
The Philadelphia Chromosome, from Negative to Positive: A Case Report of Relapsed Acute Lymphoblastic Leukemia Following Allogeneic Stem Cell Transplantation
 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
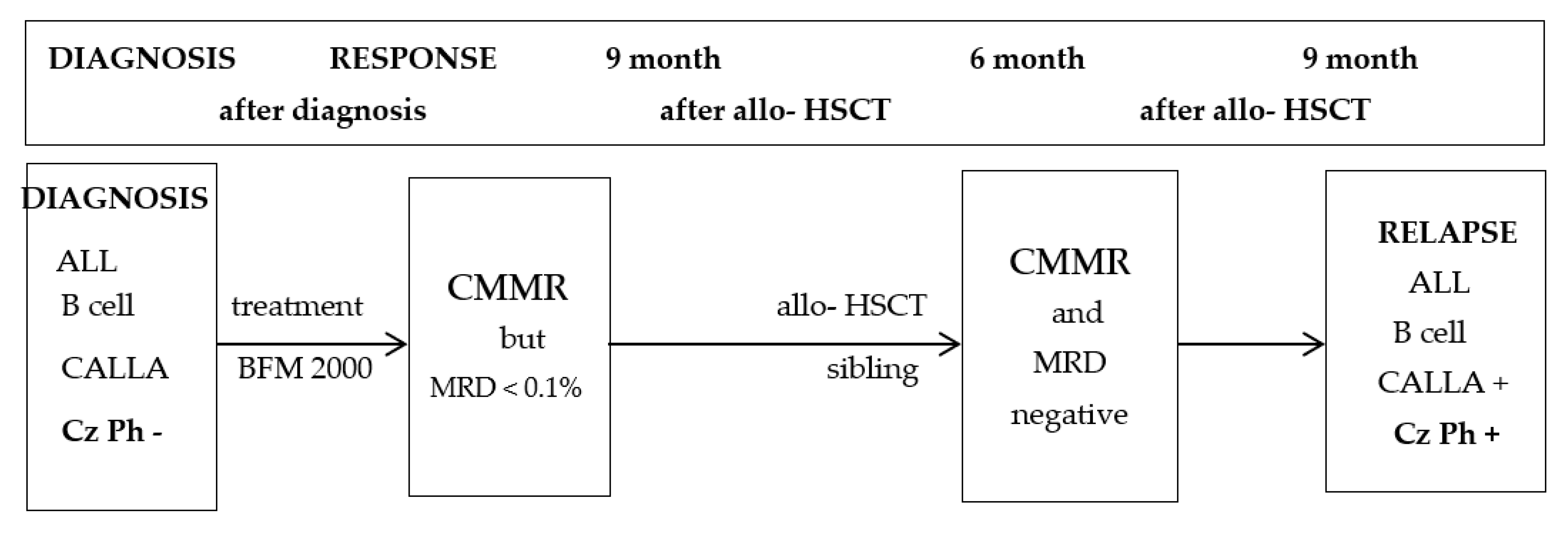
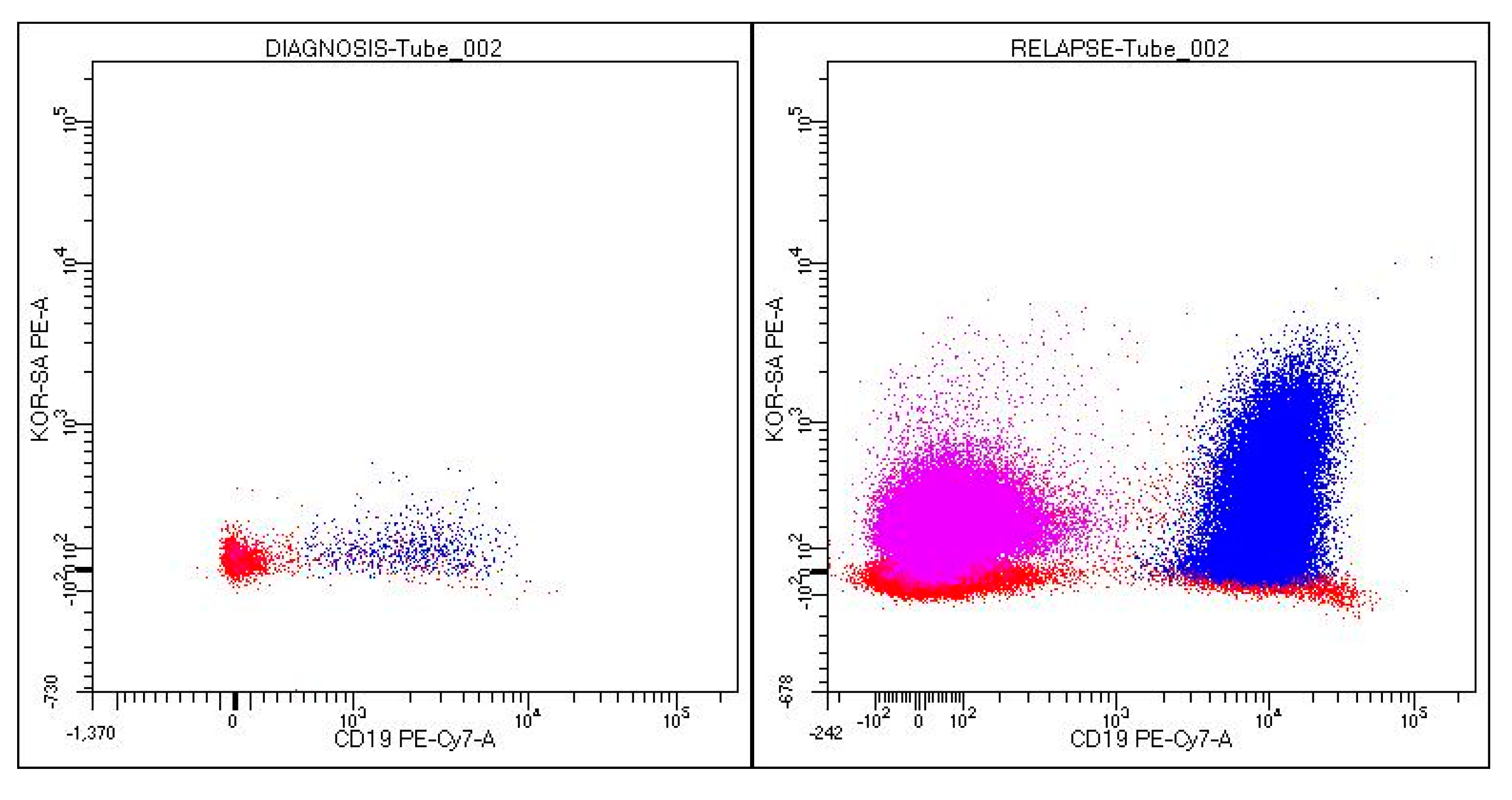
2. Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Inaba, H.; Mullighan, C.G. Pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef] [PubMed]
- Rytting, M.E.; Thomas, D.A.; O’Brien, S.M.; Ravandi-Kashani, F.; Jabbour, E.J.; Franklin, A.R.; Kadia, T.M.; Pemmaraju, N.; Daver, N.G.; Ferrajoli, A.; et al. Augmented Berlin-Frankfurt-Münster therapy in adolescents and young adults (AYAs) with acute lymphoblastic leukemia (ALL). Cancer 2014, 120, 3660–3668. [Google Scholar] [CrossRef] [PubMed]
- Sive, J.I.; Buck, G.; Fielding, A.; Lazarus, H.M.; Litzow, M.R.; Luger, S.; Marks, D.I.; McMillan, A.; Moorman, A.V.; Richards, S.M.; et al. Outcomes in older adults with acute lymphoblastic leukaemia (ALL): Results from the international MRC UKALL XII/ECOG2993 trial. Br. J. Haematol. 2012, 157, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stock, W.; Luger, S.M.; Advani, A.S.; Yin, J.; Harvey, R.C.; Mullighan, C.G.; Willman, C.L.; Fulton, N.; Laumann, K.M.; Malnassy, G.; et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: Results of CALGB 10403. Blood J. Am. Soc. Hematol. 2019, 133, 1548–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmeister, T.; Schwartz, S.; Bartram, C.R.; Gokbuget, N.; Hoelzer, D.; Thiel, E. Patients’ age and BCR-ABL frequency in adult B-precursor ALL: A retrospective analysis from the GMALL study group. Blood J. Am. Soc. Hematol. 2008, 112, 918–919. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.V. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica 2016, 101, 407–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleibetaner, B.; Gleißner, B.; Gökbuget, N.; Bartram, C.R.; Janssen, B.; Rieder, H.; Janssen, J.W.G.; Fonatsch, C.; Heyll, A.; Voliotis, D.; et al. Leading prognostic relevance of the BCR-ABL translocation in adult acute B-lineage lymphoblastic leukemia: A prospective study of the German Multicenter Trial Group and confirmed polymerase chain reaction analysis. Blood 2002, 99, 1536–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, K.; Kantarjian, H.M.; Short, N.J.; Samra, B.; Khoury, J.D.; Shamanna, R.K.; Konopleva, M.; Jain, N.; DiNardo, C.D.; Khouri, R.; et al. Prognostic factors for progression in patients with Philadelphia chromosome-positive acute lymphoblastic leukemia in complete molecular response within 3 months of therapy with tyrosine kinase inhibitors. Cancer 2021, 127, 2648–2656. [Google Scholar] [CrossRef] [PubMed]
- Bassan, R.; Intermesoli, T.; Scattolin, A.; Viero, P.; Maino, E.; Sancetta, R.; Carobolante, F.; Gianni, F.; Stefanoni, P.; Tosi, M.; et al. Minimal residual disease assessment and risk-based therapy in acute lymphoblastic leukemia. Clin. Lymphoma Myeloma Leuk. 2017, 17S, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Balduzzi, A.; Di Maio, L.; Silvestri, D.; Songia, S.; Bonanomi, S.; Rovelli, A.; Conter, V.; Biondi, A.; Cazzaniga, G.; Valsecchi, M.G. Minimal residual disease before and after transplantation for childhood acute lymphoblastic leukaemia: Is there any room for intervention? Br. J. Haematol. 2014, 164, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Kuhlen, M.; Willasch, A.M.; Dalle, J.H.; Wachowiak, J.; Yaniv, I.; Ifversen, M.; Sedlacek, P.; Guengoer, T.; Lang, P.; Bader, P.; et al. Outcome of relapse after allogeneic HSCT in children with ALL enrolled in the ALL-SCT 2003/2007 trial. Br. J. Haematol. 2018, 180, 82–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulte, D.; Gondos, A.; Brenner, H. Trends in survival after diagnosis with hematologic malignancy in adolescence or young adulthood in the United States, 1981–2005. Cancer 2009, 115, 4973–4979. [Google Scholar] [CrossRef] [PubMed]
- Boissel, N.; Auclerc, M.F.; Lhéritier, V.; Perel, Y.; Thomas, X.; Leblanc, T.; Rousselot, P.; Cayuela, J.M.; Gabert, J.; Fegueux, N.; et al. Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J. Clin. Oncol. 2003, 21, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Owaidah, T.M.; Rawas, F.I.; Al khayatt, M.F.; Elkum, N.B. Expression of CD66c and CD25 in Acute Lymphoblastic Leukemia as a Predictor of the Presence of BCR/ABL Rearrangement. Hematol. Oncol. Stem Cell Ther. 2008, 1, 34–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrente, F.; Bellesi, S.; Metafuni, E.; Puggioni, P.L.; Marietti, S.; Ciminello, A.M.; Za, T.; Sorà, F.; Fianchi, L.; Sica, S.; et al. Role of Flow-Cytometric Immunophenotyping in Prediction of BCR/ABL1 Gene Rearrangement in Adult B-Cell Acute Lymphoblastic Leukemia: FLOW-CYTOMETRY IN BCR/ABL1 ADULT B-ALL. Cytom. Clin. Cytom. 2018, 94, 468–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrušák, O.; Porwit-MacDonald, A. Antigen Expression Patterns Reflecting Genotype of Acute Leukemias. Leukemia 2002, 16, 1233–1258. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Millan, B.; Sanchéz-Martínez, D.; Roca-Ho, H.; Gutiérrez-Agüera, F.; Molina, O.; Diaz de la Guardia, R.; Torres-Ruiz, R.; Fuster, J.L.; Ballerini, P.; Suessbier, U.; et al. NG2 antigen is a therapeutic target for MLL-rearranged B-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 1557–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.C.; Chen, I.-M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome–Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terwey, T.H.; Hemmati, P.G.; Nagy, M.; Pfeifer, H.; Gökbuget, N.; Brüggemann, M.; Le Duc, T.M.; le Coutre, P.; Dörken, B.; Arnold, R. Comparison of Chimerism and Minimal Residual Disease Monitoring for Relapse Prediction after Allogeneic Stem Cell Transplantation for Adult Acute Lymphoblastic Leukemia. Biol. Blood Marrow Transplant. 2014, 20, 1522–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marinescu, E.-C.; Bumbea, H.; Iordan, I.; Dumitru, I.; Soare, D.; Ciufu, C.; Gaman, M. The Philadelphia Chromosome, from Negative to Positive: A Case Report of Relapsed Acute Lymphoblastic Leukemia Following Allogeneic Stem Cell Transplantation. Medicina 2023, 59, 671. https://doi.org/10.3390/medicina59040671
Marinescu E-C, Bumbea H, Iordan I, Dumitru I, Soare D, Ciufu C, Gaman M. The Philadelphia Chromosome, from Negative to Positive: A Case Report of Relapsed Acute Lymphoblastic Leukemia Following Allogeneic Stem Cell Transplantation. Medicina. 2023; 59(4):671. https://doi.org/10.3390/medicina59040671
Chicago/Turabian StyleMarinescu, Elena-Cristina, Horia Bumbea, Iuliana Iordan, Ion Dumitru, Dan Soare, Cristina Ciufu, and Mihaela Gaman. 2023. "The Philadelphia Chromosome, from Negative to Positive: A Case Report of Relapsed Acute Lymphoblastic Leukemia Following Allogeneic Stem Cell Transplantation" Medicina 59, no. 4: 671. https://doi.org/10.3390/medicina59040671