Hp-s1 Ganglioside Suppresses Proinflammatory Responses by Inhibiting MyD88-Dependent NF-κB and JNK/p38 MAPK Pathways in Lipopolysaccharide-Stimulated Microglial Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
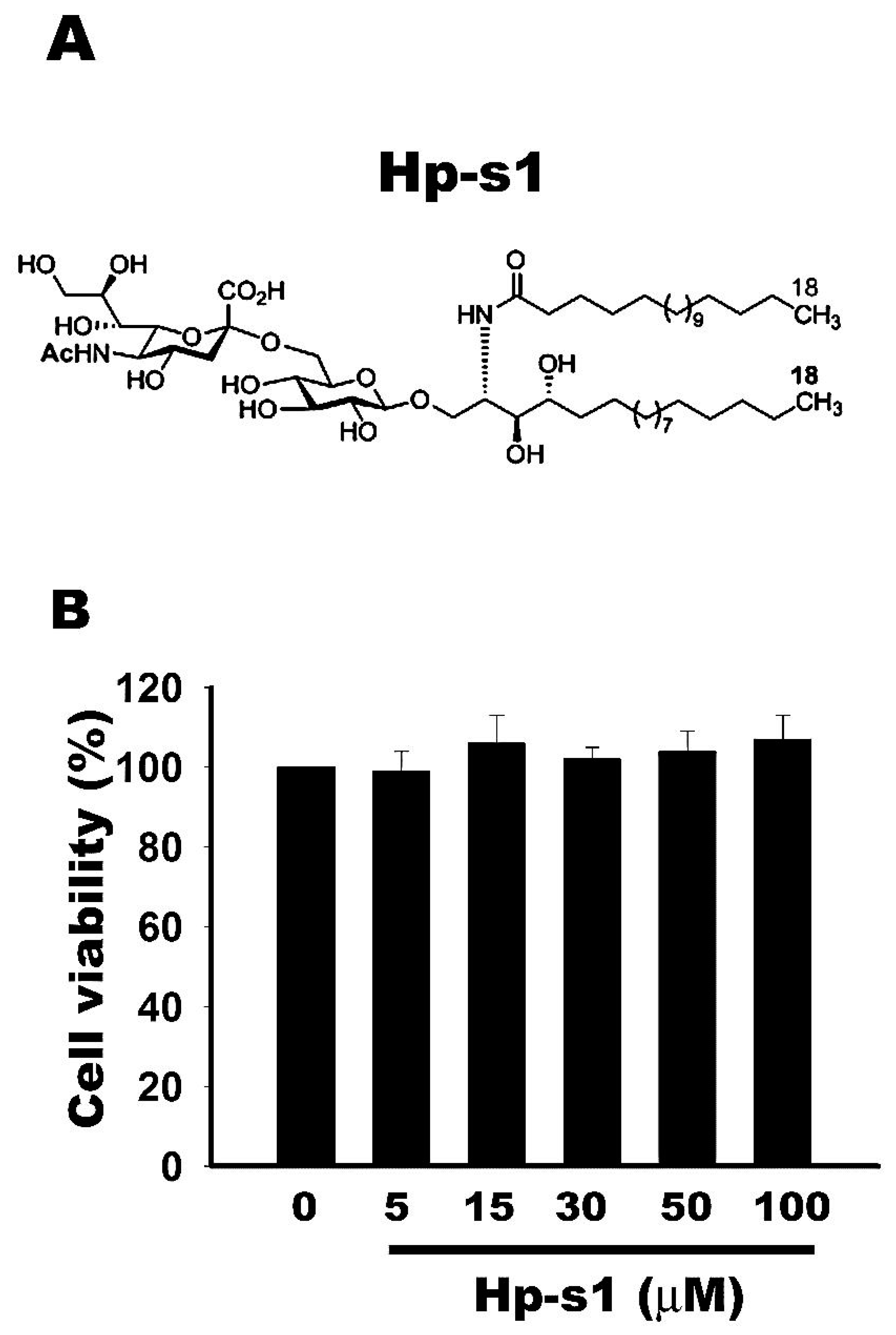
2.1. Cell Viability Is Not Affected by Hp-s1 Ganglioside
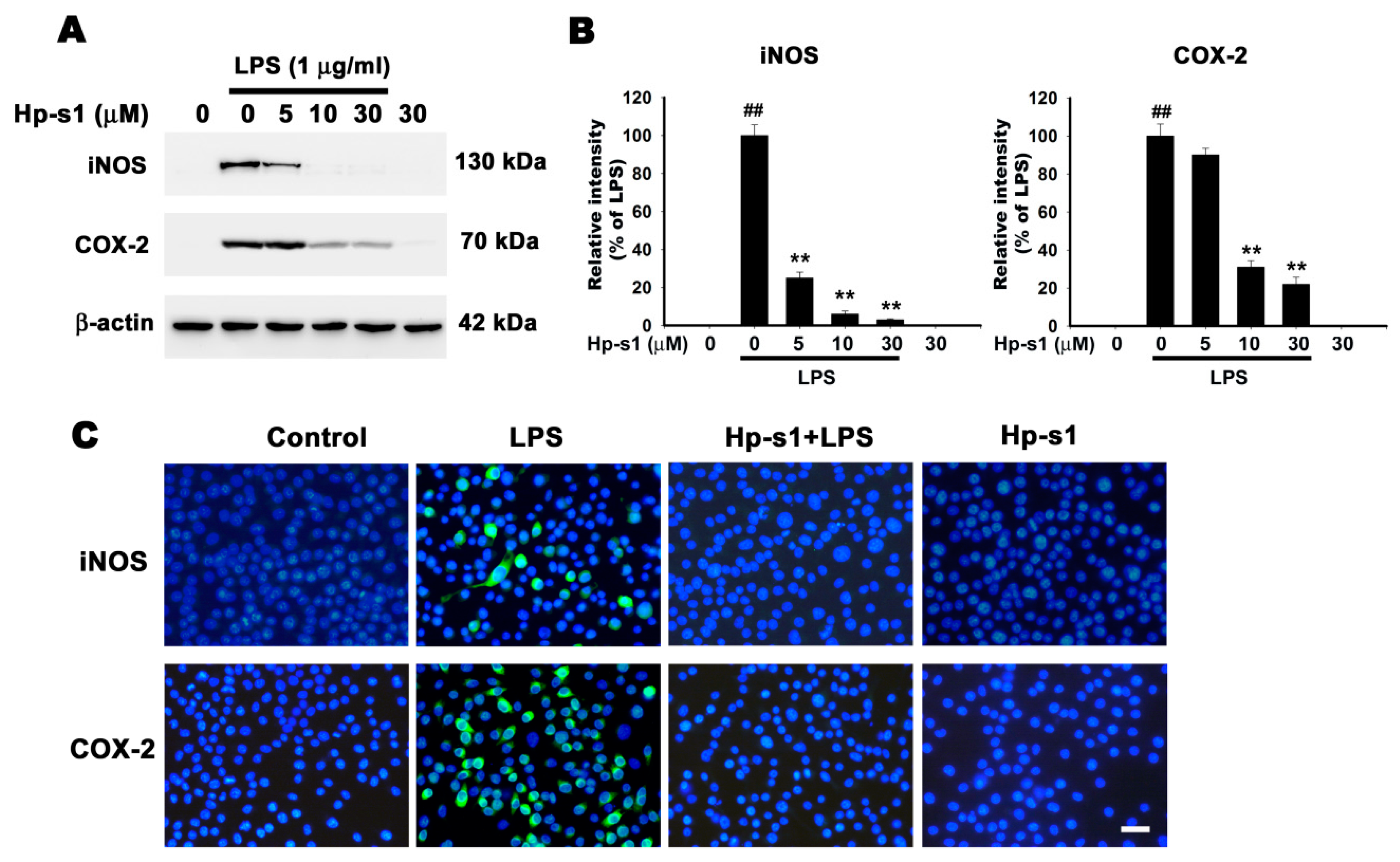
2.2. Hp-s1 Inhibits the LPS-Induced Expression of iNOS and COX-2
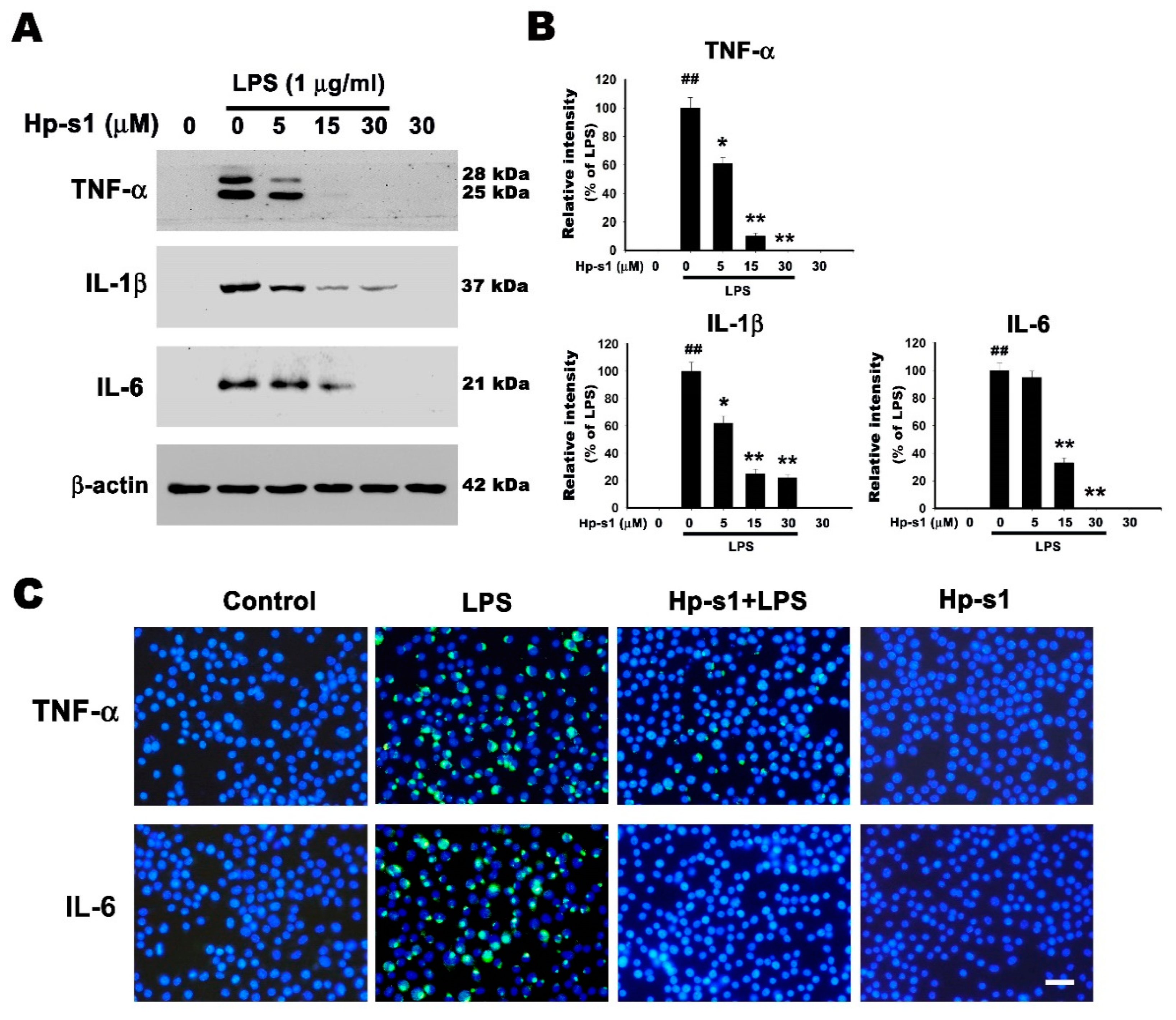
2.3. Hp-s1 Suppresses the LPS-Induced Expression of Proinflammatory Cytokines in Microglial Cells
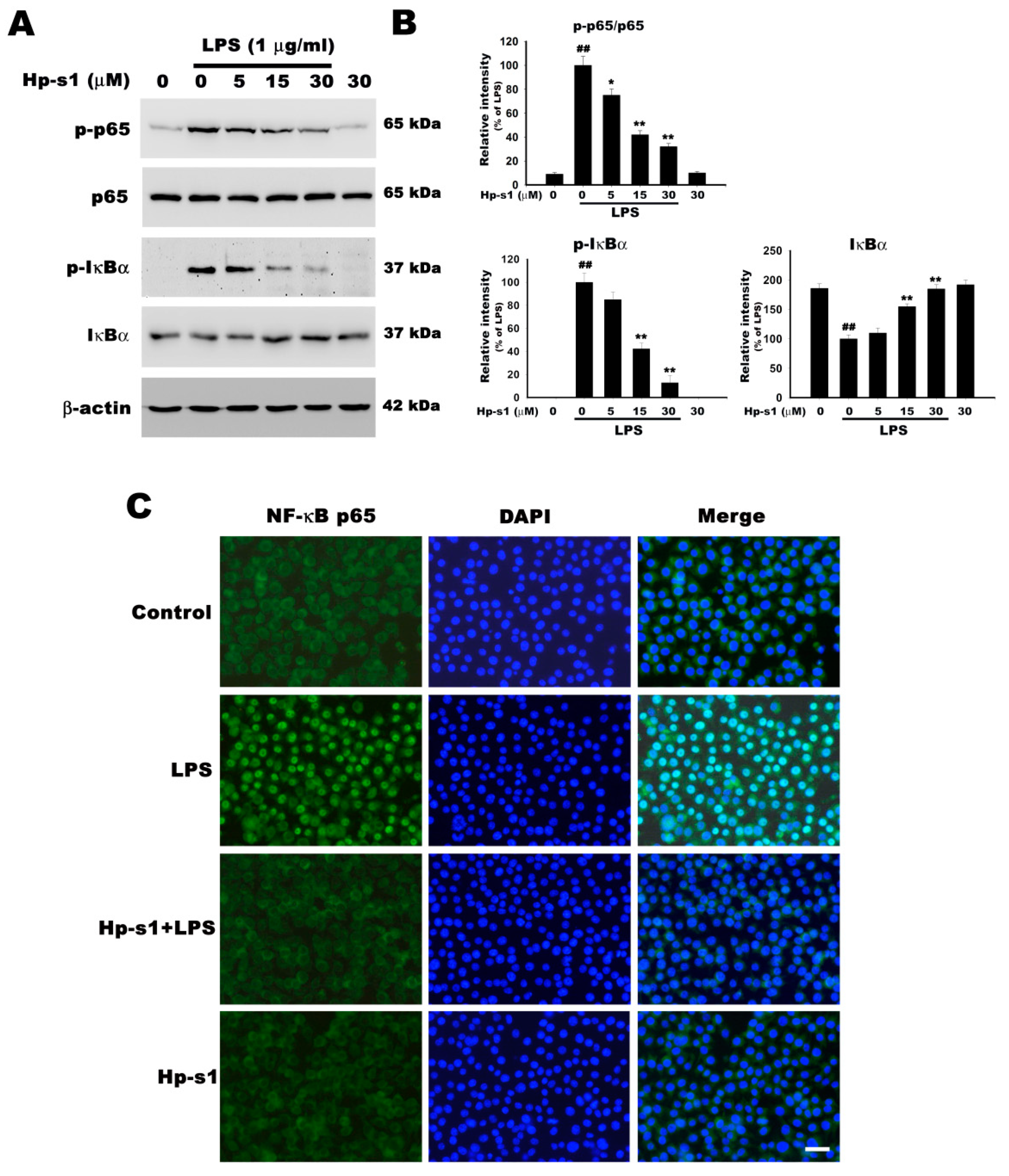
2.4. Hp-s1 Inhibits the LPS-Induced NF-κB Activation
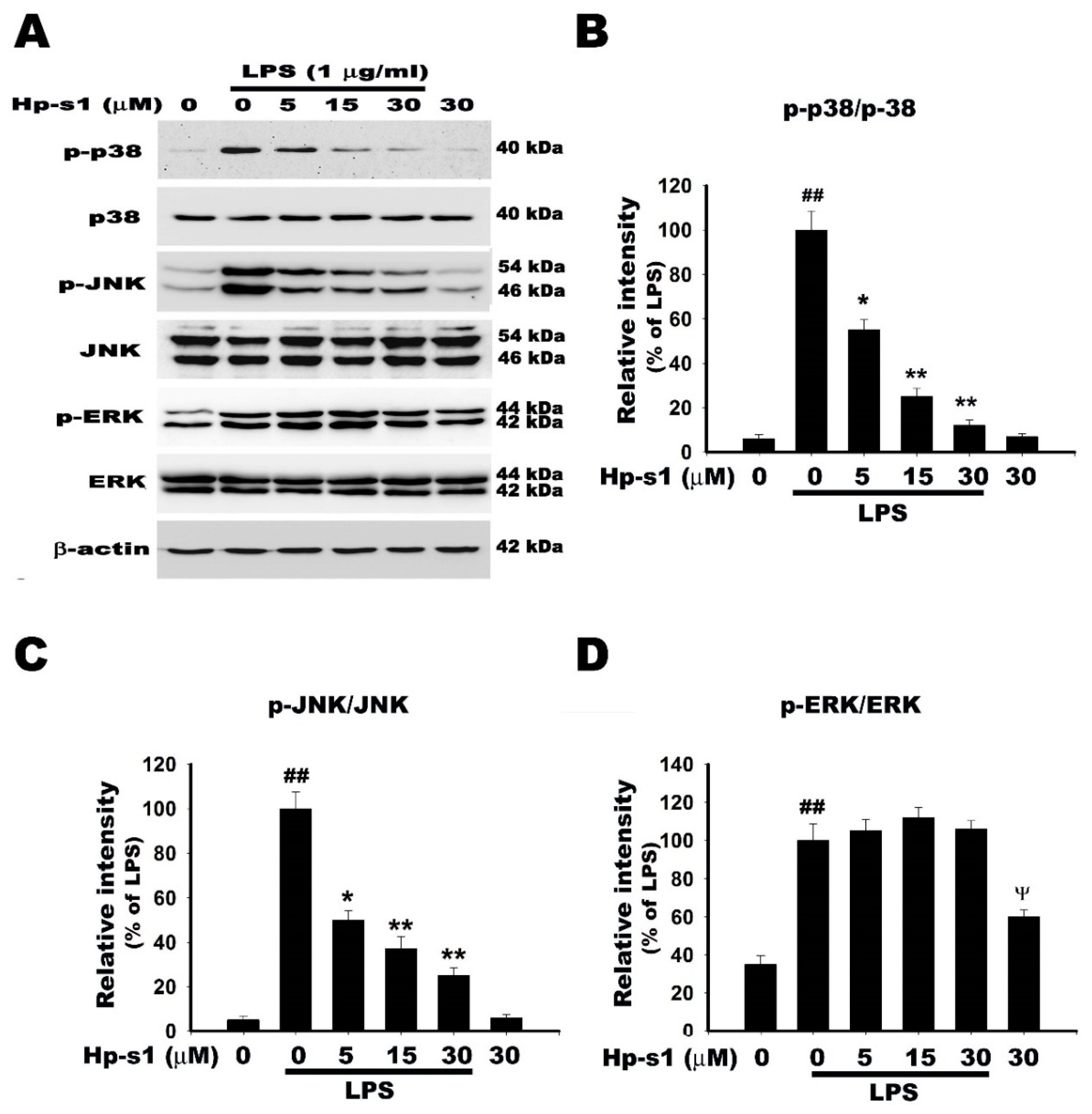
2.5. Hp-s1 Inhibits the LPS-Induced Activation of JNK and p38 MAPK
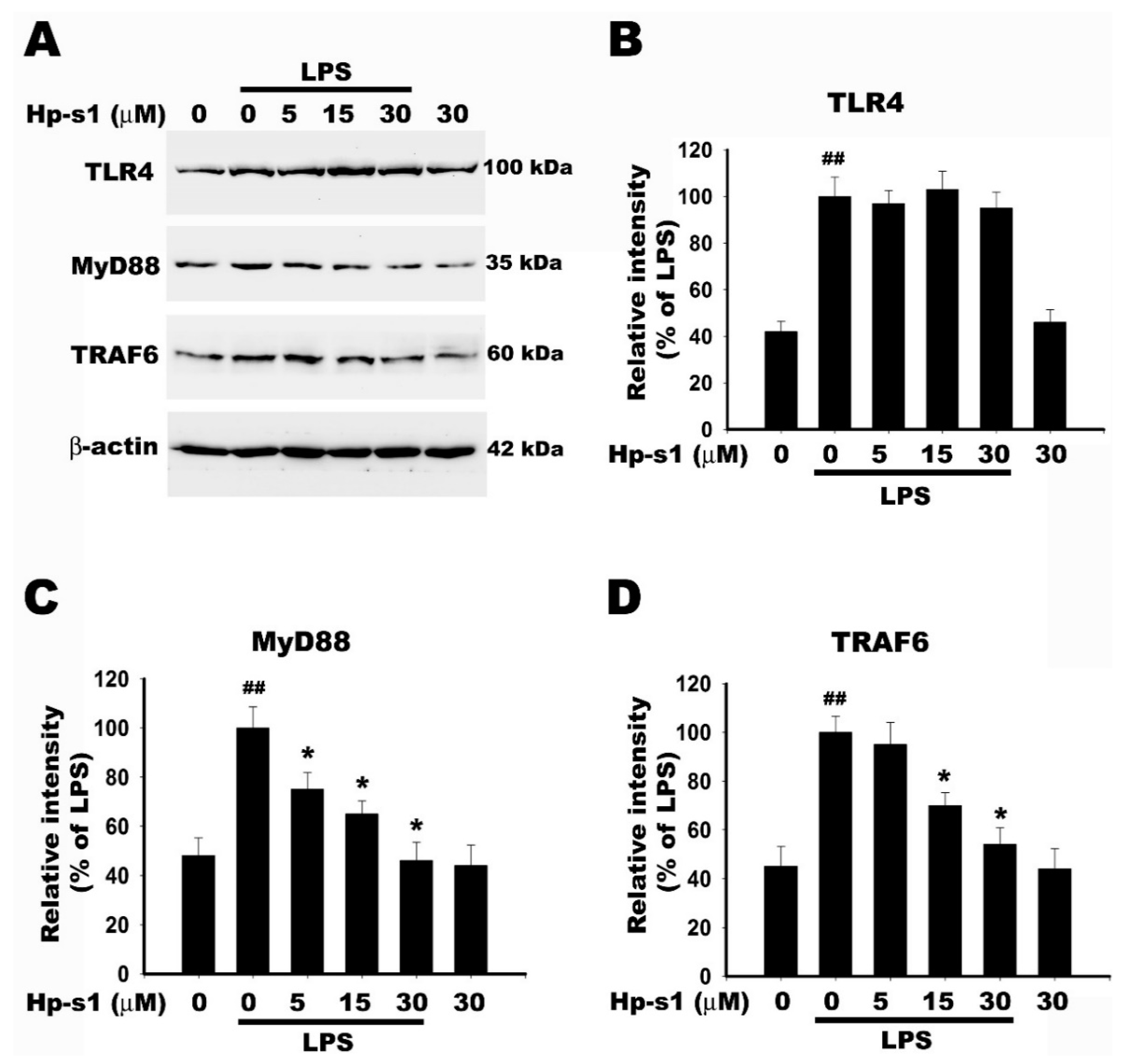
2.6. Hp-s1 Blocks the LPS-Induced MyD88/TRAF6-Dependent Signaling Pathway
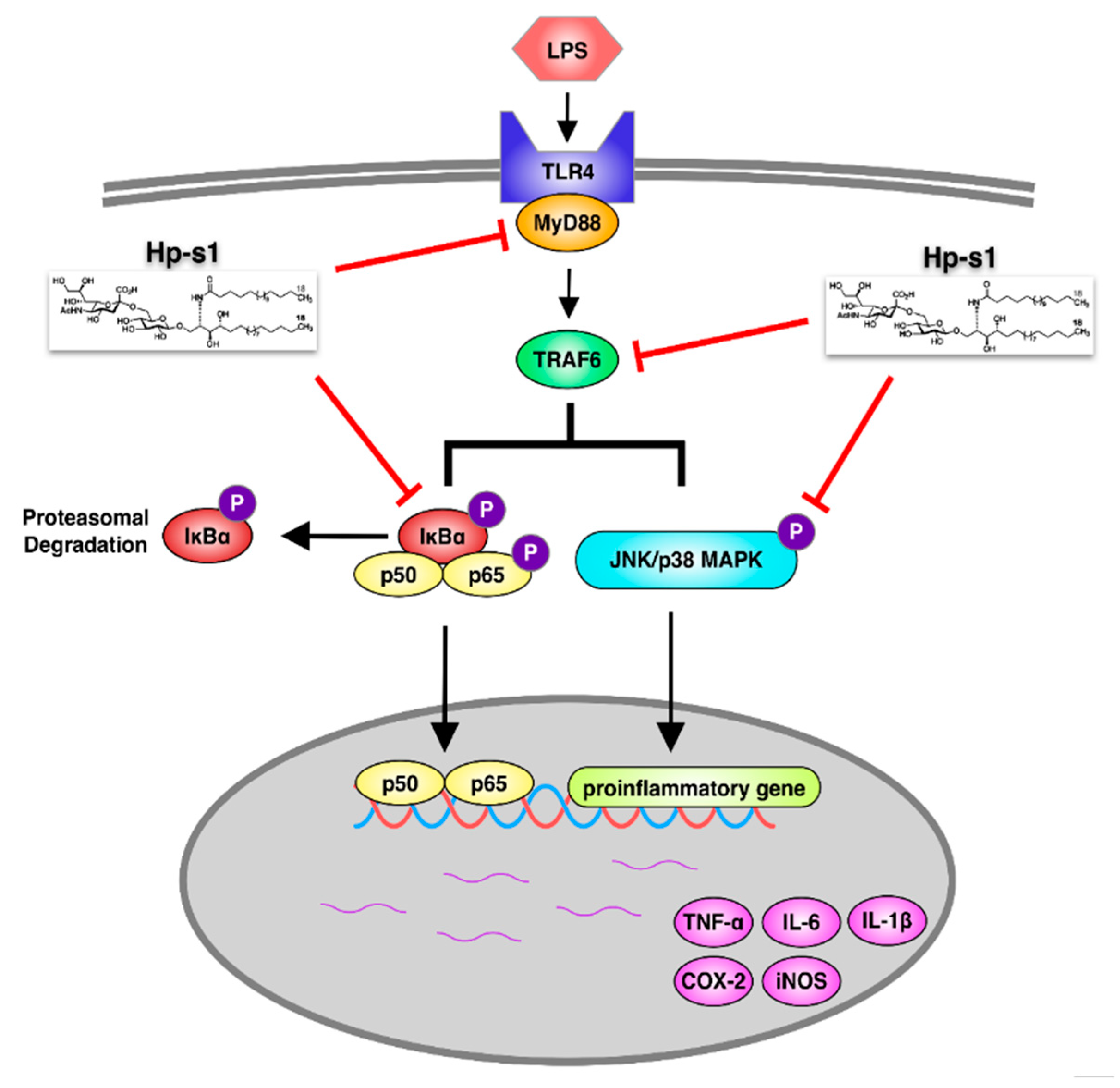
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Microglial Cell Culture
4.3. Hp-s1 Ganglioside Administration
4.4. Cell viability Assays
4.5. Immunofluorescence
4.6. Western Blot Analysis
4.7. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2016, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2012, 61, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Zhang, Y.; Chen, Y.; Zhu, J.; Yang, Y.; Zhang, H.-L. Role of Microglia in Neurological Disorders and Their Potentials as a Therapeutic Target. Mol. Neurobiol. 2016, 54, 7567–7584. [Google Scholar] [CrossRef]
- Catorce, M.N.; Gevorkian, G. LPS-induced Murine Neuroinflammation Model: Main Features and Suitability for Pre-clinical Assessment of Nutraceuticals. Curr. Neuropharmacol. 2016, 14, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Lee, S. Toll-like receptors and inflammation in the CNS. Curr. Drug Targets Inflamm. Allergy 2002, 1, 181–191. [Google Scholar]
- Bachstetter, A.D.; Van Eldik, L.J. The p38 MAP Kinase Family as Regulators of Proinflammatory Cytokine Production in Degenerative Diseases of the CNS. Aging Dis. 2010, 1, 199–211. [Google Scholar]
- Zhao, Y.; Lukiw, W.J. Bacteroidetes Neurotoxins and Inflammatory Neurodegeneration. Mol. Neurobiol. 2018, 55, 9100–9107. [Google Scholar] [CrossRef]
- Schnaar, R.L. The Biology of Gangliosides. Adv. Carbohydr. Chem. Biochem. 2019, 76, 113–148. [Google Scholar]
- Magistretti, P.J.; Geisler, F.H.; Schneider, J.S.; Li, P.A.; Fiumelli, H.; Sipione, S. Gangliosides: Treatment Avenues in Neurodegenerative Disease. Front. Neurol. 2019, 10, 859. [Google Scholar] [CrossRef] [Green Version]
- Dukhinova, M.; Veremeyko, T.; Yung, A.W.; Kuznetsova, I.S.; Lau, T.Y.; Kopeikina, E.; Chan, A.M.; Ponomarev, E.D. Fresh evidence for major brain gangliosides as a target for the treatment of Alzheimer’s disease. Neurobiol. Aging 2019, 77, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Schnaar, R.L. Gangliosides of the Vertebrate Nervous System. J. Mol. Boil. 2016, 428, 3325–3336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaeva, S.D.; Bayunova, L.; Sokolova, T.V.; Vlasova, Y.A.; Bachteeva, V.; Avrova, N.F.; Parnova, R. GM1 and GD1a gangliosides modulate toxic and inflammatory effects of E. coli lipopolysaccharide by preventing TLR4 translocation into lipid rafts. Biochim. Biophys. Acta (BBA) Mol. Cell Boil. Lipids 2015, 1851, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Pyo, H.; Joe, E.-H.; Jung, S.; Lee, S.H.; Jou, I. Gangliosides Activate Cultured Rat Brain Microglia. J. Boil. Chem. 1999, 274, 34584–34589. [Google Scholar] [CrossRef] [Green Version]
- Jou, I.; Lee, J.H.; Park, S.Y.; Yoon, H.J.; Joe, E.-H.; Park, E.J. Gangliosides Trigger Inflammatory Responses via TLR4 in Brain Glia. Am. J. Pathol. 2006, 168, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, K.; Ohmi, Y.; Kondo, Y.; Ohkawa, Y.; Tajima, O.; Furukawa, K. Regulatory function of glycosphingolipids in the inflammation and degeneration. Arch. Biochem. Biophys. 2015, 571, 58–65. [Google Scholar] [CrossRef]
- Furukawa, K.; Ohmi, Y.; Yesmin, F.; Tajima, O.; Kondo, Y.; Zhang, P.; Hashimoto, N.; Ohkawa, Y.; Bhuiyan, R.H.; Furukawa, K. Novel Molecular Mechanisms of Gangliosides in the Nervous System Elucidated by Genetic Engineering. Int. J. Mol. Sci. 2020, 21, 1906. [Google Scholar] [CrossRef] [Green Version]
- Ohmi, Y.; Tajima, O.; Ohkawa, Y.; Mori, A.; Sugiura, Y.; Furukawa, K.; Furukawa, K. Gangliosides play pivotal roles in the regulation of complement systems and in the maintenance of integrity in nerve tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 22405–22410. [Google Scholar] [CrossRef] [Green Version]
- Ohmi, Y.; Tajima, O.; Ohkawa, Y.; Yamauchi, Y.; Sugiura, Y.; Furukawa, K.; Furukawa, K. Gangliosides are essential in the protection of inflammation and neurodegeneration via maintenance of lipid rafts: Elucidation by a series of ganglioside-deficient mutant mice. J. Neurochem. 2011, 116, 926–935. [Google Scholar] [CrossRef]
- Ijuin, T.; Kitajima, K.; Song, Y.; Kitazume, S.; Inoue, S.; Haslam, S.M.; Morris, H.R.; Dell, A.; Inoue, Y. Isolation and identification of novel sulfated and nonsulfated oligosialyl glycosphingolipids from sea urchin sperm. Glycoconj. J. 1996, 13, 401–413. [Google Scholar] [CrossRef]
- Shelke, G.B.; Lih, Y.-H.; Liao, Y.-J.; Liang, C.-W.; Kuo, T.-M.; Ko, Y.-C.; Luo, S.-Y. Synthesis and Bioassay of Neurogenically Potent Gangliosides DSG-A, Hp-s1 and Their Analogues. ACS Chem. Neurosci. 2018, 9, 1264–1268. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-F.; Shih, C.-H.; Su, Y.-T.; Yao, C.-H.; Lian, J.-F.; Liao, C.-C.; Hsia, C.-W.; Shui, H.-A.; Rani, R. The total synthesis of a ganglioside Hp-s1 analogue possessing neuritogenic activity by chemoselective activation glycosylation. Org. Biomol. Chem. 2012, 10, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-F.; Yang, D.-J.; Ngo, T.H.; Shih, C.-H.; Wu, Y.-F.; Lee, C.-K.; Phraekanjanavichid, V.; Yen, S.-F.; Kao, S.-H.; Lee, H.-M.; et al. Ganglioside Hp-s1 Analogue Inhibits Amyloidogenic Toxicity in Alzheimer’s Disease Model Cells. ACS Chem. Neurosci. 2018, 10, 528–536. [Google Scholar] [CrossRef]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahimifard, M.; Maqbool, F.; Moeini-Nodeh, S.; Niaz, K.; Abdollahi, M.; Braidy, N.; Nabavi, S.M.; Nabavi, S.F. Targeting the TLR4 signaling pathway by polyphenols: A novel therapeutic strategy for neuroinflammation. Ageing Res. Rev. 2017, 36, 11–19. [Google Scholar] [CrossRef]
- Leitner, G.R.; Wenzel, T.; Marshall, N.; Gates, E.J.; Klegeris, A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targets 2019, 23, 865–882. [Google Scholar] [CrossRef]
- Azam, S.; Kim, I.-S.; Kim, J.; Haque, E.; Choi, D.-K. Regulation of Toll-Like Receptor (TLR) Signaling Pathway by Polyphenols in the Treatment of Age-Linked Neurodegenerative Diseases: Focus on TLR4 Signaling. Front. Immunol. 2019, 10, 1000. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Kopitar-Jerala, N. Innate Immune Response in Brain, NF-Kappa B Signaling and Cystatins. Front. Mol. Neurosci. 2015, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Choi, E.-J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef]
- Han, I.O.; Kim, K.-W.; Ryu, J.H.; Kim, W.-K. p38 mitogen-activated protein kinase mediates lipopolysaccharide, not interferon-gamma, -induced inducible nitric oxide synthase expression in mouse BV2 microglial cells. Neurosci. Lett. 2002, 325, 9–12. [Google Scholar] [CrossRef]
- Li, M.; Zhang, D.; Ge, X.; Zhu, X.; Zhou, Y.; Zhang, Y.; Peng, X.; Shen, A. TRAF6-p38/JNK-ATF2 axis promotes microglial inflammatory activation. Exp. Cell Res. 2019, 376, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Lee, J.H.; Yumnam, S.; Ji, E.; Kim, S.Y. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-kappaB Inhibition and Nrf2/HO-1 Activation. Cells 2019, 8, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Q.; Zhang, R.; Bhat, N.R. MAP kinase regulation of IP10/CXCL10 chemokine gene expression in microglial cells. Brain Res. 2006, 1086, 9–16. [Google Scholar] [CrossRef]
- Jeong, Y.-H.; Kim, Y.; Song, H.; Chung, Y.S.; Park, S.B.; Kim, H.-S. Anti-Inflammatory Effects of α-Galactosylceramide Analogs in Activated Microglia: Involvement of the p38 MAPK Signaling Pathway. PLoS ONE 2014, 9, e87030. [Google Scholar] [CrossRef] [Green Version]
- Duchemin, A.-M.; Ren, Q.; Mo, L.; Neff, N.H.; Hadjiconstantinou, M. GM1 ganglioside induces phosphorylation and activation of Trk and Erk in brain. J. Neurochem. 2002, 81, 696–707. [Google Scholar] [CrossRef]
- Garofalo, T.; Sorice, M.; Misasi, R.; Cinque, B.; Mattei, V.; Pontieri, G.M.; Cifone, M.G.; Pavan, A. Ganglioside GM3 activates ERKs in human lymphocytic cells. J. Lipid Res. 2002, 43, 971–978. [Google Scholar]
- Zusso, M.; Lunardi, V.; Franceschini, D.; Pagetta, A.; Lo, R.; Stifani, S.; Frigo, A.C.; Giusti, P.; Moro, S. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. J. Neuroinflamm. 2019, 16, 148. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Dunne, A.; Edjeback, M.; Gray, P.; Jefferies, C.; Wietek, C. Mal and MyD88: Adapter proteins involved in signal transduction by Toll-like receptors. J. Endotoxin Res. 2003, 9, 55–59. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- O’Neill, L.A. Signal transduction pathways activated by the IL-1 receptor/toll-like receptor superfamily. Inducible Lymphoid Org. 2002, 270, 47–61. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jin, C.Y.; Kim, C.H.; Yoo, Y.H.; Choi, S.H.; Kim, G.Y.; Yoon, H.M.; Park, H.T.; Choi, Y.H. Isorhamnetin alleviates lipopolysaccharide-induced inflammatory responses in BV2 microglia by inactivating NF-kappaB, blocking the TLR4 pathway and reducing ROS generation. Int. J. Mol. Med. 2019, 43, 682–692. [Google Scholar] [PubMed] [Green Version]
- Wang, Y.; Cui, Y.; Cao, F.; Qin, Y.; Li, W.; Zhang, J. Ganglioside GD1a suppresses LPS-induced pro-inflammatory cytokines in RAW264.7 macrophages by reducing MAPKs and NF-κB signaling pathways through TLR4. Int. Immunopharmacol. 2015, 28, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kwak, C.-H.; Ha, S.-H.; Kwon, K.-M.; Abekura, F.; Cho, S.-H.; Chang, Y.-C.; Lee, Y.-C.; Ha, K.-T.; Chung, T.-W.; et al. Ganglioside GM3 suppresses lipopolysaccharide-induced inflammatory responses in rAW 264.7 macrophage cells through NF-κB, AP-1, and MAPKs signaling. J. Cell. Biochem. 2017, 119, 1173–1182. [Google Scholar] [CrossRef]
- Walters, K.M.; Woessner, K.M. An Overview of Nonsteroidal Antiinflammatory Drug Reactions. Immunol. Allerg. Clin. N. Am. 2016, 36, 625–641. [Google Scholar] [CrossRef]
- Peppa, M.; Krania, M.; A Raptis, S. Hypertension and other morbidities with Cushing’s syndrome associated with corticosteroids: A review. Integr. Blood Press. Control. 2011, 4, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Benvenuti, S.; Brandi, M.L. Corticosteroid-induced osteoporosis: Pathogenesis and prevention. Clin. Exp. Rheumatol. 2000, 18, 64–66. [Google Scholar]
- Takenouchi, T.; Ogihara, K.; Sato, M.; Kitani, H. Inhibitory effects of U73122 and U73343 on Ca influx and pore formation induced by the activation of P2X7 nucleotide receptors in mouse microglial cell line. Biochim. Biophys. Acta (BBA) Gen. Subj. 2005, 1726, 177–186. [Google Scholar] [CrossRef]
- Nakamichi, K.; Saiki, M.; Kitani, H.; Kuboyama, Y.; Morimoto, K.; Takayama-Ito, M.; Kurane, I. Roles of NF-kappaB and MAPK signaling pathways in morphological and cytoskeletal responses of microglia to double-stranded RNA. Neurosci. Lett. 2007, 414, 222–227. [Google Scholar] [CrossRef]
- Wu, Y.-F.; Tsai, Y.-F. Assistance of the C-7,8-Picoloyl Moiety for Directing the Glycosyl Acceptors into the α-Orientation for the Glycosylation of Sialyl Donors. Org. Lett. 2017, 19, 4171–4174. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shih, J.-H.; Tsai, Y.-F.; Li, I.-H.; Chen, M.-H.; Huang, Y.-S. Hp-s1 Ganglioside Suppresses Proinflammatory Responses by Inhibiting MyD88-Dependent NF-κB and JNK/p38 MAPK Pathways in Lipopolysaccharide-Stimulated Microglial Cells. Mar. Drugs 2020, 18, 496. https://doi.org/10.3390/md18100496
Shih J-H, Tsai Y-F, Li I-H, Chen M-H, Huang Y-S. Hp-s1 Ganglioside Suppresses Proinflammatory Responses by Inhibiting MyD88-Dependent NF-κB and JNK/p38 MAPK Pathways in Lipopolysaccharide-Stimulated Microglial Cells. Marine Drugs. 2020; 18(10):496. https://doi.org/10.3390/md18100496
Chicago/Turabian StyleShih, Jui-Hu, Yow-Fu Tsai, I-Hsun Li, Ming-Hua Chen, and Yuahn-Sieh Huang. 2020. "Hp-s1 Ganglioside Suppresses Proinflammatory Responses by Inhibiting MyD88-Dependent NF-κB and JNK/p38 MAPK Pathways in Lipopolysaccharide-Stimulated Microglial Cells" Marine Drugs 18, no. 10: 496. https://doi.org/10.3390/md18100496