
Spindle Cell Rhabdomyosarcoma of the Inguinal Region Mimicking a Complicated Hernia in the Adult—An Unexpected Finding
 , ,
, ,
Abstract
:1. Introduction
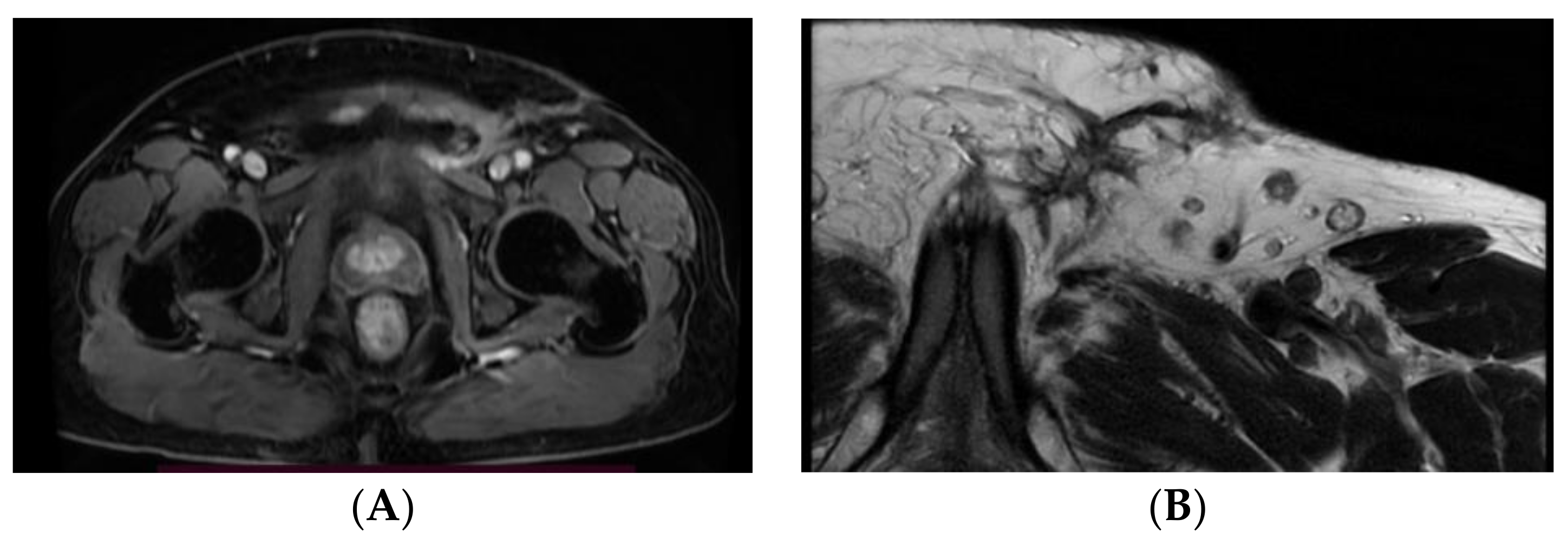
2. Detailed Case Description
3. Discussion
- -
- Spindle cell/sclerosing rhabdomyosarcoma with a somatic activating mutation of the MYOD1 gene at position Lys122 that can be homo- or heterozygous. The mutated gene interacts with the MYC oncogene. This subtype can be encountered both in children in adults, being characterized by a poor prognosis, especially in children and adolescents, with the latter group age presenting a characteristic mutation of MyoD1 p.Leu122Arg [21,27,28];
- -
- -
- -
- Cases that do not have the alterations described above, encountered most commonly around the area of the testis or within the abdominal cavity [36];
- -
- Patients with spindle cell rhabdomyosarcoma without molecular alterations [27].
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Horvai, A. Bones, Joints and Soft Tissue Tumors. In Pathologic Basis of Disease, 10th ed.; Kumar, V., Abbas, A., Aster, J., Turner, J., Eds.; Elsevier Inc.: Philadelphia, PA, USA, 2021; pp. 1171–1218. [Google Scholar]
- Villella, J.A.; Bogner, P.N.; Jani-Sait, S.N.; Block, A.M.W.; Lele, S. Rhabdomyosarcoma of the cervix in sisters with review of the literature. Gynecol. Oncol. 2005, 99, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Margioula-Siarkou, C.; Petousis, S.; Almperis, A.; Margioula-Siarkou, G.; Laganà, A.S.; Kourti, M.; Papanikolaou, A.; Dinas, K. Sarcoma Botryoides: Optimal Therapeutic Management and Prognosis of an Unfavorable Malignant Neoplasm of Female Children. Diagnostics 2023, 13, 924. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Dileo, P.; Casanova, M.; Bertulli, R.; Meazza, C.; Gandola, L.; Navarria, P.; Collini, P.; Gronchi, A.; Olmi, P.; et al. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer 2003, 98, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Yao, H.; Li, X.; Li, H.; Wu, L. Stage 1 embryonal rhabdomyosarcoma of the female genital tract: A retrospective clinical study of nine cases. World J. Surg. Oncol. 2017, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, J.; Li, Z.; Zhang, X.; Chen, J.; Feng, G. Characteristics and prognosis of embryonal rhabdomyosarcoma in children and adolescents: An analysis of 464 cases from the SEER database. Chin. Med. Assoc. Pediatr. Investig. 2020, 4, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Koscielniak, E.; Morgan, M.; Treuner, J. Soft tissue sarcoma in children: Prognosis and management. Paediatr. Drugs 2002, 4, 21–28. [Google Scholar] [CrossRef]
- Kabir, W.; Choong, P. The Epidemiology and Pathogenesis of Sarcoma. In Sarcoma, A Practical Guide to Multidisciplinary Management; Choong, P., Ed.; Springer: Singapore, 2021; pp. 11–28. [Google Scholar]
- Newton, W.A.J.; Gehan, E.A.; Webber, B.L.; Marsden, H.B.; van Unnik, A.J.; Hamoudi, A.B.; Tsokos, M.C.; Shimada, H.; Harms, D.; Schmidt, D.; et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995, 76, 1073–1085. [Google Scholar]
- Parham, D.M.; Ellison, D.A. Rhabdomyosarcomas in Adults and Children: An Update. Arch. Pathol. Lab. Med. 2006, 130, 1454–1465. [Google Scholar] [CrossRef]
- Cavazzana, A.O.; Schmidt, D.; Ninfo, V.; Harms, D.; Tollot, M.; Carli, M.; Treuner, J.; Betto, R.; Salviati, G. Spindle cell rhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcoma. Am. J. Surg. Pathol. 1992, 16, 229–235. [Google Scholar] [CrossRef]
- Rubin, B.P.; Hasserjian, R.P.; Singer, S.; Janecka, I.; Fletcher, J.A.; Fletcher, C.D.M. Spindle cell rhabdomyosarcoma (so-called) in adults: Report of two cases with emphasis on differential diagnosis. Am. J. Surg. Pathol. 1998, 22, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Mentzel, T.; Katenkamp, D. Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch. 2000, 436, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.J.; Nodit, L. Spindle cell rhabdomyosarcoma: A brief diagnostic review and differential diagnosis. Arch. Pathol. Lab. Med. 2013, 137, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Zhang, L.; Sung, Y.S.; Huang, S.C.; Chen, C.L.; Bisogno, G.; Zin, A.; Agaram, N.P.; LaQuaglia, M.P.; Wexler, L.H.; et al. A Molecular Study of Pediatric Spindle and Sclerosing Rhabdomyosarcoma: Identification of Novel and Recurrent VGLL2-related Fusions in Infantile Cases. Am. J. Surg. Pathol. 2016, 40, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.; Szuhai, K. Spindle cell/sclerosing rhabdomyosarcoma. In WHO Classification of Tumours Editorial Board—Soft Tissue and Bone Tumours, 5th ed.; WHO Press: Geneve, Switzerland; International Agency for Research on Cancer: Lyon, France, 2020; pp. 211–213. [Google Scholar]
- Weiss, S.; Goldblum, J. Soft Tissue Tumors, 5th ed.; Applied Pathology; Elsevier-Saunders: Philadelphia, PA, USA, 1988; Volume 6, pp. 153–240. [Google Scholar]
- Jakkampudi, A.; Kaliyath, S.; Hegde, P.; Mathias, M.; Shetty, V. Spindle cell rhabdomyosarcoma in the adult: A rare case report. J. Oral Maxillofac. Pathol. 2022, 26 (Suppl. S1), S103. [Google Scholar]
- Parham, D.M.B.F. Embryonal rhabdomyosarcoma. In WHO Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone; Fletcher, C.D., Bridge, J.A., Hogendoorn, P.C.M.F., Eds.; IARC Press: Lyon, France, 2013; pp. 127–129. [Google Scholar]
- Agaram, N.P.; LaQuaglia, M.P.; Alaggio, R.; Zhang, L.; Fujisawa, Y.; Ladanyi, M.; Wexler, L.H.; Antonescu, C.R. MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: An aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod. Pathol. 2019, 32, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Little, D.J.; Ballo, M.T.; Zagars, G.K.; Pisters, P.W.T.; Patel, S.R.; El-Naggar, A.K.; Garden, A.S.; Benjamin, R.S. Adult rhabdomyosarcoma: Outcome following multimodality treatment. Cancer 2002, 95, 377–388. [Google Scholar] [CrossRef]
- Nascimento, A.F.; Fletcher, C.D.M. Spindle cell rhabdomyosarcoma in adults. Am. J. Surg. Pathol. 2005, 29, 1106–1113. [Google Scholar] [CrossRef]
- Hawkins, W.G.; Hoos, A.; Antonescu, C.R.; Urist, M.J.; Leung, D.H.; Gold, J.S.; Woodruff, J.M.; Lewis, J.J.; Brennan, M.F. Clinicopathologic analysis of patients with adult rhabdomyosarcoma. Cancer 2001, 91, 794–803. [Google Scholar] [CrossRef]
- Stock, N.; Chibon, F.; Nguyen Binh, M.B.; Terrier, P.; Michels, J.J.; Valo, I.; Robin, Y.M.; Guillou, L.; Ranchère-Vince, D.; Decouvelaere, A.V.; et al. Adult-type rhabdomyosarcoma: Analysis of 57 cases with clinicopathologic description, identification of 3 morphologic patterns and prognosis. Am. J. Surg. Pathol. 2009, 33, 1850–1859. [Google Scholar] [CrossRef]
- Hartmann, S.; Lessner, G.; Mentzel, T.; Kübler, A.C.; Müller-Richter, U.D.A. An adult spindle cell rhabdomyosarcoma in the head and neck region with long-term survival: A case report. J. Med. Case Rep. 2014, 8, 208. [Google Scholar] [CrossRef] [PubMed]
- Łomiak, M.; Świtaj, T.; Spałek, M.; Radzikowska, J.; Chojnacka, M.; Falkowski, S.; Wągrodzki, M.; Kukwa, W.; Szumera-Ciećkiewicz, A.; Rutkowski, P.; et al. Diagnosis and treatment of rhabdomyosarcomas. Oncol. Clin. Pract. 2023. [Google Scholar] [CrossRef]
- Kohsaka, S.; Shukla, N.; Ameur, N.; Ito, T.; Ng, C.K.Y.; Wang, L.; Lim, D.; Marchetti, A.; Viale, A.; Pirun, M.; et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat. Genet. 2014, 46, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.; Perrin, V.; Guillemot, D.; Reynaud, S.; Coindre, J.M.; Karanian, M.; Guinebretière, J.M.; Freneaux, P.; Le Loarer, F.; Bouvet, M.; et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J. Pathol. 2018, 245, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; Zhang, L.; Sung, Y.S.; Cavalcanti, M.S.; Torrence, D.; Wexler, L.; Francis, G.; Sommerville, S.; Swanson, D.; Dickson, B.C.; et al. Expanding the Spectrum of Intraosseous Rhabdomyosarcoma: Correlation between Two Distinct Gene Fusions and Phenotype. Am. J. Surg. Pathol. 2019, 43, 695. [Google Scholar] [CrossRef]
- Chrisinger, J.S.A.; Wehrli, B.; Dickson, B.C.; Fasih, S.; Hirbe, A.C.; Shultz, D.B.; Zadeh, G.; Gupta, A.A.; Demicco, E.G. Epithelioid and spindle cell rhabdomyosarcoma with FUS-TFCP2 or EWSR1-TFCP2 fusion: Report of two cases. Virchows Arch. 2020, 477, 725–732. [Google Scholar] [CrossRef]
- Wong, D.D.; van Vliet, C.; Gaman, A.; Giardina, T.; Amanuel, B. Rhabdomyosarcoma with FUS re-arrangement: Additional case in support of a novel subtype. Pathology 2019, 51, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Dashti, N.K.; Wehrs, R.N.; Thomas, B.C.; Nair, A.; Davila, J.; Buckner, J.C.; Martinez, A.P.; Sukov, W.R.; Halling, K.C.; Howe, B.M.; et al. Spindle cell rhabdomyosarcoma of bone with FUS–TFCP2 fusion: Confirmation of a very recently described rhabdomyosarcoma subtype. Histopathology 2018, 73, 514–520. [Google Scholar] [CrossRef]
- Tagami, Y.; Sugita, S.; Kubo, T.; Iesato, N.; Emori, M.; Takada, K.; Tsujiwaki, M.; Segawa, K.; Sugawara, T.; Kikuchi, T.; et al. Spindle cell rhabdomyosarcoma in a lumbar vertebra with FUS-TFCP2 fusion. Pathol. Res. Pract. 2019, 215, 152399. [Google Scholar] [CrossRef]
- Le Loarer, F.; Cleven, A.H.G.; Bouvier, C.; Castex, M.P.; Romagosa, C.; Moreau, A.; Salas, S.; Bonhomme, B.; Gomez-Brouchet, A.; Laurent, C.; et al. A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod. Pathol. 2020, 33, 404–419. [Google Scholar] [CrossRef]
- Leuschner, I.; Newton, W.; Schmidt, D.; Sachs, N.; Asmar, L.; Hamoudi, A.; Harms, D.; Maurer, H.M. Spindle Cell Variants of Embryonal Rhabdomyosarcoma in the Paratesticular Region A Report of the Intergroup Rhabdomyosarcoma Study. Am. J. Surg. Pathol. 1993, 17, 858. [Google Scholar] [CrossRef] [PubMed]
- Kaçar, A.; Demir, H.A.; Durak, H.; Dervişoǧlu, S. Spindle cell rhabdomyosarcoma displaying CD34 positivity: A potential diagnostic pitfall; report of two pediatric cases. Turk. Patoloji Derg. 2013, 29, 221–226. [Google Scholar] [PubMed]
- Zhao, Z.; Yin, Y.; Zhang, J.; Qi, J.; Zhang, D.; Ma, Y.; Wang, Y.; Li, S.; Zhou, J. Spindle cell/sclerosing rhabdomyosarcoma: Case series from a single institution emphasizing morphology, immunohistochemistry and follow-up. Int. J. Clin. Exp. Pathol. 2015, 8, 13814. [Google Scholar] [PubMed]
- Smith, M.H.; Atherton, D.; Reith, J.D.; Islam, N.M.; Bhattacharyya, I.; Cohen, D.M. Rhabdomyosarcoma, Spindle Cell/Sclerosing Variant: A Clinical and Histopathological Examination of this Rare Variant with Three New Cases from the Oral Cavity. Head. Neck Pathol. 2017, 11, 494. [Google Scholar] [CrossRef]
- Meza, J.L.; Anderson, J.; Pappo, A.S.; Meyer, W.H. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: The children’s oncology group. J. Clin. Oncol. 2006, 24, 3844–3851. [Google Scholar] [CrossRef] [PubMed]
- Borinstein, S.C.; Steppan, D.; Hayashi, M.; Loeb, D.M.; Isakoff, M.S.; Binitie, O.; Brohl, A.S.; Bridge, J.A.; Stavas, M.; Shinohara, E.T.; et al. Consensus and controversies regarding the treatment of rhabdomyosarcoma. Pediatr. Blood Cancer 2018, 65, e26809. [Google Scholar] [CrossRef] [PubMed]
- Ladra, M.M.; Szymonifka, J.D.; Mahajan, A.; Friedmann, A.M.; Yeap, B.Y.; Goebel, C.P.; MacDonald, S.M.; Grosshans, D.R.; Rodriguez-Galindo, C.; Marcus, K.J.; et al. Preliminary results of a phase II trial of proton radiotherapy for pediatric rhabdomyosarcoma. J. Clin. Oncol. 2014, 32, 3762–3770. [Google Scholar] [CrossRef]
- Benkhaled, S.; Mané, M.; Jungels, C.; Shumelinsky, F.; Aubain, N.D.; Saint Van Gestel, D. Successful treatment of synchronous chemoresistant pulmonary metastasis from pleomorphic rhabdomyosarcoma with stereotaxic body radiation therapy: A case report and a review of the literature. Cancer Treat. Res. Commun. 2021, 26, 100282. [Google Scholar] [CrossRef]
- Esnaola, N.F.; Rubin, B.P.; Baldini, E.H.; Vasudevan, N.; Demetri, G.D.; Fletcher, C.D.M.; Singer, S. Response to Chemotherapy and Predictors of Survival in Adult Rhabdomyosarcoma. Ann. Surg. 2001, 234, 215. [Google Scholar] [CrossRef]
- Kojima, Y.; Hashimoto, K.; Ando, M.; Yonemori, K.; Yamamoto, H.; Kodaira, M.; Yunokawa, M.; Shimizu, C.; Tamura, K.; Hosono, A.; et al. Comparison of dose intensity of vincristine, dactinomycin, and cyclophosphamide chemotherapy for child and adult rhabdomyosarcoma: A retrospective analysis. Cancer Chemother. Pharmacol. 2012, 70, 391–397. [Google Scholar] [CrossRef]
- Raney, R.B.; Maurer, H.M.; Anderson, J.R.; Andrassy, R.J.; Donaldson, S.S.; Qualman, S.J.; Wharam, M.D.; Wiener, E.S.; Crist, W.M. The Intergroup Rhabdomyosarcoma Study Group (IRSG): Major Lessons From the IRS-I Through IRS-IV Studies as Background for the Current IRS-V Treatment Protocols. Sarcoma 2001, 5, 925281. [Google Scholar] [CrossRef]
- Simon, J.H.; Paulino, A.C.; Ritchie, J.M.; Mayr, N.A.; Buatti, J.M. Presentation, Prognostic Factors and Patterns of Failure in Adult Rhabdomyosarcoma. Sarcoma 2003, 7, 147. [Google Scholar] [CrossRef]
- Keskin, S.; Ekenel, M.; Basaran, M.; Kilicaslan, I.; Tunc, M.; Bavbek, S. Clinicopathological characteristics and treatment outcomes of adult patients with paratesticular rhabdomyosarcoma (PRMS): A 10-year single-centre experience. Can. Urol. Assoc. J. 2012, 6, 42. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prognosis | Type |
|---|---|
| Superior prognosis | Botryoid rhabdomyosarcoma |
| Spindle cell rhabdomyosarcoma | |
| Intermediate prognosis | Embryonal rhabdomyosarcoma |
| Poor prognosis | Alveolar rhabdomyosarcoma |
| Undifferentiated rhabdomyosarcoma |
| Immunohistochemistry Marker | Result |
|---|---|
| Vimentin | Diffuse and intense positive in the tumoral proliferation |
| Myogenic determination gene (MyoD1) | Diffuse positive in the tumoral proliferation |
| Myogenin (Myf-4) | Focal positive in the tumoral proliferation |
| Desmin | Focal positive in the tumoral proliferation |
| Actin | Positive in the tumoral proliferation |
| Epithelial membrane antigen (EMA) | Negative in the tumoral proliferation |
| CD34 | Negative in the tumoral proliferation; the presence of internal control |
| Ki67 | Positive in approximately 25% of the tumoral cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigorean, V.T.; Serescu, R.; Anica, A.; Coman, V.E.; Bedereag, Ş.I.; Sfetea, R.C.; Liţescu, M.; Pleşea, I.E.; Florea, C.G.; Burleanu, C.; et al. Spindle Cell Rhabdomyosarcoma of the Inguinal Region Mimicking a Complicated Hernia in the Adult—An Unexpected Finding. Medicina 2023, 59, 1515. https://doi.org/10.3390/medicina59091515
Grigorean VT, Serescu R, Anica A, Coman VE, Bedereag ŞI, Sfetea RC, Liţescu M, Pleşea IE, Florea CG, Burleanu C, et al. Spindle Cell Rhabdomyosarcoma of the Inguinal Region Mimicking a Complicated Hernia in the Adult—An Unexpected Finding. Medicina. 2023; 59(9):1515. https://doi.org/10.3390/medicina59091515
Chicago/Turabian StyleGrigorean, Valentin Titus, Radu Serescu, Andrei Anica, Violeta Elena Coman, Ştefan Iulian Bedereag, Roxana Corina Sfetea, Mircea Liţescu, Iancu Emil Pleşea, Costin George Florea, Cosmin Burleanu, and et al. 2023. "Spindle Cell Rhabdomyosarcoma of the Inguinal Region Mimicking a Complicated Hernia in the Adult—An Unexpected Finding" Medicina 59, no. 9: 1515. https://doi.org/10.3390/medicina59091515