Anti-Diabetic Activity of 2,3,6-Tribromo-4,5-Dihydroxybenzyl Derivatives from Symphyocladia latiuscula through PTP1B Downregulation and α-Glucosidase Inhibition



Abstract
:1. Introduction
2. Results
2.1. Inhibitory Activity of Methanol Extract and Solvent Soluble Fractions on Tyrosine Phosphatase 1B (PTP1B) and α-Glucosidase
2.2. Inhibitory Activity of Bromophenols on PTP1B and α-Glucosidase
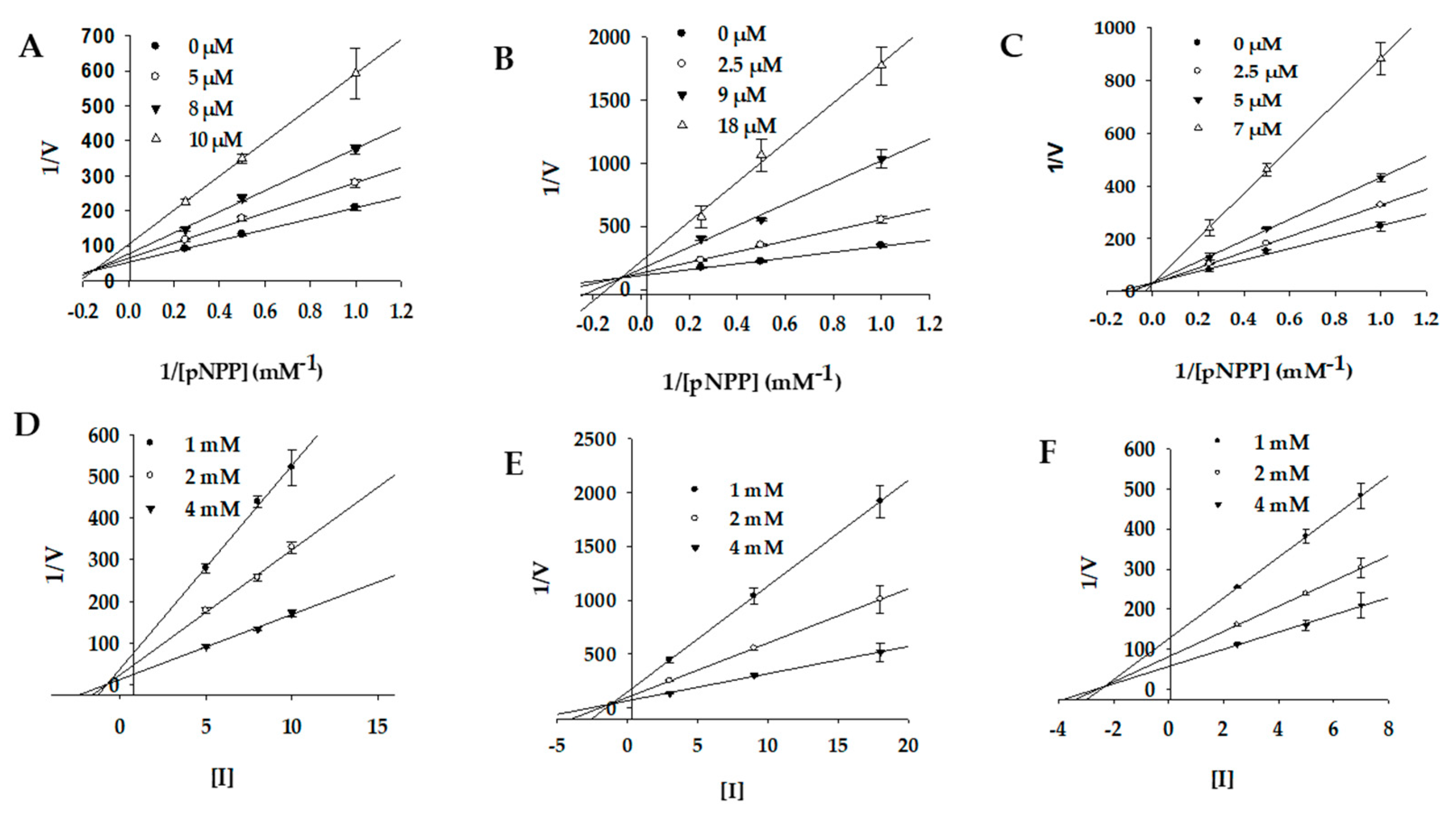
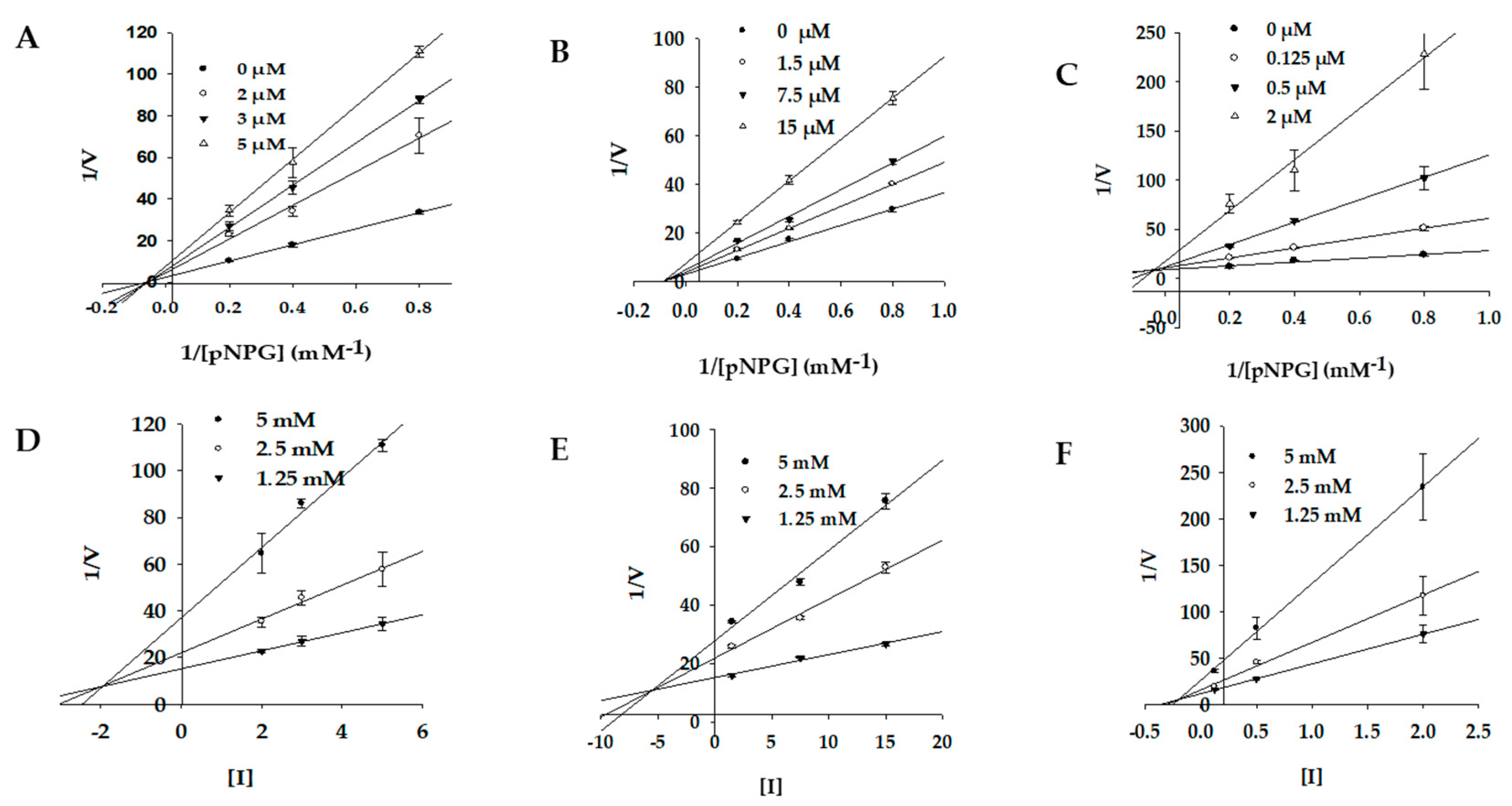
2.3. Enzyme Kinetics of PTP1B and α-Glucosidase Inhibition
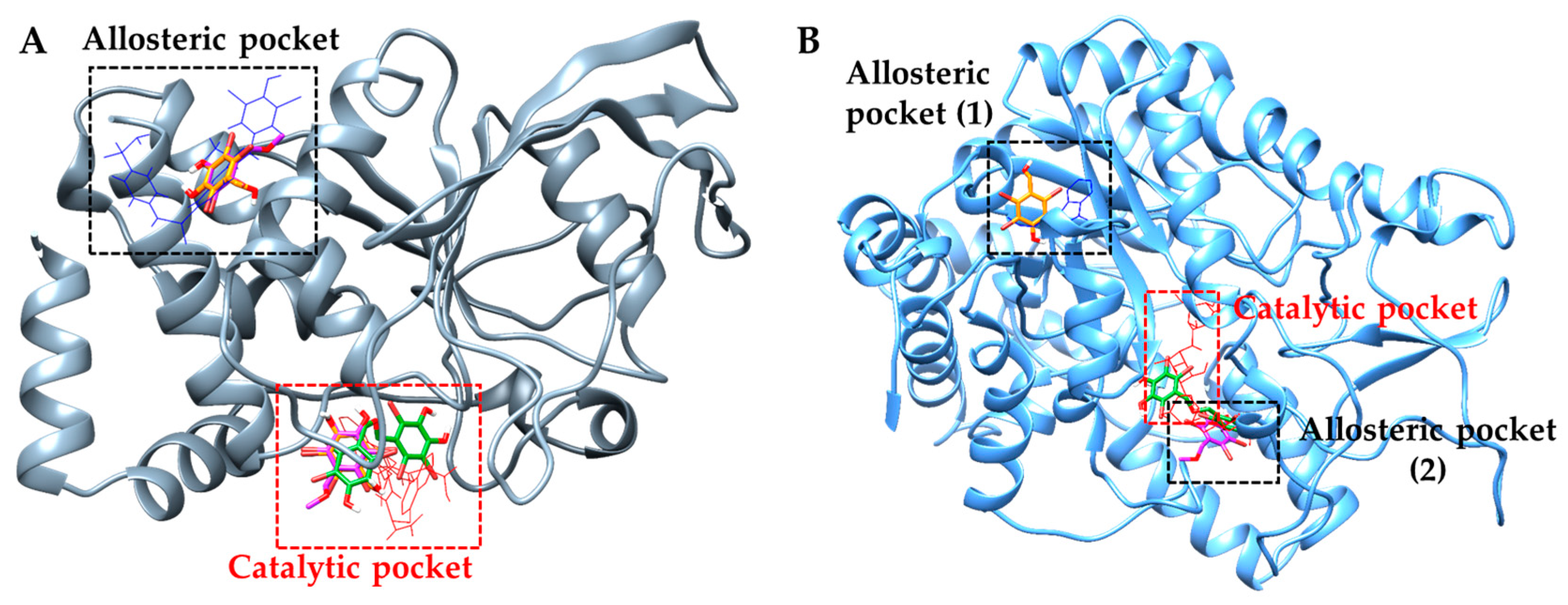
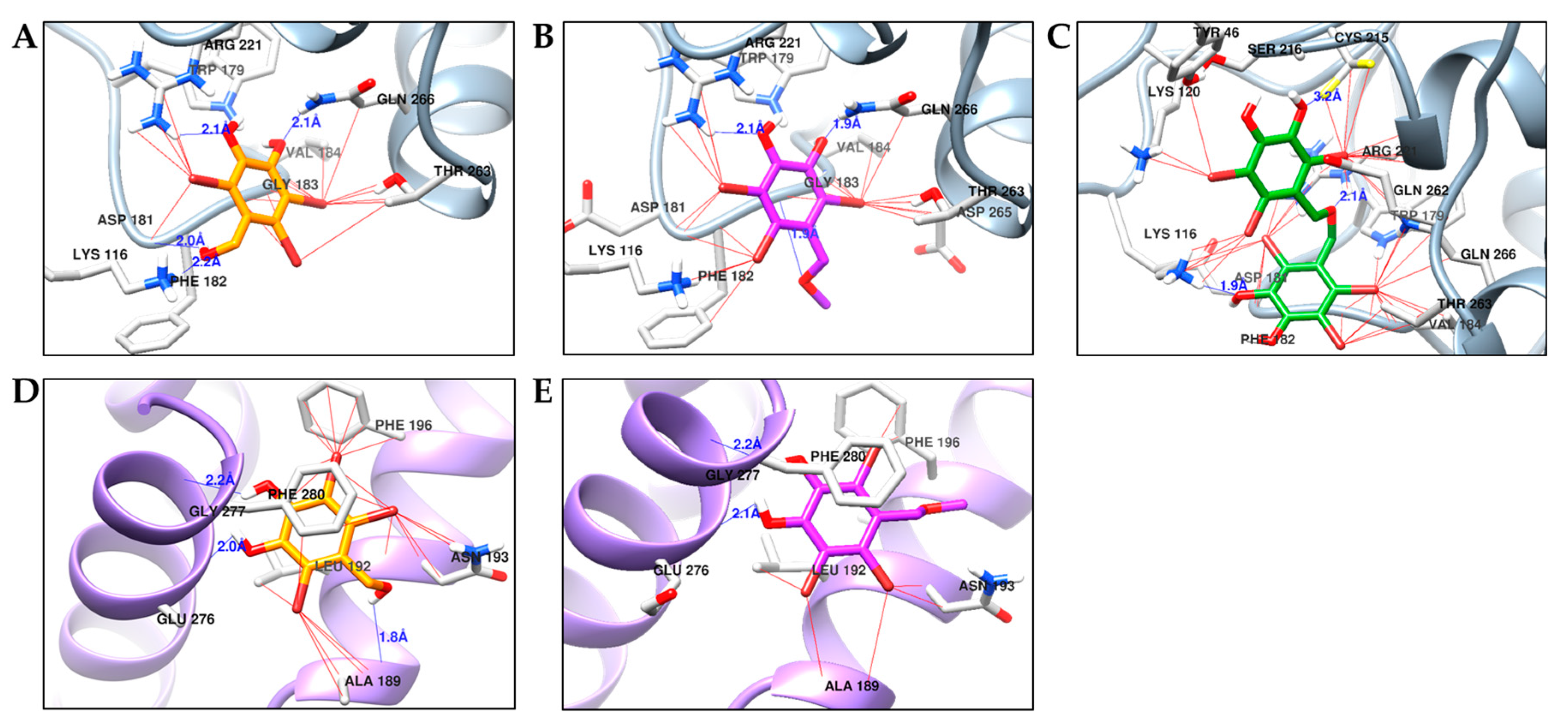
2.4. Molecular Docking Simulation of PTP1B Inhibition
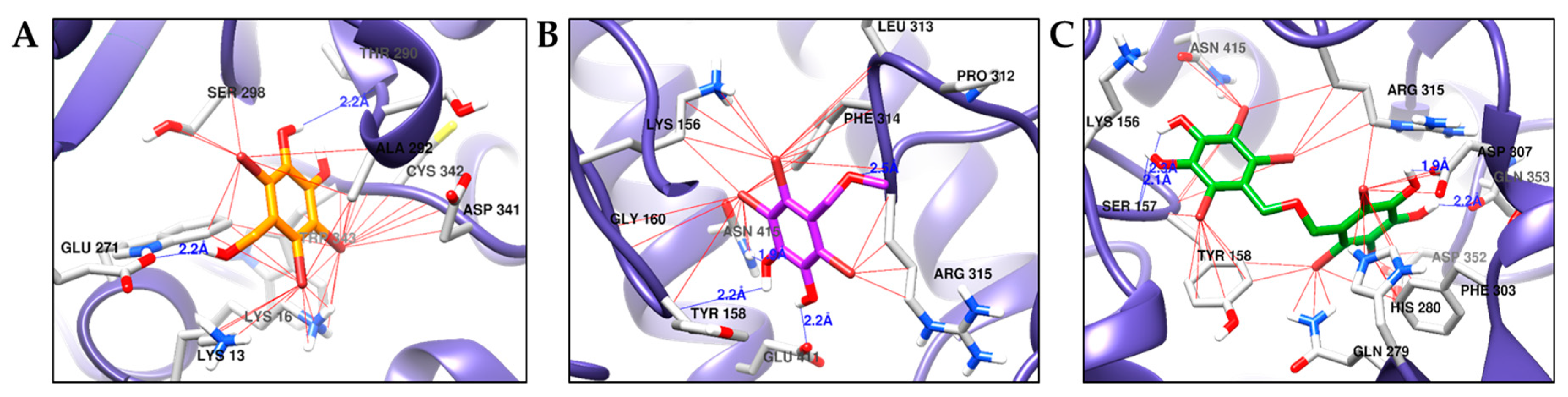
2.5. Molecular Docking Simulation of α-Glucosidase Inhibition
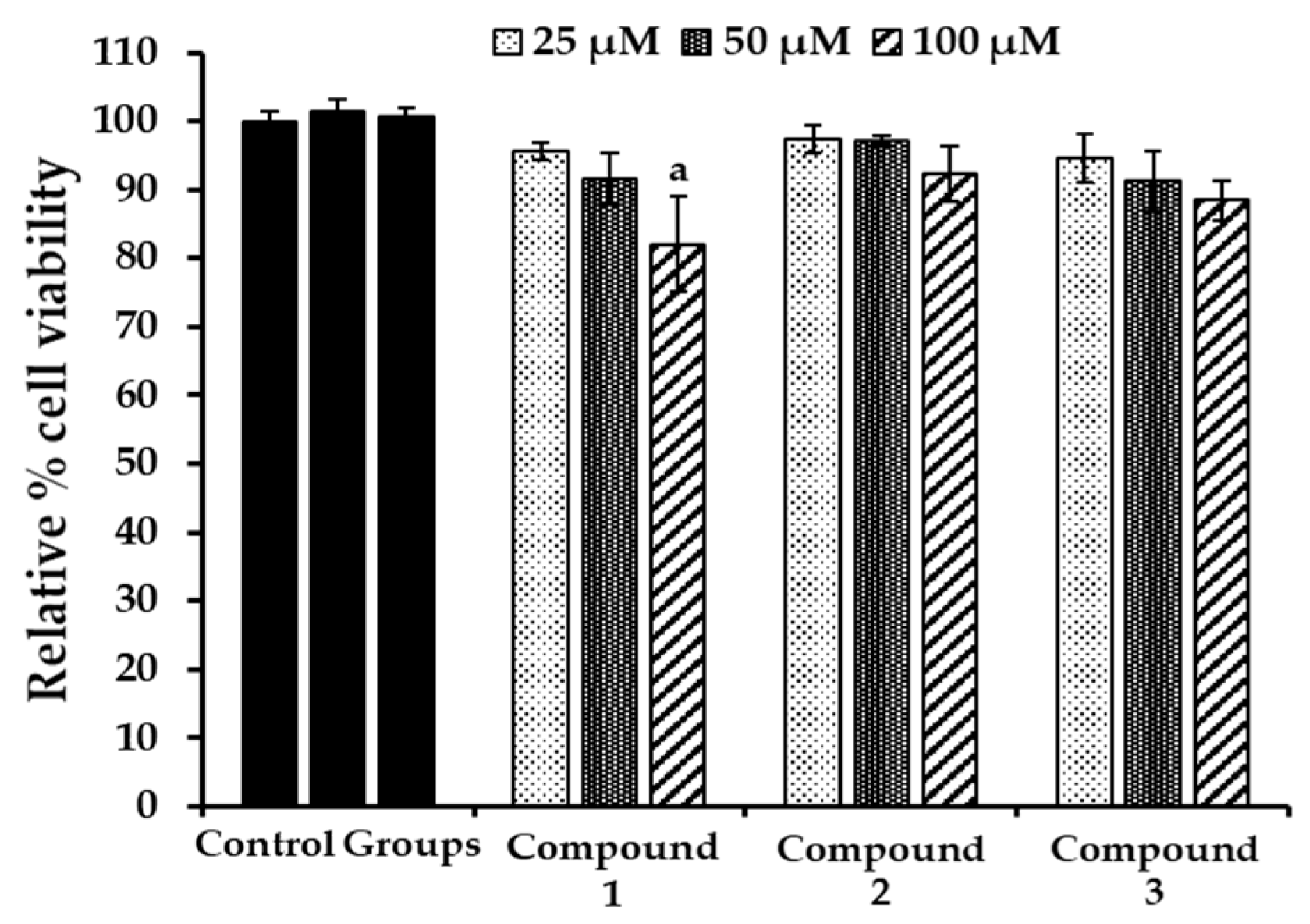
2.6. Evaluation of Cytotoxicity in HepG2 Cells
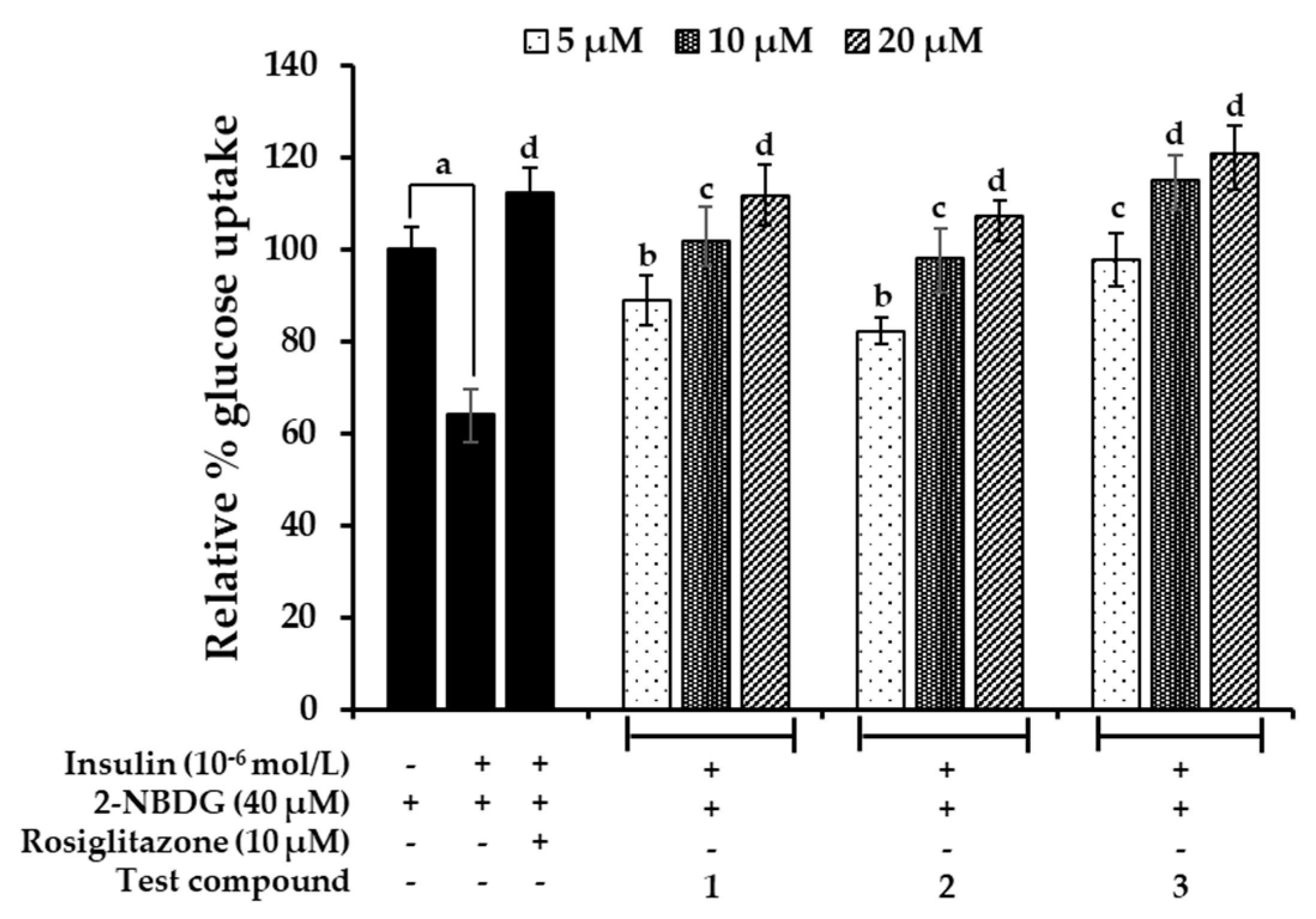
2.7. Effects on Glucose Uptake in Insulin-Resistant HepG2 Cells
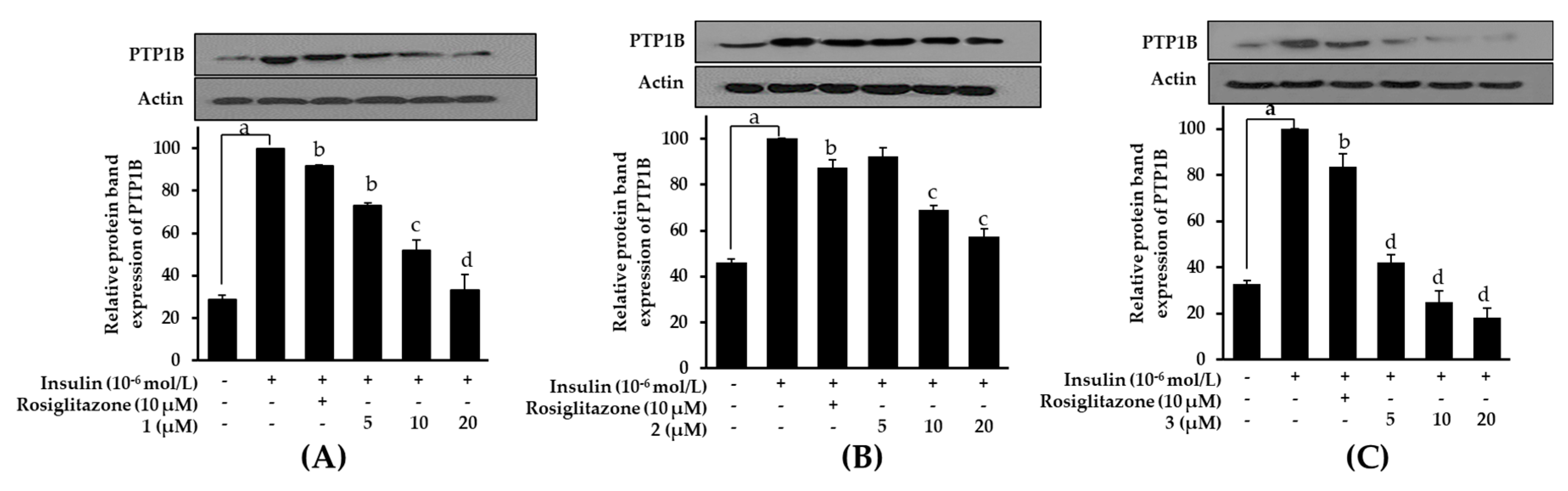
2.8. Effects on PTP1B Expression Level in Insulin-Resistant HepG2 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Algal Material
4.3. Extraction and Fractionation
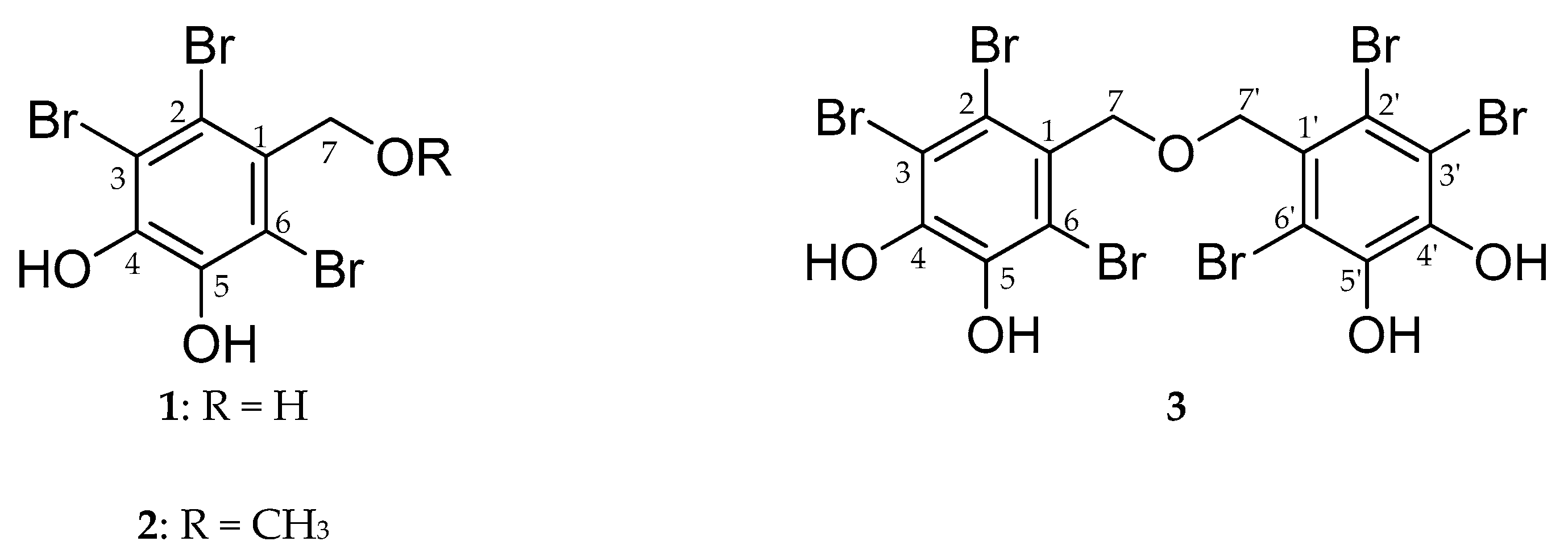
4.4. Isolation of Bromophenol Derivatives from Symphyocladia latiuscula
4.5. Protein Tyrosine Phosphate 1B (PTP1B) Inhibitory Assay
4.6. α-Glucosidase Inhibitory Assay
4.7. Kinetic Study Against PTP1B and α-Glucosidase
4.8. Molecular Docking Simulation of PTP1B and α-Glucosidase Inhibition
4.9. Cell Culture, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) Assay, and Insulin Resistance Induction
4.10. Glucose Uptake Assay
4.11. Preparation of Cell Lysates and Western Blot Analysis
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Andreoulakis, E.; Hyphantis, T.; Kandylis, D.; Iacovides, A. Depression in diabetes mellitus: A comprehensive review. Hippokratia 2012, 16, 205–214. [Google Scholar] [PubMed]
- Association, A.D. Classification and diagnosis of diabetes: Standards of medical care in diabetes—2018. Diabetes Care 2018, 41, S13–S27. [Google Scholar] [CrossRef] [PubMed]
- Kenner, K.A.; Anyanwu, E.; Olefsky, J.M.; Kusari, J. Protein-tyrosine phosphatase 1B is a negative regulator of insulin-and insulin-like growth factor-I-stimulated signaling. J. Biol. Chem. 1996, 271, 19810–19816. [Google Scholar] [CrossRef]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.C. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.S.; Kulkarni, V.M. 3D-QSAR CoMFA and CoMSIA on protein tyrosine phosphatase 1B inhibitors. Bioorg. Med. Chem. 2002, 10, 2267–2282. [Google Scholar] [CrossRef]
- Lund, I.K.; Andersen, H.S.; Iversen, L.F.; Olsen, O.H.; Møller, K.B.; Pedersen, A.K.; Ge, Y.; Holsworth, D.D.; Newman, M.J.; Axe, F.U. Structure-based design of selective and potent inhibitors of protein-tyrosine phosphatase β. J. Biol. Chem. 2004, 279, 24226–24235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, X.; Kim, S.K. Application of marine biomaterials for nutraceuticals and functional foods. Food Sci. Biotechnol. 2012, 21, 625–631. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Hu, G.; Yu, J.; Zhu, X.; Lin, Y.; Chen, S.; Yuan, J. Statistical research on the bioactivity of new marine natural products discovered during the 28 years from 1985 to 2012. Mar. Drugs 2015, 13, 202. [Google Scholar] [CrossRef]
- Cabrita, M.T.; Vale, C.; Rauter, A.P. Halogenated compounds from marine algae. Mar. Drugs 2010, 8, 2301–2317. [Google Scholar] [CrossRef]
- Gribble, G.W. Biological activity of recently discovered halogenated marine natural products. Mar. Drugs 2015, 13, 4044–4136. [Google Scholar] [CrossRef]
- Mayer, A.; Rodríguez, A.; Taglialatela-Scafati, O.; Fusetani, N. Marine pharmacology in 2009–2011: Marine compounds with antibacterial, antidiabetic, antifungal, anti-Inflammatory, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Mar. Drugs 2013, 11, 2510. [Google Scholar] [PubMed]
- Eguchi, K.; Kato, H.; Fujiwara, Y.; Losung, F.; Mangindaan, R.E.; de Voogd, N.J.; Takeya, M.; Tsukamoto, S. Bastadins, brominated-tyrosine derivatives, suppress accumulation of cholesterol ester in macrophages. Bioorg. Med. Chem. Lett. 2015, 25, 5389–5392. [Google Scholar] [CrossRef]
- Asolkar, R.N.; Singh, A.; Jensen, P.R.; Aalbersberg, W.; Carte, B.K.; Feussner, K.D.; Subramani, R.; DiPasquale, A.; Rheingold, A.L.; Fenical, W. Marinocyanins, cytotoxic bromo-phenazinone meroterpenoids from a marine bacterium from the streptomycete clade MAR4. Tetrahedron 2017, 73, 2234–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerebtzoff, G.; Li-Blatter, X.; Fischer, H.; Frentzel, A.; Seelig, A. Halogenation of drugs enhances membrane binding and permeation. Chembiochem 2004, 5, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Gentry, C.L.; Egleton, R.D.; Gillespie, T.; Abbruscato, T.J.; Bechowski, H.B.; Hruby, V.J.; Davis, T.P. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides 1999, 20, 1229–1238. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, Z.; Liu, Y.; Lu, Y.; Chen, K.; Zhu, W. Halogen bond: Its role beyond drug-target binding affinity for drug discovery and development. J. Chem. Inf. Model. 2014, 54, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Mendez, L.; Henriquez, G.; Sirimulla, S.; Narayan, M. Looking back, looking forward at halogen bonding in drug discovery. Molecules 2017, 22, 1397. [Google Scholar] [CrossRef] [PubMed]
- Kurata, K. Bis (2, 3, 6-tribromo-4, 5-dihydroxybenzyl) ether from the red alga, Symphyocladia latiuscula. Phytochemistry 1980, 19, 141–142. [Google Scholar] [CrossRef]
- Park, H.J.; Kurokawa, M.; Shiraki, K.; Nakamura, N.; Choi, J.S.; Hattori, M. Antiviral activity of the marine alga Symphyocladia latiuscula against herpes simplex virus (HSV-1) in vitro and its therapeutic efficacy against HSV-1 infection in mice. Biol. Pharm. Bull. 2005, 28, 2258–2262. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, G.; Xiao, L.; Xu, X.; Liu, X.; Xu, P.; Lin, X. Bis (2, 3-dibromo-4, 5-dihydroxybenzyl) ether, a marine algae derived bromophenol, inhibits the growth of botrytis cinerea and interacts with DNA molecules. Mar. Drugs 2014, 12, 3838–3851. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, S.E.; Hossain, M.A.; Kim, M.Y.; Kim, M.N.; Chung, H.Y.; Choi, J.S.; Yoo, Y.H.; Kim, N.D. 2, 3, 6-Tribromo-4, 5-dihydroxybenzyl methyl ether induces growth inhibition and apoptosis in MCF-7 human breast cancer cells. Arch. Pharm. Res. 2007, 30, 1132–1137. [Google Scholar] [CrossRef]
- Choi, J.S.; Park, H.J.; Jung, H.A.; Chung, H.Y.; Jung, J.H.; Choi, W.C. A cyclohexanonyl bromophenol from the red alga Symphyocladia latiuscula. J. Nat. Prod. 2000, 63, 1705–1706. [Google Scholar] [CrossRef]
- Wang, W.; Okada, Y.; Shi, H.; Wang, Y.; Okuyama, T. Structures and aldose reductase inhibitory effects of bromophenols from the red alga Symphyocladia latiuscula. J. Nat. Prod. 2005, 68, 620–622. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, H.; Mitani, T.; Kawabata, J.; Takahashi, K. Inhibitory potencies of bromophenols from Rhodomelaceae algae against α-glucosidase activity. Fisheries Sci. 1999, 65, 300–303. [Google Scholar] [CrossRef]
- Xu, X.; Yang, H.; Khalil, Z.G.; Yin, L.; Xiao, X.; Neupane, P.; Bernhardt, P.V.; Salim, A.A.; Song, F.; Capon, R.J. Chemical diversity from a Chinese marine red alga, Symphyocladia latiuscula. Mar. Drugs 2017, 15, 374. [Google Scholar] [CrossRef]
- Lin, X.; Liu, M. Bromophenols from marine algae with potential anti-diabetic activities. J. Ocean Univ. China 2012, 11, 533–538. [Google Scholar] [CrossRef]
- Park, H.J.; Chung, H.Y.; Kim, J.; Choi, J.S. Antioxidant activity of 2, 3, 6-tribromo-4, 5-dihydroxy benzyl methyl ether from Symphyocladia latiuscula. Fish. Aquat. Sci. 1999, 2, 1–7. [Google Scholar]
- Taha, M.; Shah, S.A.A.; Afifi, M.; Imran, S.; Sultan, S.; Rahim, F.; Khan, K.M. Synthesis, α-glucosidase inhibition and molecular docking study of coumarin based derivatives. Bioorg. Chem. 2018, 77, 586–592. [Google Scholar] [CrossRef]
- Pye, C.R.; Bertin, M.J.; Lokey, R.S.; Gerwick, W.H.; Linington, R.G. Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. USA 2017, 114, 5601–5606. [Google Scholar] [CrossRef] [Green Version]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef]
- Whitfield, F.B.; Helidoniotis, F.; Shaw, K.J.; Svoronos, D. Distribution of bromophenols in species of marine algae from eastern Australia. J. Agric. Food Chem. 1999, 47, 2367–2373. [Google Scholar] [CrossRef]
- Ezzat, S.; Bishbishy, M.; Habtemariam, S.; Salehi, B.; Sharifi-Rad, M.; Martins, N.; Sharifi-Rad, J. Looking at marine-derived bioactive molecules as upcoming anti-diabetic agents: A special emphasis on PTP1B inhibitors. Molecules 2018, 23, 3334. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, P.S.; Jayasri, M.A. Marine algae as a prospective source for antidiabetic compounds—A brief review. Curr. Diabetes Rev. 2018, 14, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, H.; Mitani, T.; Kawabata, J.; Takahashi, K. Two new bromophenols from the red alga Odonthalia corymbifera. J. Nat. Prod. 1999, 62, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, X.; Gao, L.; Cui, C.; Li, C.; Li, J.; Wang, B. Extraction and PTP1B inhibitory activity of bromophenols from the marine red alga Symphyocladia latiuscula. Chin. J. Oceanol. Limn. 2011, 29, 686–690. [Google Scholar] [CrossRef]
- Zhang, R.; Yu, R.; Xu, Q.; Li, X.; Luo, J.; Jiang, B.; Wang, L.; Guo, S.; Wu, N.; Shi, D. Discovery and evaluation of the hybrid of bromophenol and saccharide as potent and selective protein tyrosine phosphatase 1B inhibitors. Eur. J. Med. Chem. 2017, 134, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Samuele, A.; Crespan, E.; Garbelli, A.; Bavagnoli, L.; Maga, G. The power of enzyme kinetics in the drug development process. Curr. Pharm. Biotechnol. 2013, 14, 551–560. [Google Scholar] [CrossRef]
- Barford, D.; Flint, A.J.; Tonks, N.K. Crystal structure of human protein tyrosine phosphatase 1B. Science 1994, 263, 1397–1404. [Google Scholar] [CrossRef]
- Shen, K.; Keng, Y.F.; Wu, L.; Guo, X.L.; Lawrence, D.S.; Zhang, Z.Y. Acquisition of a specific and potent PTP1B inhibitor from a novel combinatorial library and screening procedure. J. Biol. Chem. 2001, 276, 47311–47319. [Google Scholar] [CrossRef]
- Seong, S.H.; Roy, A.; Jung, H.A.; Jung, H.J.; Choi, J.S. Protein tyrosine phosphatase 1B and α-glucosidase inhibitory activities of Pueraria lobata root and its constituents. J. Ethnopharmacol. 2016, 194, 706–716. [Google Scholar] [CrossRef]
- Sarmiento, M.; Zhao, Y.; Gordon, S.J.; Zhang, Z.Y. Molecular basis for substrate specificity of protein-tyrosine phosphatase 1B. J. Biol. Chem. 1998, 273, 26368–26374. [Google Scholar] [CrossRef]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Hu, X.; Xu, X.; Zhang, G.; Gong, D. Inhibitory mechanism of two allosteric inhibitors, oleanolic acid and ursolic acid on α-glucosidase. Int. J. Biol. Macromol. 2018, 107, 1844–1855. [Google Scholar] [CrossRef] [PubMed]
- Şöhretoğlu, D.; Sari, S.; Özel, A.; Barut, B. α-Glucosidase inhibitory effect of Potentilla astracanica and some isoflavones: Inhibition kinetics and mechanistic insights through in vitro and in silico studies. Int. J. Biol. Macromol. 2017, 105, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Ismail, N.H.; Lalani, S.; Fatmi, M.Q.; Atia Tul, W.; Siddiqui, S.; Khan, K.M.; Imran, S.; Choudhary, M.I. Synthesis of novel inhibitors of α-glucosidase based on the benzothiazole skeleton containing benzohydrazide moiety and their molecular docking studies. Eur. J. Med. Chem. 2015, 92, 387–400. [Google Scholar] [CrossRef]
- Nagarajan, A.; Petersen, M.C.; Nasiri, A.R.; Butrico, G.; Fung, A.; Ruan, H.B.; Kursawe, R.; Caprio, S.; Thibodeau, J.; Bourgeois-Daigneault, M.C.; et al. MARCH1 regulates insulin sensitivity by controlling cell surface insulin receptor levels. Nat. Commun. 2016, 7, 12639. [Google Scholar] [CrossRef] [Green Version]
- Panzhinskiy, E.; Ren, J.; Nair, S. Pharmacological inhibition of protein tyrosine phosphatase 1B: A promising strategy for the treatment of obesity and type 2 diabetes mellitus. Curr. Med. Chem. 2013, 20, 2609–2625. [Google Scholar] [CrossRef]
- Panzhinskiy, E.; Ren, J.; Nair, S. Protein tyrosine phosphatase 1B and insulin resistance: Role of endoplasmic reticulum stress/reactive oxygen species/nuclear factor kappa B axis. PLoS ONE 2013, 8, e77228. [Google Scholar] [CrossRef]
- Cui, L.; Na, M.; Oh, H.; Bae, E.Y.; Jeong, D.G.; Ryu, S.E.; Kim, S.; Kim, B.Y.; Oh, W.K.; Ahn, J.S. Protein tyrosine phosphatase 1B inhibitors from Morus root bark. Bioorg. Med. Chem. Lett. 2006, 16, 1426–1429. [Google Scholar] [CrossRef]
- Jung, H.A.; Paudel, P.; Seong, S.H.; Min, B.S.; Choi, J.S. Structure-related protein tyrosine phosphatase 1B inhibition by naringenin derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 2274–2280. [Google Scholar] [CrossRef]
- Li, T.; Zhang, X.; Song, Y.; Liu, J. A microplate-based screening method for α-glucosidase inhibitors. Chin. J. Clin. Pharmacol. Therapeut. 2005, 10, 1128–1134. [Google Scholar]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef] [Green Version]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, G.; Pan, J.; Wang, Y. α-Glucosidase inhibition by luteolin: Kinetics, interaction and molecular docking. Int. J. Biol. Macromol. 2014, 64, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Liu, T.; Chen, C.; Li, M.Y.; Wang, Z.Y.; Chen, R.S.; Wei, G.X.; Wang, X.Y.; Luo, D.Q. Fumosorinone, a novel PTP1B inhibitor, activates insulin signaling in insulin-resistance HepG2 cells and shows anti-diabetic effect in diabetic KKAy mice. Toxicol. Appl. Pharmacol. 2015, 285, 61–70. [Google Scholar] [CrossRef]
- Paudel, P.; Yu, T.; Seong, S.H.; Kuk, E.B.; Jung, H.A.; Choi, J.S. Protein tyrosine phosphatase 1B inhibition and glucose uptake potentials of mulberrofuran G, albanol B, and kuwanon G from root bark of Morus alba L. in insulin-resistant HepG2 cells: An in vitro and in silico study. Int. J. Mol. Sci. 2018, 19, 1542. [Google Scholar] [CrossRef]
- Bhakta, H.K.; Paudel, P.; Fujii, H.; Sato, A.; Park, C.H.; Yokozawa, T.; Jung, H.A.; Choi, J.S. Oligonol promotes glucose uptake by modulating the insulin signaling pathway in insulin-resistant HepG2 cells via inhibiting protein tyrosine phosphatase 1B. Arch. Pharm. Res. 2017, 40, 1314–1327. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Samples | Yield (%) a | IC50 Values (µg/mL) b ± SD | |
|---|---|---|---|
| PTP 1B | α-Glucosidase | ||
| MeOH | 29.62 * | 9.01 ± 0.33 e | 91.58 ± 9.66 f |
| CH2Cl2 | 20.89 | 6.95 ± 0.94 f | 16.76 ± 1.90 h |
| EtOAc | 4.56 | 2.79 ± 0.11 g | 6.71 ± 0.15 i |
| n-BuOH | 10.55 | 8.72 ± 0.39 e | 26.15 ± 0.14 g |
| H2O | 57.36 | 55.03 ± 1.35 d | 867.70 ± 42.20 d |
| Acarbose c | - | - | 121.33 ± 2.24 e |
| Ursolic acid c | - | 6.53 ± 0.23 f | - |
| Compounds | PTP1B (n = 3) | α-Glucosidase (n = 3) | ||||
|---|---|---|---|---|---|---|
| IC50 (µM) a | Inhibition Type b | Ki (µM) c | IC50 (µM) a | Inhibition Type b | Ki (µM) c | |
| 1 | 7.74 ± 0.14 f | Mixed-type | 1.19 | 2.63 ± 0.11 g | Noncompetitive | 1.92 |
| 2 | 8.50 ± 0.45 e | Mixed-type | 2.40 | 7.24 ± 0.02 f | Noncompetitive | 5.54 |
| 3 | 5.29 ± 0.08 g | Competitive | 2.25 | 1.92 ± 0.02 h | Mixed-type | 0.21 |
| Acarbose d | - | - | - | 212.66 ± 0.35 e | - | - |
| Ursolic acid d | 8.66 ± 0.82 e | - | - | - | - | - |
| Compounds | Binding Energy (kcal/mol) | H-bond Interactions | Other Interactions |
|---|---|---|---|
| 1 | −5.96 | Lys116, Asp181, Arg221, Gln226 | Thr263, Val184, Gln266, Gly183, Arg221, Phe182, Lys116, Trp179 |
| −5.83 | Ala189, Glu276, Gly277 | Ala189, Leu192, Asn193, Phe196, Phe280, Glu276, Gly277 | |
| 2 | −5.80 | Arg211, Gln266, Gly183 | Arg211, Gln266, Trp179, Thr283, Asp265, Asp181, Lys116, Phe182, Val184 |
| −5.83 | Glu276, Gly277 | Ala189, Leu192, Asn193, Phe280, Glu276, Gly277, Phe196 | |
| 3 | −6.86 | Lys116, Cys215, Arg221 | Tyr46, Lys120, Lys116, Asp181, Phe182, Val184, Thr263, Gln266, Trp179, Gln262, Arg221, Cys215, Ser216 |
| Compound A a, b | −7.78 | Asp48, Lys116, Lys120, Asp181 | Ala217, Tyr46, Met258, Gln262, Ala217 |
| Compound B a, c | −11.30 | Glu276, Lys279, Phe280, Asn193 | Leu192, Phe196, Ile281, Phe280, Ala189, Lys197 |
| Compounds a | Binding Energy (kcal/mol) | H-Bond Interactions | Other Interactions |
|---|---|---|---|
| 1 | −5.94 | Thr290, Glu271 | Lys13, Lys16, Trp343, Cys342, Ala292, Thr290, Leu297, Ser298, Glu271 |
| 2 | −5.61 | Tyr158, Arg315, Glu411, Asn415 | Lys156, Tyr158, Gly160, Pro312, Leu313, Phe314, Arg315, Glu411, Asn415 |
| 3 | −8.06 | Ser157, Asp307, Gln353 | Tyr158, Lys156, Ser157, Asn415, Arg315, Asp307, Gln353, Phe303, Asp352, His280, Gln279 |
| Compound C b | −8.60 | Asp69, His112, Tyr158, Gln182, Asp215, Arg213, Ser240, Asp242, Glu277, His280, Asp307, Asp352, Arg442 | Tyr72, Lys156, Phe178, Val216, Gln279, Phe303, Arg315, Gln353, His351, Glu411 |
| Compound D c | −6.85 | Glu296, His295 | Trp15, Lys16, Asn259, Ala292, Thr290, Arg294, Leu297, Ser291, Ser298, Asp341, Cys342, Trp343 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paudel, P.; Seong, S.H.; Park, H.J.; Jung, H.A.; Choi, J.S. Anti-Diabetic Activity of 2,3,6-Tribromo-4,5-Dihydroxybenzyl Derivatives from Symphyocladia latiuscula through PTP1B Downregulation and α-Glucosidase Inhibition. Mar. Drugs 2019, 17, 166. https://doi.org/10.3390/md17030166
Paudel P, Seong SH, Park HJ, Jung HA, Choi JS. Anti-Diabetic Activity of 2,3,6-Tribromo-4,5-Dihydroxybenzyl Derivatives from Symphyocladia latiuscula through PTP1B Downregulation and α-Glucosidase Inhibition. Marine Drugs. 2019; 17(3):166. https://doi.org/10.3390/md17030166
Chicago/Turabian StylePaudel, Pradeep, Su Hui Seong, Hye Jin Park, Hyun Ah Jung, and Jae Sue Choi. 2019. "Anti-Diabetic Activity of 2,3,6-Tribromo-4,5-Dihydroxybenzyl Derivatives from Symphyocladia latiuscula through PTP1B Downregulation and α-Glucosidase Inhibition" Marine Drugs 17, no. 3: 166. https://doi.org/10.3390/md17030166