12.7 mmol) and Et3N (2.7 mL, 19.1 mmol, 1.5 equiv) in DCM (38 mL), which was cooled at –50 °C under nitrogen, was added dropwise mesyl chloride (1.25 mL, 15.9 mmol, 1.25 equiv) in DCM (5.0 mL). Transfer of mesyl chloride was completed by rinsing with DCM (3 × 0.2 mL). The mixture was then allowed to stir for 4 h at −50 °C and then allowed to warm up slowly to −5 °C. TLC monitoring (elution with DCM) showed completion of the reaction, and distilled water (50 mL) was added to quench the reaction. Extraction was done with DCM, and organic layers were washed with brine and dried over Na2SO4. DCM was removed under reduced pressure, and the crude product was purified by two successive column chromatographies on silica gel (15 g and then 40 g, 0%–3% acetone in petroleum ether) to provide 35 as an ochre oil (2.89 g, 59%). Rf = 0.61 (petroleum ether/acetone 80:20).
1H NMR (400 MHz, CDCl3): δ 5.52 (dtt, 1H, J = 10.9, 7.3, 1.4 Hz), 5.39 (dtt, 1H, J = 10.9, 7.3, 1.5 Hz), 4.22 (t, 2H, J = 6.6 Hz), 3.33–3.25 (m, which could be analyzed as a br dddd at 3.29 ppm with J = 7.2, 6.8, 6.5, 5.3 Hz, 1H, CHN3), 3.01 (s, 3H), 2.35–2.22 (m, 2H), 2.05 (br dt (apparent q), 2H, J = 7.2, 7.0 Hz), 1.79–1.70 (m, 2H), 1.56–1.22 (m, 20H with 2H at 1.56–1.47 ppm), 0.89 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 133.07 (CH=CH), 124.60 (CH=CH), 70.17 (CH2), 62.94 (CH), 37.37 (CH3), 33.99 (CH2), 32.29 (CH2), 31.72 (CH2), 29.47 (CH2), 29.31 (CH2), 29.16 (CH2), 29.12 (CH2), 29.08 (CH2), 28.99 (CH2), 27.40 (CH2), 26.13 (CH2), 25.41 (CH2), 22.58 (CH2), 14.07 (CH3).
3.2.25. (R)-4-((((S,Z)-12-Azidooctadec-9-en-1-yl)oxy)methyl)-2,2-dimethyl-1,3-dioxolane (36)
To a stirred mixture at rt of 35 (2.48 g, 6.4 mmol), n-Bu4NBr (515.8 mg, 1.6 mmol, 0.25 equiv), DMSO (12.8 mL) and 50% aqueous NaOH (0.67 mL, 2 equiv) was added (R)-solketal 18 (1002.8 mg, ≥95% pure, 7.21 mmol, 1.13 equiv). The resulting mixture was then stirred overnight for 15 h at 35 °C. TLC monitoring showed completion of the reaction, and distilled water (40 mL) was added. Extraction was done with petroleum ether/EtOAc (80:20), and organic layers were washed again with brine and dried over Na2SO4. Solvent was removed under reduced pressure, and the crude product was purified by two successive column chromatographies on silica gel (11 g and then 30 g, 0%–0.5% acetone in petroleum ether) to afford 36 as an ochre oil (1.41 g, 52%). Rf = 0.56 (petroleum ether/acetone 95:5).
1H NMR (400 MHz, CDCl3): δ 5.52 (dtt, 1H, J = 10.9, 7.2, 1.5 Hz), 5.38 (dtt, 1H, J = 10.9, 7.2, 1.5 Hz), 4.27 (tt, 1H, J = 6.4, 5.7 Hz), 4.06 (dd, 1H, J = 8.2, 6.4 Hz), 3.73 (dd, 1H, J = 8.2, 6.4 Hz), 3.54–3.39 (m, 4H with 1H dd at 3.52 ppm, J = 9.9, 5.7 Hz and 1H dd at 3.42 ppm, J = 9.9, 5.6 Hz), 3.33–3.25 (m, 1H, CHN3), 2.36–2.20 (m, 2H), 2.04 (br td (apparent q), 2H, J = 7.1, 7.0 Hz), 1.62–1.40 (m, 7H, 2 CH2 + CH3 q at 1.43 ppm, J = 0.6 Hz), 1.40–1.24 (m, 21H, 9 CH2 + CH3 q at 1.37 ppm, J = 0.6 Hz), 0.89 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 133.18 (CH=CH), 124.51 (CH=CH), 109.37 (CMe2), 74.76 (CH), 71.88 (CH2), 71.83 (CH2), 66.94 (CH2), 62.95 (CH), 33.98 (CH2), 32.28 (CH2), 31.72 (CH2), 29.55 (CH2), 29.52 (CH2), 29.48 (CH2), 29.42 (CH2), 29.26 (CH2), 29.08 (CH2), 27.44 (CH2), 26.78 (CH3), 26,13 (CH2), 26.05 (CH2), 25.43 (CH3), 22.58 (CH2), 14.07 (CH3).
HRMS (ESI, m/z) calculated for C24H45N3O3Na [M + Na]+: 446.3358, found: 446.3365.
3.2.26. (S)-3-(((S,Z)-12-Azidooctadec-9-en-1-yl)oxy)propane-1,2-diol (10)
To a solution of 36 (610.7 mg, 1.44 mmol) in methanol (5.8 mL), was added p-toluenesulfonic acid monohydrate (13.7 mg, 0.072 mmol, 0.05 equiv) and distilled water (0.58 mL). The flask was then purged with nitrogen, stoppered and dipped in a preheated bath at 60 °C. Stirring was maintained for 4 h at 60 °C. TLC monitoring showed completion of the reaction, and sodium bicarbonate (6.7 mg, 0.08 mmol, 0.055 equiv) was added to the mixture, and stirring was continued for 1 h at 60 °C. Methanol was removed under reduced pressure, and the crude product was purified by column chromatography on silica gel (5 g, 0%–5% acetone in petroleum ether and then petroleum ether + 5% acetone + 12% methanol) to provide 10 as an ochre oil (539.1 mg, 97%). Rf = 0.03 (petroleum ether/acetone 80:20).
IR (KBr) ν 3396, 2928, 2855, 2100, 1464, 1254, 1125, 1037 cm−1.
1H NMR (400 MHz, CDCl3): δ 5.52 (dtt, 1H, J = 10.9, 7.3, 1.4 Hz), 5.38 (dtt, 1H, J = 10.9, 7.2, 1.5 Hz), 3.91–3.83 (m, which could be analyzed as a br tt at 3.86 ppm with J = 5.3, 4.1 Hz, exchange with D2O improved a little bit the resolution), 3.77–3.68 (m, 1H (dd at 3.71 ppm after exchange with D2O, J = 11.4, 3.9 Hz)), 3.65 (dd, 1H, J = 11.4, 5.2 Hz, unchanged after exchange with D2O), 3.56–3.42 (m, 4H with 1H dd at 3.54 ppm, J = 9.7, 4.0 Hz), 3.33–3.25 (m, which could be analyzed as a dddd at 3.29 ppm with J = 7.6, 6.6, 6.4, 5.2 Hz, 1H, CHN3), 2.72 (envelope from 2.85 to 2.59 ppm, 1H, OH), 2.37–2.20 (m, 3H: 1 OH as a partly overlapped envelope, which topped at 2.32 ppm + 1 CH2, which was centered at 2.29 ppm), 2.05 (br dt (apparent q), 2H, J = 7.1, 6.9 Hz), 1.62–1.40 (m, 5H), 1.40–1.23 (m, 17H), 0.89 (t, 3H, J = 6.9 Hz, CH3).
13C NMR (100 MHz, CDCl3): δ 133.16 (CH=CH), 124.53 (CH=CH), 72.52 (CH2), 71.83 (CH2), 70.43 (CH), 64.29 (CH2), 62.95 (CH), 33.97 (CH2), 32.28 (CH2), 31.72 (CH2), 29.57 (CH2), 29.51 (CH2), 29.46 (CH2), 29.41 (CH2), 29.24 (CH2), 29.08 (CH2), 27.43 (CH2), 26.13 (CH2), 26.07 (CH2), 22.58 (CH2), 14.07 (CH3).
[α]D21: −17.7; [α]57821: −18.7; [α]54621: −21.3; [α]43621: −37.9; [α]36521: −62.5 (c 2.26, CHCl3).
HRMS (ESI, m/z) calculated for C21H41N3O3Na [M + Na]+: 406.3046, found: 406.3046, calculated for C21H40N3O3Na2 [M − H + 2Na]+: 428.2865, found: 428.2882, calculated for C21H41NO3Na [M – N2 + Na]+: 378.2984, found: 378.2998.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}