
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) and Comorbidities: Linked by Vascular Pathomechanisms and Vasoactive Mediators?
{kind=link}
Abstract
:1. Introduction
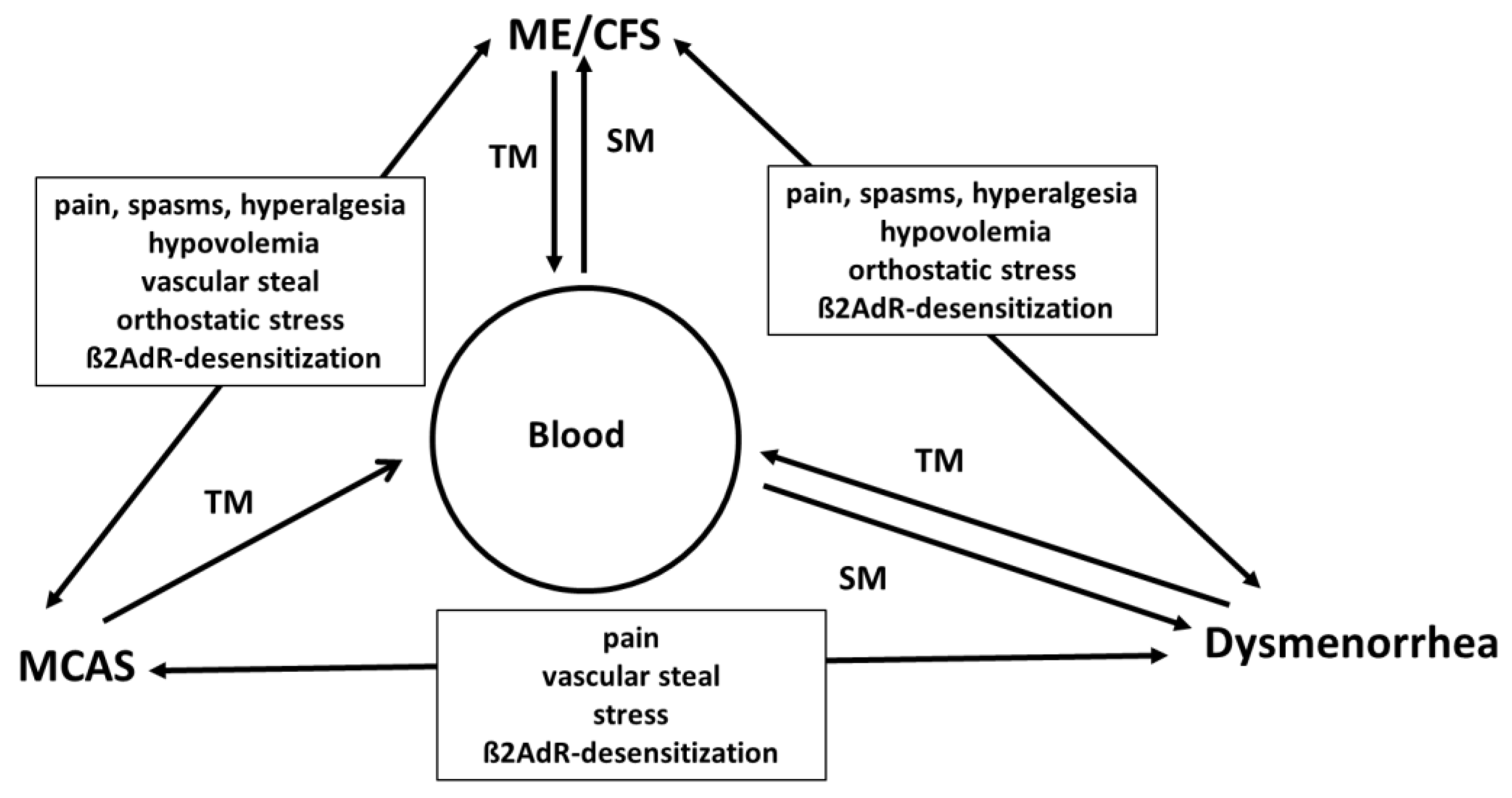
2. Common Pathomechanisms in ME/CFS, MCA and Dysmenorrhea
2.1. Excessive Generation of Tissue Inflammatory Mediators and Spillover into the Systemic Circulation Causes Symptoms
2.2. Association of MCA and ME/CFS and Common Pathomechanisms
2.3. Link of Endometriosis/Dysmenorrhea and ME/CFS
2.4. The Potential Role of Dysfunctional β2AdR
2.5. Mutual Triggering and Disease Initiation
2.6. Disease Specific Symptoms versus Common Mechanisms
3. Association with Postural Tachycardia Syndrome, Reduced Cerebral Blood Flow and Orthostatic Intolerance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carruthers, B.M.; Van De Sande, M.I.; De Meirleir, K.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; Powles, A.C.; Speight, N.; Vallings, R.; et al. Myalgic encephalomyelitis: International Consensus Criteria. J. Intern. Med. 2011, 270, 327–338. [Google Scholar] [CrossRef]
- Jason, L.A.; Holtzman, C.S.; Sunnquist, M.; Cotler, J. The development of an instrument to assess post-exertional malaise in patients with myalgic encephalomyelitis and chronic fatigue syndrome. J. Health Psychol. 2021, 26, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.L.; Weitzer, D.J. Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)—A Systemic Review and Comparison of Clinical Presentation and Symptomatology. Medicina 2021, 57, 418. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Cholevas, C.; Polyzoidis, K.; Politis, A. Long-COVID syndrome-associated brain fog and chemofog: Luteolin to the rescue. Biofactors 2021, 47, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.; Scheibenbogen, C. A Unifying Hypothesis of the Pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): Recognitions from the finding of autoantibodies against β2-adrenergic receptors. Autoimmun. Rev. 2020, 19, 102527. [Google Scholar] [CrossRef] [PubMed]
- Boneva, R.S.; Lin, J.-M.S.; Wieser, F.; Nater, U.M.; Ditzen, B.; Taylor, R.N.; Unger, E.R. Endometriosis as a Comorbid Condition in Chronic Fatigue Syndrome (CFS): Secondary Analysis of Data From a CFS Case-Control Study. Front. Pediatr. 2019, 7, 195. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.J.; Lee, S.H.; Park, J.W.; Han, M.; Park, M.J.; Han, S.J. Dysfunctional signaling underlying endometriosis: Current state of knowledge. J. Mol. Endocrinol. 2018, 60, R97–R113. [Google Scholar] [CrossRef]
- Signorile, P.G.; Cassano, M.; Viceconte, R.; Marcattilj, V.; Baldi, A. Endometriosis: A Retrospective Analysis of Clinical Data from a Cohort of 4083 Patients, with Focus on Symptoms. In Vivo 2022, 36, 874–883. [Google Scholar] [CrossRef]
- DiBenedetti, D.; Soliman, A.M.; Gupta, C.; Surrey, E.S. Patients’ perspectives of endometriosis-related fatigue: Qualitative interviews. J. Patient-Rep. Outcomes 2020, 4, 33. [Google Scholar] [CrossRef]
- Rowe, P.C.; Underhill, R.A.; Friedman, K.J.; Gurwitt, A.; Medow, M.S.; Schwartz, M.S.; Speight, N.; Stewart, J.M.; Vallings, R.; Rowe, K.S. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Diagnosis and Management in Young People: A Primer. Front. Pediatr. 2017, 5, 121. [Google Scholar] [CrossRef]
- Yong, S.J.; Liu, S. Proposed subtypes of post-COVID-19 syndrome (or long-COVID) and their respective potential therapies. Rev. Med. Virol. 2022, 32, e2315. [Google Scholar] [CrossRef] [PubMed]
- Dixit, N.M.; Churchill, A.; Nsair, A.; Hsu, J.J. Post-Acute COVID-19 Syndrome and the cardiovascular system: What is known? Am. Heart J. Plus Cardiol. Res. Pract. 2021, 5, 100025. [Google Scholar] [CrossRef]
- Wirth, K.J.; Löhn, M. Orthostatic Intolerance after COVID-19 Infection: Is Disturbed Microcirculation of the Vasa Vasorum of Capacitance Vessels the Primary Defect? Medicina 2022, 58, 1807. [Google Scholar] [CrossRef] [PubMed]
- Blitshteyn, S.; Brinth, L.; Hendrickson, J.E.; Martinez-Lavin, M. Autonomic dysfunction and HPV immunization: An overview. Immunol. Res. 2018, 66, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, T.; Gu, S.; Carneiro-Leão, L.; Gutsche, A.; Maurer, M.; Siebenhaar, F. Idiopathic mast cell activation syndrome is more often suspected than diagnosed—A prospective real-life study. Allergy 2022, 77, 2794–2802. [Google Scholar] [CrossRef]
- Glynne, P.; Tahmasebi, N.; Gant, V.; Gupta, R. Long COVID following mild SARS-CoV-2 infection: Characteristic T cell alterations and response to antihistamines. J. Investig. Med. 2022, 70, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Mashauri, H.L. COVID-19 Histamine theory: Why antihistamines should be incorporated as the basic component in COVID-19 management? Health Sci. Rep. 2023, 6, e1109. [Google Scholar] [CrossRef]
- Qu, C.; Fuhler, G.M.; Pan, Y. Could Histamine H1 Receptor Antagonists Be Used for Treating COVID-19? Int. J. Mol. Sci. 2021, 22, 5672. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.D.; Lambert, N.; Downs, C.A.; Abrahim, H.; Hughes, T.D.; Rahmani, A.M.; Burton, C.W.; Chakraborty, R. Antihistamines for Postacute Sequelae of SARS-CoV-2 Infection. J. Nurse Pract. 2022, 18, 335–338. [Google Scholar] [CrossRef]
- Hamilton, M.J. Nonclonal Mast Cell Activation Syndrome: A Growing Body of Evidence. Immunol. Allergy Clin. N. Am. 2018, 38, 469–481. [Google Scholar] [CrossRef]
- Arun, S.; Storan, A.; Myers, B. Mast cell activation syndrome and the link with long COVID. Br. J. Hosp. Med. 2022, 83, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, L.B.; Brook, J.B.; Walters, A.S.; Goris, A.; Afrin, L.B.; Molderings, G.J. Mast cell activation symptoms are prevalent in Long-COVID. Int. J. Infect. Dis. 2021, 112, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, B.; Szyłło, K.; Romanowicz, H. Endometriosis: Epidemiology, Classification, Pathogenesis, Treatment and Genetics (Review of Literature). Int. J. Mol. Sci. 2021, 22, 10554. [Google Scholar] [CrossRef] [PubMed]
- Knific, T.; Fishman, D.; Vogler, A.; Gstöttner, M.; Wenzl, R.; Peterson, H.; Rižner, T.L. Multiplex analysis of 40 cytokines do not allow separation between endometriosis patients and controls. Sci. Rep. 2019, 9, 16738. [Google Scholar] [CrossRef]
- Ylikorkala, O.; Dawood, M.Y. New concepts in dysmenorrhea. Am. J. Obstet. Gynecol. 1978, 130, 833–847. [Google Scholar] [CrossRef]
- Barcikowska, Z.; Rajkowska-Labon, E.; Grzybowska, M.E.; Hansdorfer-Korzon, R.; Zorena, K. Inflammatory Markers in Dysmenorrhea and Therapeutic Options. Int. J. Environ. Res. Public Health 2020, 17, 1191. [Google Scholar] [CrossRef]
- Guimarães, I.; Póvoa, A.M. Primary Dysmenorrhea: Assessment and Treatment. Rev. Bras. Ginecol. Obstet. 2020, 42, 501–507. [Google Scholar] [CrossRef]
- Iacovides, S.; Avidon, I.; Baker, F.C. What we know about primary dysmenorrhea today: A critical review. Hum. Reprod. Update 2015, 21, 762–778. [Google Scholar] [CrossRef]
- Hashimoto, K.; Hirose, M.; Furukawa, S.; Hayakawa, H.; Kimura, E. Changes in Hemodynamics and Bradykinin Concentration in Coronary Sinus Blood in Experimental Coronary Artery Occlusion. Jpn. Heart J. 1977, 18, 679–689. [Google Scholar] [CrossRef]
- Koch, M.; Wendorf, M.; Dendorfer, A.; Wolfrum, S.; Schulze, K.; Spillmann, F.; Schultheiss, H.-P.; Tschöpe, C. Cardiac kinin level in experimental diabetes mellitus: Role of kininases. Am. J. Physiol. Circ. Physiol. 2003, 285, H418–H423. [Google Scholar] [CrossRef]
- Linz, W.; Wiemer, G.; Schölkens, B.A. Role of Kinins in the Pathophysiology of Myocardial Ischemia: In Vitro and In Vivo Studies. Diabetes 1996, 45, S51–S58. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.-L.; Chen, S.-R.; Scicli, G.M.; Carretero, O.A. Cardiac interstitial bradykinin release during ischemia is enhanced by ischemic preconditioning. Am. J. Physiol. Circ. Physiol. 2000, 279, H116–H121. [Google Scholar] [CrossRef] [PubMed]
- Staszewska-Barczak, J.; Ferreira, S.H.; Vane, J.R. An excitatory nociceptive cardiac reflex elicited by bradykinin and potentiated by prostaglandins and myocardial ischaemia. Cardiovasc. Res. 1976, 10, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, C.L.; Carretero, O.A.; Mindroiu, T.; Longhurst, J.C. Bradykinin release from contracting skeletal muscle of the cat. J. Appl. Physiol. 1990, 69, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.J.; Scheibenbogen, C. Pathophysiology of skeletal muscle disturbances in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). J. Transl. Med. 2021, 19, 162. [Google Scholar] [CrossRef] [PubMed]
- Bynke, A.; Julin, P.; Gottfries, C.-G.; Heidecke, H.; Scheibenbogen, C.; Bergquist, J. Autoantibodies to beta-adrenergic and muscarinic cholinergic receptors in Myalgic Encephalomyelitis (ME) patients—A validation study in plasma and cerebrospinal fluid from two Swedish cohorts. Brain Behav. Immun. Health 2020, 7, 100107. [Google Scholar] [CrossRef] [PubMed]
- Malkova, A.; Shoenfeld, Y. Autoimmune autonomic nervous system imbalance and conditions: Chronic fatigue syndrome, fibromyalgia, silicone breast implants, COVID and post-COVID syndrome, sick building syndrome, post-orthostatic tachycardia syndrome, autoimmune diseases and autoimmune/inflammatory syndrome induced by adjuvants. Autoimmun. Rev. 2023, 22, 103230. [Google Scholar] [CrossRef]
- Wirth, K.J.; Scheibenbogen, C. Dyspnea in Post-COVID Syndrome following Mild Acute COVID-19 Infections: Potential Causes and Consequences for a Therapeutic Approach. Medicina 2022, 58, 419. [Google Scholar] [CrossRef]
- Wirth, K.J.; Scheibenbogen, C.; Paul, F. An attempt to explain the neurological symptoms of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Transl. Med. 2021, 19, 471. [Google Scholar] [CrossRef]
- Loebel, M.; Grabowski, P.; Heidecke, H.; Bauer, S.; Hanitsch, L.G.; Wittke, K.; Meisel, C.; Reinke, P.; Volk, H.-D.; Fluge, Ø.; et al. Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome. Brain Behav. Immun. 2016, 52, 32–39. [Google Scholar] [CrossRef]
- Abe, N.; Toyama, H.; Ejima, Y.; Saito, K.; Tamada, T.; Yamauchi, M.; Kazama, I. α1-Adrenergic Receptor Blockade by Prazosin Synergistically Stabilizes Rat Peritoneal Mast Cells. BioMed Res. Int. 2020, 2020, 3214186. [Google Scholar] [CrossRef] [PubMed]
- MC van Campen, C.; Rowe, P.C.; Visser, F.C. Reductions in Cerebral Blood Flow Can Be Provoked by Sitting in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients. Healthcare 2020, 8, 394. [Google Scholar] [CrossRef]
- Petra, A.I.; Panagiotidou, S.; Stewart, J.M.; Conti, P.; Theoharides, T.C. Spectrum of mast cell activation disorders. Expert Rev. Clin. Immunol. 2014, 10, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Tsilioni, I.; Ren, H. Recent advances in our understanding of mast cell activation—Or should it be mast cell mediator disorders? Expert Rev. Clin. Immunol. 2019, 15, 639–656. [Google Scholar] [CrossRef] [PubMed]
- Rueff, A.; Dray, A. Sensitization of peripheral afferent fibres in the in vitro neonatal rat spinal cord-tail by bradykinin and prostaglandins. Neuroscience 1993, 54, 527–535. [Google Scholar] [CrossRef]
- Lim, E.-J.; Ahn, Y.-C.; Jang, E.-S.; Lee, S.-W.; Lee, S.-H.; Son, C.-G. Systematic review and meta-analysis of the prevalence of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). J. Transl. Med. 2020, 18, 100. [Google Scholar] [CrossRef]
- Luo, L.; Zhang, Y.; Huang, T.; Zhou, F.; Xiong, C.; Liu, Y.; Zhai, P.; Wang, G.; Tan, J.; Jiao, C.; et al. A description of the current status of chronic fatigue syndrome and associated factors among university students in Wuhan, China. Front. Psychiatry 2022, 13, 1047014. [Google Scholar] [CrossRef]
- Deumer, U.-S.; Varesi, A.; Floris, V.; Savioli, G.; Mantovani, E.; López-Carrasco, P.; Rosati, G.M.; Prasad, S.; Ricevuti, G. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): An Overview. J. Clin. Med. 2021, 10, 4786. [Google Scholar] [CrossRef]
- Novak, P.; Giannetti, M.P.; Weller, E.; Hamilton, M.J.; Castells, M. Mast cell disorders are associated with decreased cerebral blood flow and small fiber neuropathy. Ann. Allergy Asthma Immunol. 2021, 128, 299–306.e1. [Google Scholar] [CrossRef]
- Freitag, H.; Szklarski, M.; Lorenz, S.; Sotzny, F.; Bauer, S.; Philippe, A.; Kedor, C.; Grabowski, P.; Lange, T.; Riemekasten, G.; et al. Autoantibodies to Vasoregulative G-Protein-Coupled Receptors Correlate with Symptom Severity, Autonomic Dysfunction and Disability in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Clin. Med. 2021, 10, 3675. [Google Scholar] [CrossRef]
- Wallukat, G.; Hohberger, B.; Wenzel, K.; Fürst, J.; Schulze-Rothe, S.; Wallukat, A.; Hönicke, A.-S.; Müller, J. Functional autoantibodies against G-protein coupled receptors in patients with persistent Long-COVID-19 symptoms. J. Transl. Autoimmun. 2021, 4, 100100. [Google Scholar] [CrossRef] [PubMed]
- Dotan, A.; David, P.; Arnheim, D.; Shoenfeld, Y. The autonomic aspects of the post-COVID19 syndrome. Autoimmun. Rev. 2022, 21, 103071. [Google Scholar] [CrossRef] [PubMed]
- Szewczykowski, C.; Mardin, C.; Lucio, M.; Wallukat, G.; Hoffmanns, J.; Schröder, T.; Raith, F.; Rogge, L.; Heltmann, F.; Moritz, M.; et al. Long COVID: Association of Functional Autoantibodies against G-Protein-Coupled Receptors with an Impaired Retinal Microcirculation. Int. J. Mol. Sci. 2022, 23, 7209. [Google Scholar] [CrossRef]
- Mikkelsen, E.; Sakr, A.M.; Jespersen, L.T. Studies on the Effect of Histamine in Isolated Human Pulmonary Arteries and Veins. Acta Pharmacol. Toxicol. 2009, 54, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Schoeffter, P.; Godfraind, T. Histamine Receptors in the Smooth Muscle of Human Internal Mammary Artery and Saphenous Vein. Basic Clin. Pharmacol. Toxicol. 1989, 64, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Diana, J.; Schwinghamer, J.; Young, S. Direct effect of histamine on arterial and venous resistance in isolated dog hindlimb. Am. J. Physiol. Content 1968, 214, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Dachman, W.D.; Bedarida, G.; Blaschke, T.F.; Hoffman, B.B. Histamine-induced venodilation in human beings involves both H1 and H2 receptor subtypes. J. Allergy Clin. Immunol. 1994, 93, 606–614. [Google Scholar] [CrossRef]
- Müller-Schweinitzer, E. On the pharmacology of venous smooth muscle from dog and man. Folia Haematol. Int. Mag. Klin. Morphol. Blutforsch. 1979, 106, 690–704. [Google Scholar]
- Tsuru, H.; Kohno, S.; Iwata, M.; Shigei, T. Characterization of histamine receptors in isolated rabbit veins. Experiment 1987, 243, 696–702. [Google Scholar]
- Bergner, M.; Gräser, T.; Handschuk, L.; Tiedt, N. Vasomotor tone of isolated porcine coronary veins in response to acetylcholine, noradrenaline, and histamine. Biomed. Biochim. Acta 1988, 47, 775–779. [Google Scholar]
- Yoshiuchi, K.; Farkas, J.; Natelson, B.H. Patients with chronic fatigue syndrome have reduced absolute cortical blood flow. Clin. Physiol. Funct. Imaging 2006, 26, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Van Campen, C.M.C.; Rowe, P.C.; Visser, F.C. Cerebral blood flow remains reduced after tilt testing in myalgic encephalomyelitis/chronic fatigue syndrome patients. Clin. Neurophysiol. Pract. 2021, 6, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.Y.; Barnden, L.R.; Kwiatek, R.A.; Bhuta, S.; Hermens, D.F.; Lagopoulos, J. Neuroimaging characteristics of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): A systematic review. J. Transl. Med. 2020, 18, 335. [Google Scholar] [CrossRef]
- Shan, Z.Y.; Finegan, K.; Bhuta, S.; Ireland, T.; Staines, D.R.; Marshall-Gradisnik, S.M.; Barnden, L.R. Brain function characteristics of chronic fatigue syndrome: A task fMRI study. NeuroImage Clin. 2018, 19, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sadato, N.; Okada, T.; Mizuno, K.; Sasabe, T.; Tanabe, H.C.; Saito, D.N.; Onoe, H.; Kuratsune, H.; Watanabe, Y. Reduced responsiveness is an essential feature of chronic fatigue syndrome: A fMRI study. BMC Neurol. 2006, 6, 9. [Google Scholar] [CrossRef]
- Stanasila, L.; Diviani, D. Protein-Protein Interactions at the Adrenergic Receptors. Curr. Drug Targets 2012, 13, 15–27. [Google Scholar] [CrossRef]
- Hoeijmakers, J.G.; Merkies, I.S.; Faber, C.G. Small fiber neuropathies: Expanding their etiologies. Curr. Opin. Neurol. 2022, 35, 545–552. [Google Scholar] [CrossRef]
- Bragée, B.; Michos, A.; Drum, B.; Fahlgren, M.; Szulkin, R.; Bertilson, B.C. Signs of Intracranial Hypertension, Hypermobility, and Craniocervical Obstructions in Patients With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Neurol. 2020, 11, 828. [Google Scholar] [CrossRef]
- Hulens, M.; Bruyninckx, F.; Dankaerts, W.; Rasschaert, R.; De Mulder, P.; Stalmans, I.; Vansant, G.; Bervoets, C. High Prevalence of Perineural Cysts in Patients with Fibromyalgia and Chronic Fatigue Syndrome. Pain Med. 2021, 22, 883–890. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wirth, K.J.; Löhn, M. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) and Comorbidities: Linked by Vascular Pathomechanisms and Vasoactive Mediators? Medicina 2023, 59, 978. https://doi.org/10.3390/medicina59050978
Wirth KJ, Löhn M. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) and Comorbidities: Linked by Vascular Pathomechanisms and Vasoactive Mediators? Medicina. 2023; 59(5):978. https://doi.org/10.3390/medicina59050978
Chicago/Turabian StyleWirth, Klaus J., and Matthias Löhn. 2023. "Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) and Comorbidities: Linked by Vascular Pathomechanisms and Vasoactive Mediators?" Medicina 59, no. 5: 978. https://doi.org/10.3390/medicina59050978