Astaxanthin Inhibits p70 S6 Kinase 1 Activity to Sensitize Insulin Signaling


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
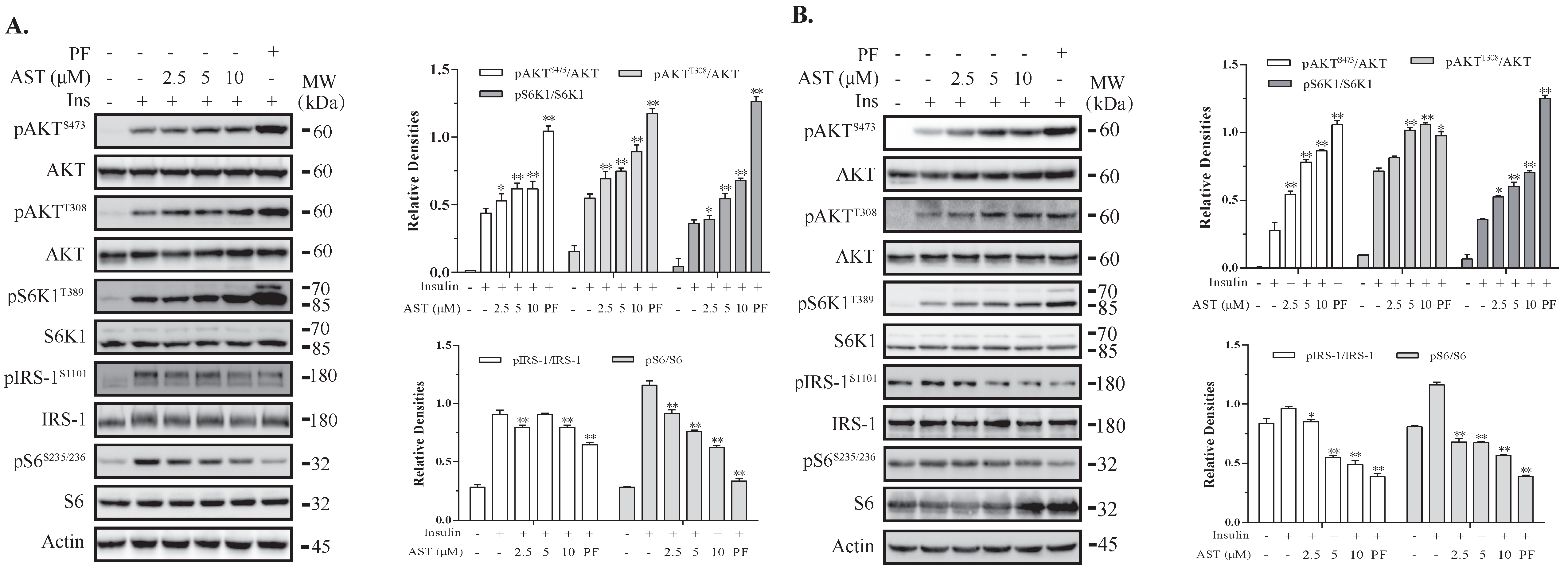
2.1. AST Induces Feedback Activation of the PI-3 Kinase Pathway
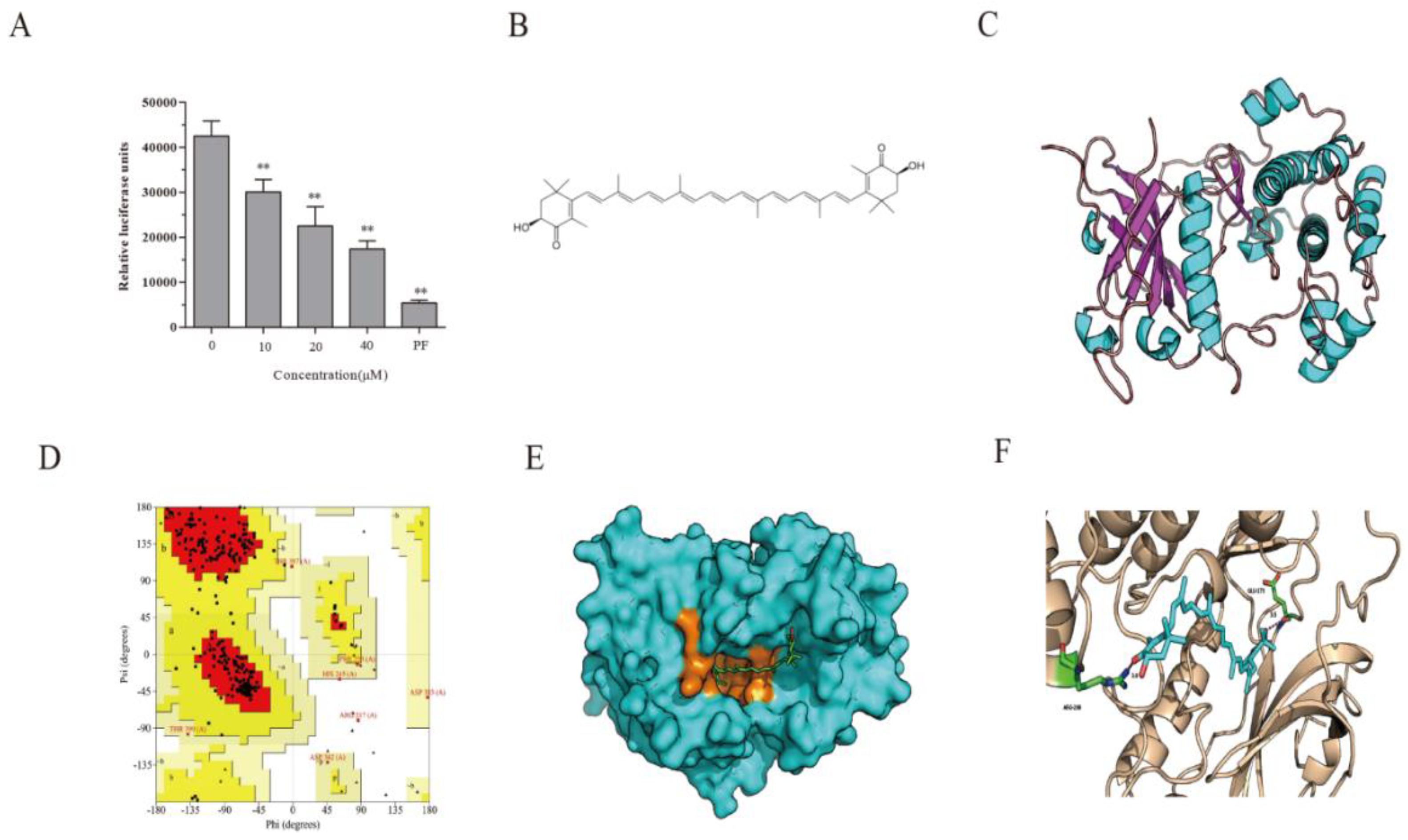
2.2. AST Inhibits S6K1 Activity
2.3. S6K1 Structure Prediction and Model Validation
2.4. Docking Fitting
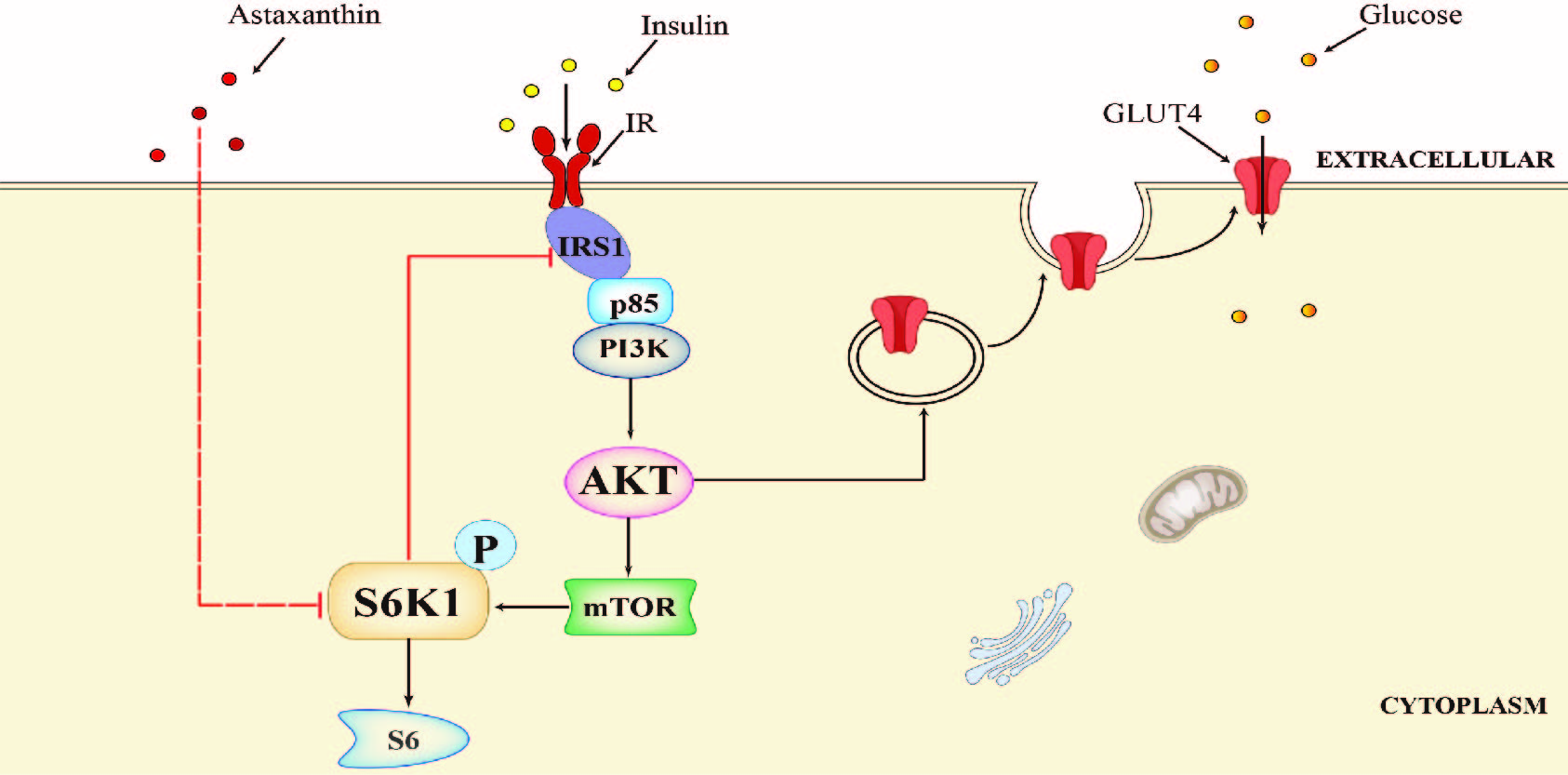
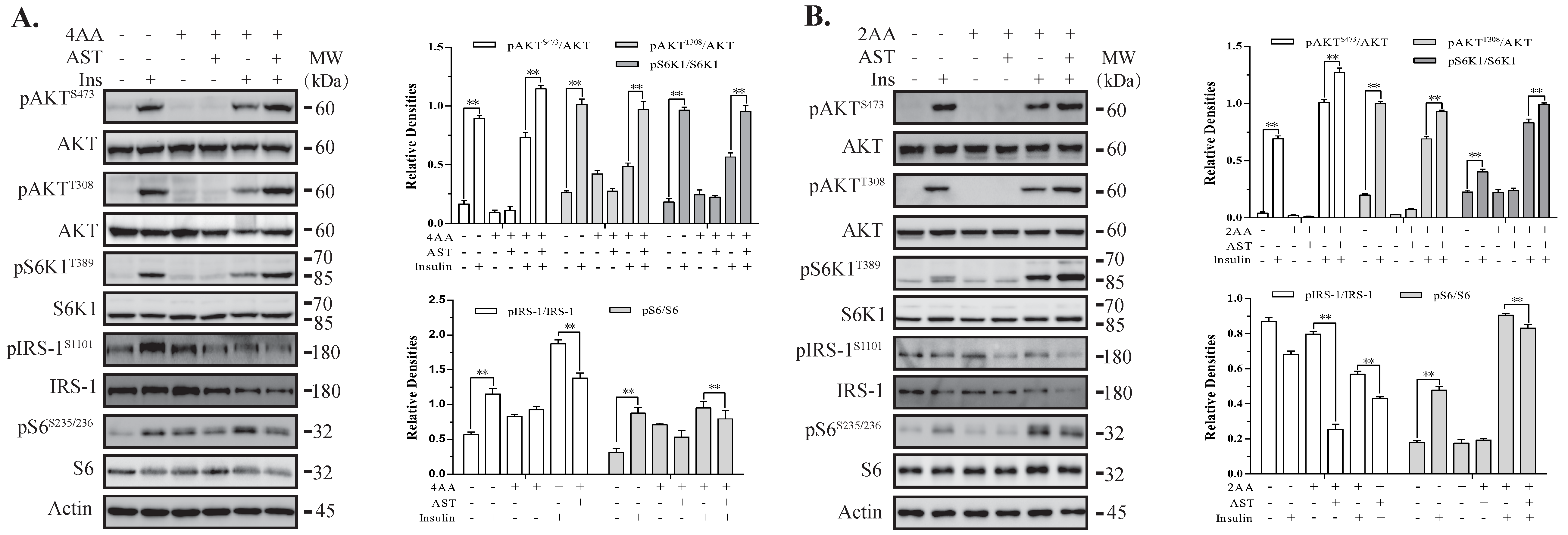
2.5. AST Sensitizes The IR by Feedback Activating The PI-3 Kinase Pathway
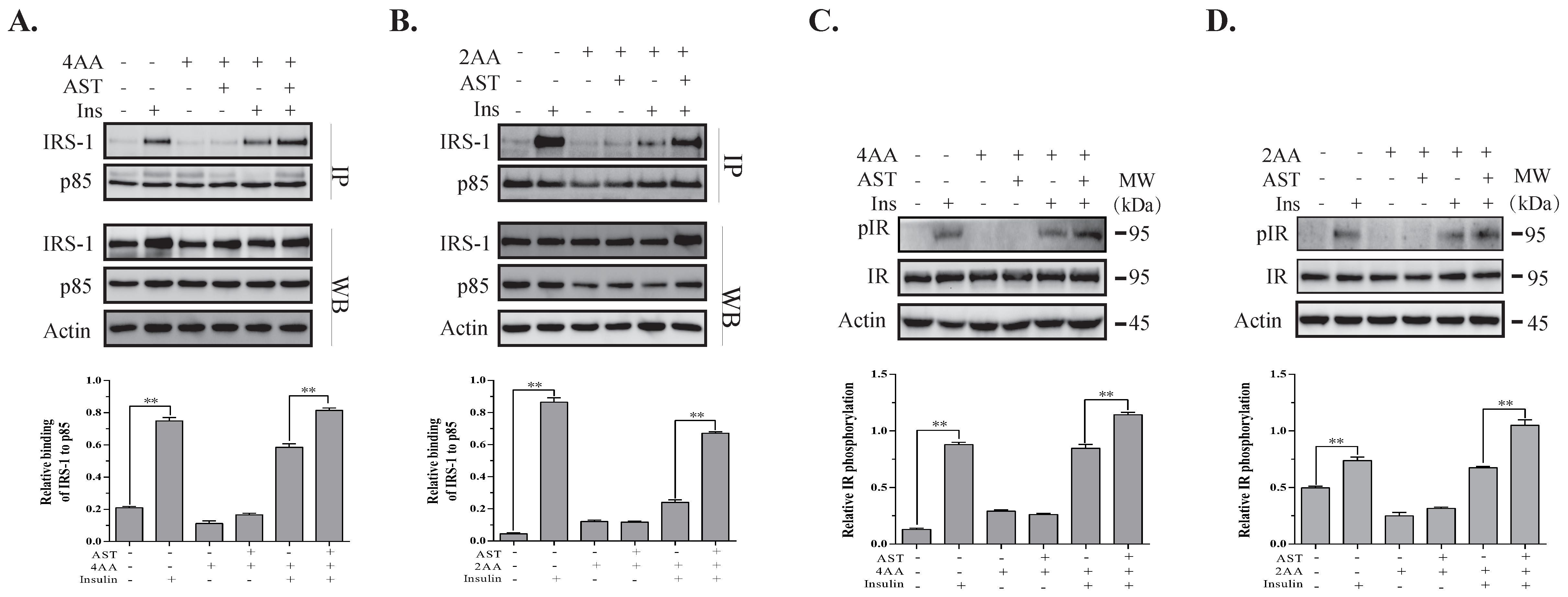
2.6. AST Increases IR Tyrosine Phosphorylation and IRS-1 Binding to the p85 Subunit of PI-3 Kinase
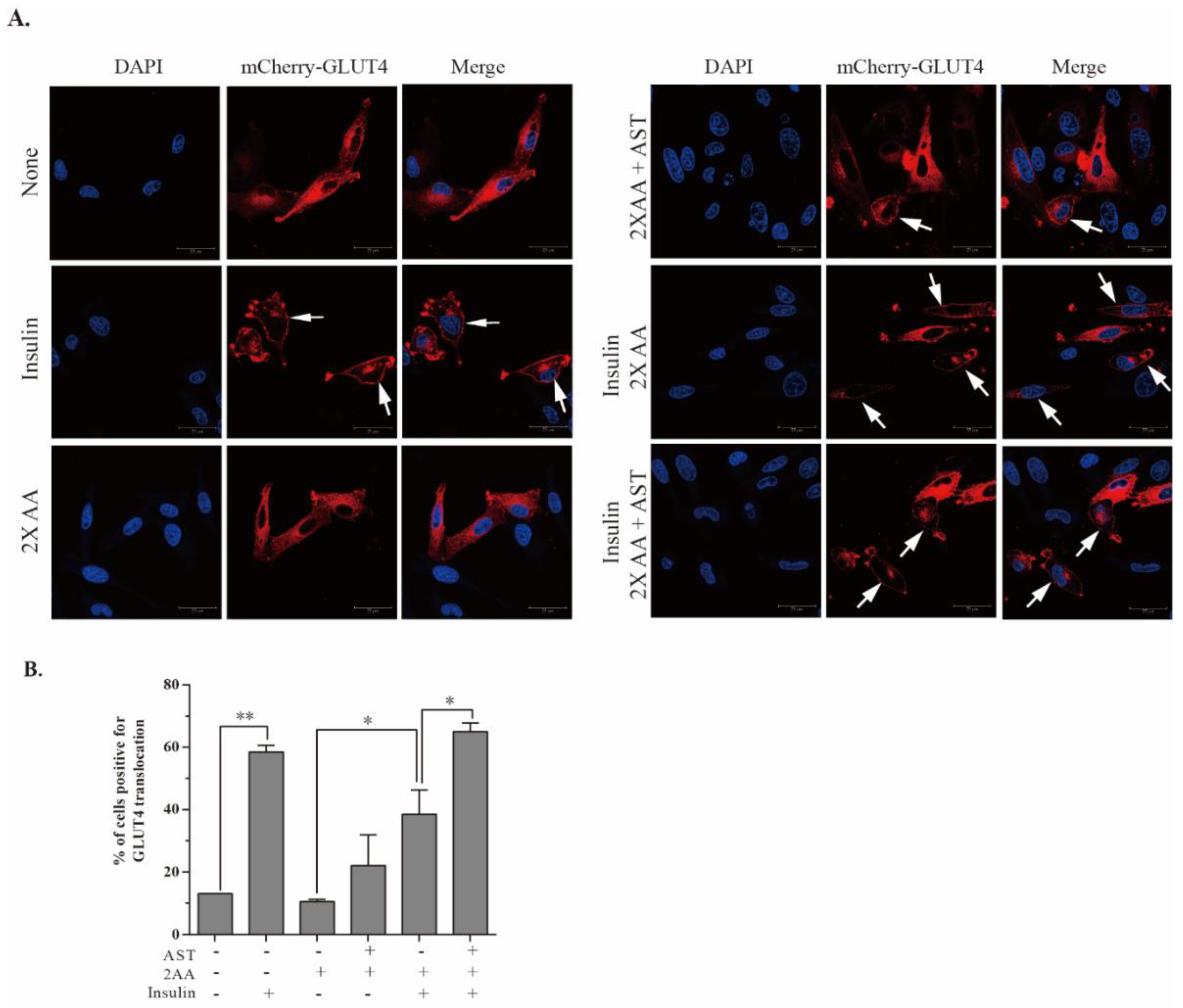
2.7. AST Promotes GLUT4 Translocation to The Plasma Membrane
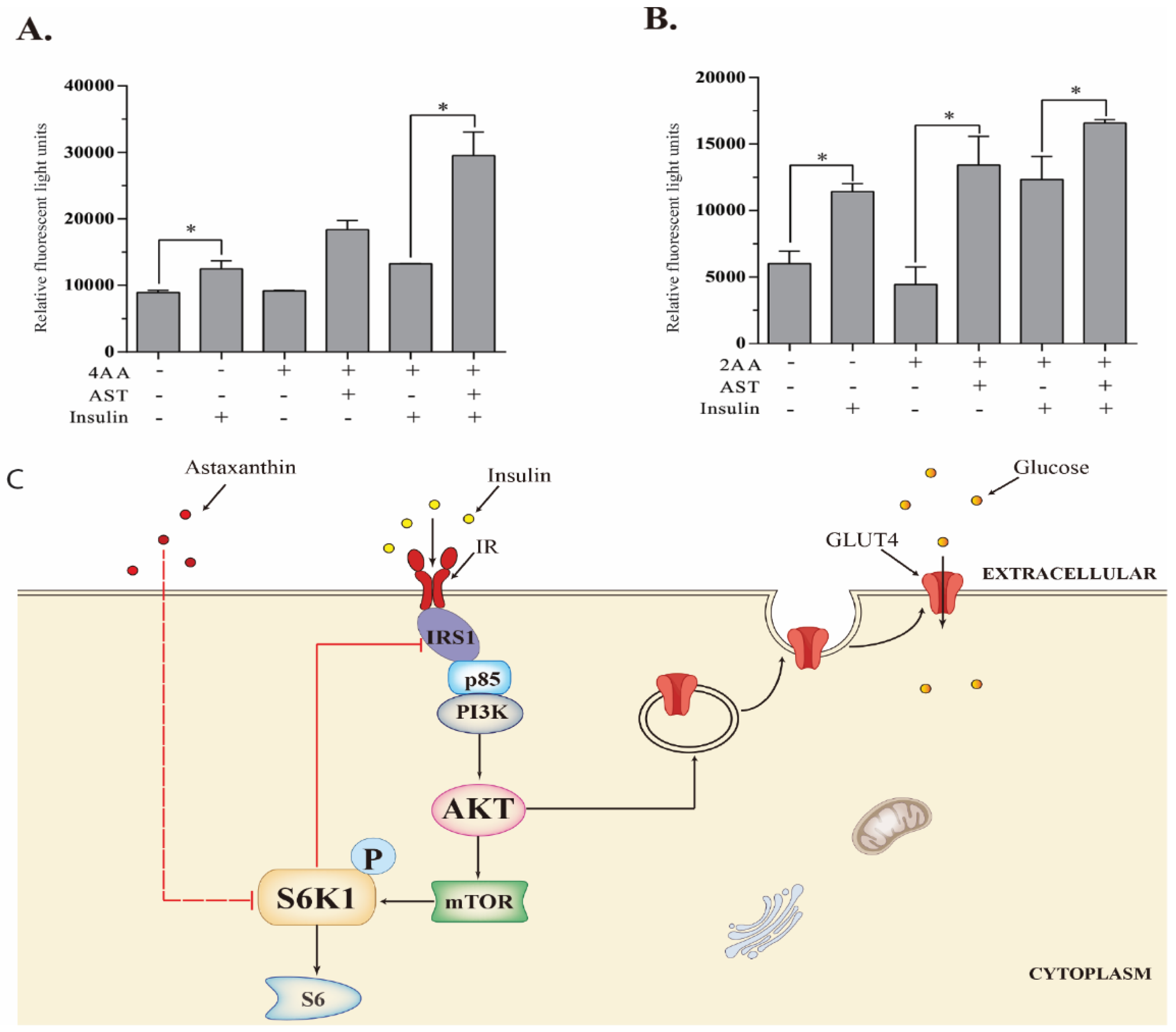
2.8. AST Improves Glucose Uptake
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Differentiation
4.3. Western Blot
4.4. Homology Modeling
4.5. Docking Modeling
4.6. In Vitro S6K1 Assay
4.7. Co-Immunoprecipitation
4.8. GLUT4 Translocation
4.9. Glucose Uptake
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes mellitus statistics on prevalence and mortality: Facts and fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.Z.; Magliano, D.J.; Herman, W.H.; Shaw, J.E. Diabetes: A 21st century challenge. Lancet Diabetes Endocrinol. 2014, 2, 56–64. [Google Scholar] [CrossRef]
- Saeedi, P.; Salpea, P.; Karuranga, S.; Petersohn, I.; Malanda, B.; Gregg, E.W.; Unwin, N.; Wild, S.H.; Williams, R. Mortality attributable to diabetes in 20–79 years old adults, 2019 estimates: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2020, 162, 108086. [Google Scholar] [CrossRef] [Green Version]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9 (th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- Nathan, D.M. Diabetes: Advances in Diagnosis and Treatment. JAMA 2015, 314, 1052–1062. [Google Scholar] [CrossRef]
- Backholer, K.; Peeters, A.; Herman, W.H.; Shaw, J.E.; Liew, D.; Ademi, Z.; Magliano, D.J. Diabetes prevention and treatment strategies: Are we doing enough? Diabetes Care 2013, 36, 2714–2719. [Google Scholar] [CrossRef] [Green Version]
- Cefalu, W.T.; Buse, J.B.; Tuomilehto, J.; Fleming, G.A.; Ferrannini, E.; Gerstein, H.C.; Bennett, P.H.; Ramachandran, A.; Raz, I.; Rosenstock, J.; et al. Update and next steps for real-world translation of interventions for type 2 diabetes prevention: Reflections from a diabetes care editors’ expert forum. Diabetes Care 2016, 39, 1186–1201. [Google Scholar] [CrossRef] [Green Version]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, T1–T23. [Google Scholar] [CrossRef]
- Guo, S. Molecular Basis of Insulin Resistance: The role of IRS and Foxo1 in the control of diabetes mellitus and its complications. Drug Discov. Today Dis. Mech. 2013, 10, e27–e33. [Google Scholar] [CrossRef] [Green Version]
- Bogan, J.S. Regulation of glucose transporter translocation in health and diabetes. Annu. Rev. Biochem. 2012, 81, 507–532. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, S.H.; D’Alessio, D.; Thomas, G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006, 3, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004, 431, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kusudo, T.; Takeuchi, T.; Yamashita, Y.; Kontani, Y.; Okamatsu, Y.; Saito, M.; Mori, N.; Yamashita, H. Evodiamine inhibits insulin-stimulated mTOR-S6K activation and IRS1 serine phosphorylation in adipocytes and improves glucose tolerance in obese/diabetic mice. PLoS ONE 2013, 8, e83264. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sun, J.; Prinz, R.A.; Li, Y.; Xu, X. Gingerenone A Sensitizes the Insulin Receptor and Increases Glucose Uptake by Inhibiting the Activity of p70 S6 Kinase. Mol. Nutr. Food Res. 2018, 62, 1800709. [Google Scholar] [CrossRef]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential health-promoting effects of astaxanthin: A high-value carotenoid mostly from microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar] [CrossRef]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications--a review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef]
- Rammuni, M.N.; Ariyadasa, T.U.; Nimarshana, P.H.V.; Attalage, R.A. Comparative assessment on the extraction of carotenoids from microalgal sources: Astaxanthin from H. pluvialis and beta-carotene from D. salina. Food Chem. 2019, 277, 128–134. [Google Scholar] [CrossRef]
- Landon, R.; Gueguen, V.; Petite, H.; Letourneur, D.; Pavon-Djavid, G.; Anagnostou, F. Impact of Astaxanthin on Diabetes Pathogenesis and Chronic Complications. Mar. Drugs 2020, 18, 357. [Google Scholar] [CrossRef]
- Kogure, K. Novel antioxidative activity of astaxanthin and its synergistic effect with vitamin E. J. Nutr. Sci. Vitaminol. 2019, 65, S109–S112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Y.; Nagashimada, M.; Zhuge, F.; Zhan, L.; Nagata, N.; Tsutsui, A.; Nakanuma, Y.; Kaneko, S.; Ota, T. Astaxanthin prevents and reverses diet-induced insulin resistance and steatohepatitis in mice: A comparison with vitamin E. Sci. Rep. 2015, 5, 17192. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, K.; Naito, Y.; Hasegawa, G.; Nakamura, N.; Takahashi, J.; Yoshikawa, T. Astaxanthin protects beta-cells against glucose toxicity in diabetic db/db mice. Redox Rep. 2002, 7, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Farruggia, C.; Kim, M.; Bae, M.; Lee, Y.; Pham, T.X.; Yang, Y.; Han, M.J.; Park, Y.; Lee, J. Astaxanthin exerts anti-inflammatory and antioxidant effects in macrophages in NRF2-dependent and independent manners. J. Nutr. Biochem. 2018, 62, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Sorrenti, V.; Davinelli, S.; Scapagnini, G.; Willcox, B.J.; Allsopp, R.C.; Willcox, D.C. Astaxanthin as a putative geroprotector: Molecular basis and focus on brain aging. Mar. Drugs 2020, 18, 351. [Google Scholar] [CrossRef]
- Arunkumar, E.; Bhuvaneswari, S.; Anuradha, C.V. An intervention study in obese mice with astaxanthin, a marine carotenoid--effects on insulin signaling and pro-inflammatory cytokines. Food Funct. 2012, 3, 120–126. [Google Scholar] [CrossRef]
- Bhuvaneswari, S.; Anuradha, C.V. Astaxanthin prevents loss of insulin signaling and improves glucose metabolism in liver of insulin resistant mice. Can. J. Physiol. Pharmacol. 2012, 90, 1544–1552. [Google Scholar] [CrossRef]
- Ni, Y.; Zhuge, F.; Nagashimada, M.; Ota, T. Novel Action of Carotenoids on Non-Alcoholic Fatty Liver Disease: Macrophage Polarization and Liver Homeostasis. Nutrients 2016, 8, 391. [Google Scholar] [CrossRef] [Green Version]
- Urakaze, M.; Kobashi, C.; Satou, Y.; Takagi, M.; Shigeta, K.; Toshima, M.; Takahashi, J.; Nishida, M. Clinical Study of Astaxanthin on Glucose Tolerance in Nondiabetic Subjects. Diabetes 2018, 67. [Google Scholar] [CrossRef]
- Ishiki, M.; Nishida, Y.; Ishibashi, H.; Wada, T.; Fujisaka, S.; Takikawa, A.; Urakaze, M.; Sasaoka, T.; Usui, I.; Tobe, K. Impact of divergent effects of astaxanthin on insulin signaling in L6 cells. Endocrinology 2013, 154, 2600–2612. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ding, C.; Zhang, S.; Xu, Y. Neuroprotective effects of astaxanthin against oxygen and glucose deprivation damage via the PI3K/Akt/GSK3beta/Nrf2 signalling pathway in vitro. J. Cell. Mol. Med. 2020, 24, 8977–8985. [Google Scholar] [CrossRef] [PubMed]
- Zick, Y. Insulin resistance: A phosphorylation-based uncoupling of insulin signaling. Trends Cell Biol. 2001, 11, 437–441. [Google Scholar] [CrossRef]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roohbakhsh, A.; Karimi, G.; Iranshahi, M. Carotenoids in the treatment of diabetes mellitus and its complications: A mechanistic review. Biomed. Pharm. 2017, 91, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Murillo, A.G.; Fernandez, M.L. Potential of Dietary Non-Provitamin A Carotenoids in the Prevention and Treatment of Diabetic Microvascular Complications. Adv. Nutr. 2016, 7, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wu, C.; Kim, J.; Kim, B.; Lee, S.J. Astaxanthin reduces hepatic lipid accumulations in high-fat-fed C57BL/6J mice via activation of peroxisome proliferator-activated receptor (PPAR) alpha and inhibition of PPAR gamma and Akt. J. Nutr. Biochem. 2016, 28, 9–18. [Google Scholar] [CrossRef]
- Choi, C.I. Astaxanthin as a Peroxisome Proliferator-Activated Receptor (PPAR) Modulator: Its therapeutic implications. Mar. Drugs 2019, 17, 242. [Google Scholar] [CrossRef] [Green Version]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2012, 441, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Doscas, M.E.; Williamson, A.J.; Usha, L.; Bogachkov, Y.; Rao, G.S.; Xiao, F.; Wang, Y.; Ruby, C.; Kaufman, H.; Zhou, J.; et al. Inhibition of p70 S6 kinase (S6K1) activity by A77 1726 and its effect on cell proliferation and cell cycle progress. Neoplasia 2014, 16, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Shum, M.; Bellmann, K.; St-Pierre, P.; Marette, A. Pharmacological inhibition of S6K1 increases glucose metabolism and Akt signalling in vitro and in diet-induced obese mice. Diabetologia 2016, 59, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Osterlie, M.; Bjerkeng, B.; Liaaen-Jensen, S. Plasma appearance and distribution of astaxanthin E/Z and R/S isomers in plasma lipoproteins of men after single dose administration of astaxanthin. J. Nutr. Biochem. 2000, 11, 482–490. [Google Scholar] [CrossRef]
- Tremblay, F.; Brule, S.; Um, S.H.; Li, Y.; Masuda, K.; Roden, M.; Sun, X.J.; Krebs, M.; Polakiewicz, R.D.; Thomas, G. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 14056–14061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dann, S.G.; Selvaraj, A.; Thomas, G. mTOR Complex1-S6K1 signaling: At the crossroads of obesity, diabetes and cancer. Trends Mol. Med. 2007, 13, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sun, J.; Doscas, M.E.; Ye, J.; Williamson, A.J.; Li, Y.; Li, Y.; Prinz, R.A.; Xu, X. Control of hyperglycemia in male mice by leflunomide: Mechanisms of action. J. Endocrinol. 2018, 237, 43–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boura-Halfon, S.; Zick, Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E581–E591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.; Holman, G.D. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E29–E37. [Google Scholar] [CrossRef]
- Stockli, J.; Fazakerley, D.J.; James, D.E. GLUT4 exocytosis. J. Cell Sci. 2011, 124, 4147–4159. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, R.; Teruel, T.; Lorenzo, M. Akt mediates insulin induction of glucose uptake and up-regulation of GLUT4 gene expression in brown adipocytes. FEBS Lett. 2001, 494, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Penumathsa, S.V.; Thirunavukkarasu, M.; Zhan, L.; Maulik, G.; Menon, V.P.; Bagchi, D.; Maulik, N. Resveratrol enhances GLUT-4 translocation to the caveolar lipid raft fractions through AMPK/Akt/eNOS signalling pathway in diabetic myocardium. J. Cell. Mol. Med. 2008, 12, 2350–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, T.; Zhao, Y.; Zhang, X.; Lin, X. Astaxanthin inhibits acetaldehyde-induced cytotoxicity in SH-SY5Y cells by modulating Akt/CREB and p38MAPK/ERK signaling pathways. Mar. Drugs 2016, 14, 56. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.C.; Chen, J.C.; Wang, T.J.; Zheng, H.Y.; Chen, W.C.; Chang, P.Y.; Lin, Y.W. Astaxanthin down-regulates Rad51 expression via inactivation of AKT kinase to enhance mitomycin C-induced cytotoxicity in human non-small cell lung cancer cells. Biochem. Pharmacol. 2016, 105, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Kavitha, K.; Kowshik, J.; Kishore, T.K.K.; Baba, A.B.; Nagini, S. Astaxanthin inhibits NF-κB and Wnt/β-catenin signaling pathways via inactivation of Erk/MAPK and PI3K/Akt to induce intrinsic apoptosis in a hamster model of oral cancer. Biochim. Biophys. Acta 2013, 1830, 4433–4444. [Google Scholar] [CrossRef] [PubMed]
- Rho, Y.H.; Oeser, A.; Chung, C.P.; Milne, G.L.; Stein, C.M. Drugs Used in the Treatment of Rheumatoid Arthritis: Relationship between current use and cardiovascular risk factors. Arch. Drug Inf. 2009, 2, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Coblyn, J.S.; Shadick, N.; Helfgott, S. Leflunomide-associated weight loss in rheumatoid arthritis. Arthritis Rheum 2001, 44, 1048–1051. [Google Scholar] [CrossRef]
- Zebisch, K.; Voigt, V.; Wabitsch, M.; Brandsch, M. Protocol for effective differentiation of 3T3-L1 cells to adipocytes. Anal. Biochem. 2012, 425, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.N.D.; Schwede, T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009, 4, 1–13. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Ma, B.; Chen, J.; Jeong, Y.; Xu, X. Astaxanthin Inhibits p70 S6 Kinase 1 Activity to Sensitize Insulin Signaling. Mar. Drugs 2020, 18, 495. https://doi.org/10.3390/md18100495
Li C, Ma B, Chen J, Jeong Y, Xu X. Astaxanthin Inhibits p70 S6 Kinase 1 Activity to Sensitize Insulin Signaling. Marine Drugs. 2020; 18(10):495. https://doi.org/10.3390/md18100495
Chicago/Turabian StyleLi, Chunmei, Bixia Ma, Junhong Chen, Yoonhwa Jeong, and Xiulong Xu. 2020. "Astaxanthin Inhibits p70 S6 Kinase 1 Activity to Sensitize Insulin Signaling" Marine Drugs 18, no. 10: 495. https://doi.org/10.3390/md18100495