
Molecular Pathogenesis of Endotheliopathy and Endotheliopathic Syndromes, Leading to Inflammation and Microthrombosis, and Various Hemostatic Clinical Phenotypes Based on “Two-Activation Theory of the Endothelium” and “Two-Path Unifying Theory” of Hemostasis


Abstract
:1. Introduction
2. Genesis of Endotheliopathy
2.1. Causes of Endotheliopathy
2.1.1. Complement System
2.1.2. Endothelial Molecular Pathogenesis
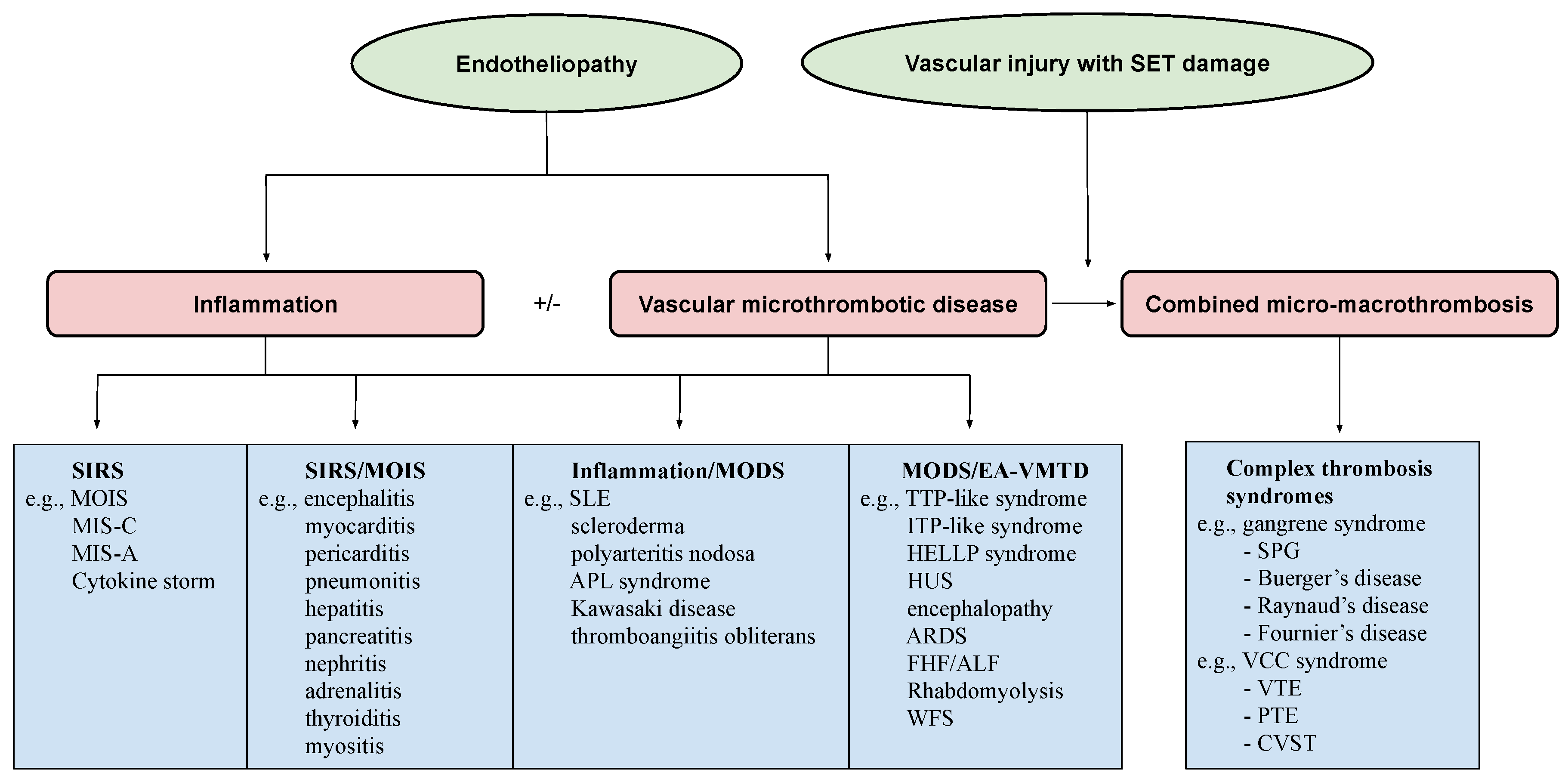
2.2. Genesis of Clinical Phenotypes in Endotheliopathy
2.2.1. Hemostasis Based on the Blood-Vessel Model
2.2.2. Vascular Injury Provoking Thrombosis
- The well-known role of ULVWF from ECs in the early phase of hemostasis in external bodily injury;
- The lack of participation of TF in hemostasis producing septic microthrombosis [4];
- TF unavailable in the EC injury but available from the SET/EVT in local bleeding vascular injury;
- Distinctly different disseminated microthrombosis occurring in the microvasculature in sepsis compared with localized macrothrombosis formed after fibrin clots occurring in a local large-vessel injury;
- The irrefutable hemostatic fundamental: “The hemostasis and thrombogenesis can be activated only by a vascular injury”.
- Damaged ECs from the blood-vessel wall following a vascular injury release ULVWF/FVIII in partnership, which are the essential components, along with platelets, in the formation of microthrombi strings. The process of ULVWF interacting with platelets is called “microthrombogenesis”;
- Damaged SET from the blood-vessel wall following a local vascular injury releases TF into circulation, which activates FVIIa and leads to the formation of fibrin clots via the extrinsic cascade with the sequential activation of FIX, FX, FV, FII, and fibrinogen to fibrin. The process is called “fibrinogenesis”;
- A local traumatic vascular injury involving ECs and SET produces both microthrombi strings and fibrin clots. The proposed theory is that the forming process of macrothrombosis must be the result of the “unifying mechanism of microthrombi strings and fibrin meshes”. This process is called “macrothrombogenesis”, in which neutrophil extracellular traps (NETosis) passively participate;
- The understanding of this unifying mechanism is very important in the understanding of arterial and venous combined micro–macrothrombosis, which is a process that should be called micro–macrothrombogenesis.
3. Endotheliopathic Syndromes
3.1. Consumptive Thrombocytopenia
3.2. Arterial Endotheliopathy vs. Venous Endotheliopathy
3.3. TTP-like Syndrome and ITP-like Syndrome
- TTP-like syndrome and ITP-like syndrome (e.g., most cases of ITP) are associated with thrombocytopenia, complement activation [45,46,47], and elevated VWF antigen/FVIII and ADAMTS13 insufficiency [10,48,49,50,51,52]. Interpretation: These changes are consistent with the activated complement system promoting endotheliopathy that leads to the exocytosis of ULVWF/FVIII, consumptive thrombocytopenia, and microthrombosis. However, microthrombosis occurs in the microvasculature in arterial endotheliopathy, as seen in TTP-like syndrome (i.e., aEA-VMTD), but it is commonly “silent” in venous circulation, as seen in ITP-like syndrome in venous endotheliopathy (i.e., vEA-VMTD);
- Despite thrombocytopenia, both are associated with the thrombophilic state [53,54,55,56]. Interpretation: TTP-like syndrome is already in the “microthrombotic state” within the microvasculature, and ITP-like syndrome is likely in “silent” microthrombotic state because the microthrombosis occurring in the venous system does not produce MAHA and MODS. However, acute venous thromboembolism (VTE), which is venous combined micro–macrothrombosis, can develop in ITP-like syndrome if additional venous vascular injury occurs, and especially in the intensive care unit (ICU) [14];
- Both have shown a beneficial response to therapeutic plasma exchange (TPE), intravenous immunoglobulins (IVIG), and rituximab [57,58,59,60,61,62,63,64]. Interpretation: The therapeutic benefit of these plasma therapies and anti-immune therapy suggests that both aEA-VMTD (TTP-like syndrome) and vEA-VMTD (ITP-like syndrome) share the same pathogenesis due to activated complement-associated endothewliopathy.
3.4. Endotheliopathic Inflammatory Syndromes
- Organ-designated endotheliitis, which is called multiorgan inflammatory syndrome (MOIS) [13] (e.g., myocarditis, pericarditis, encephalitis, pneumonitis, thyroiditis, hepatitis, nephritis, pancreatitis, adrenalitis, myositis, lymphadenitis, and others);
- Systemic inflammatory response syndrome (SIRS) [6];
- Cytokine storm/cytokine release syndrome [65];
- Multisystem inflammatory syndrome in child (MIS-C) [66];
- Multisystem inflammatory syndrome in adult (MIS-A) [67];
- Kawasaki disease [68];
- Polyarteritis nodosa [69];
- Acute necrotizing fasciitis [70];
- Thromboangiitis obliterans [71];
- Localized endothelial inflammatory syndromes (e.g., Sjogren’s syndrome, temporal arteritis, Crohn’s disease, Grave’s diseases, Hashimoto’s thyroiditis, Alzheimer’s disease, amyotrophic lateral sclerosis, myasthenia gravis, diabetic endotheliitis, rheumatoid endotheliitis, and others);
- Focal, local, regional, or disseminated endotheliitis/angiitis/vasculitis;
- Angiodysplasia/hemangiomatosis/telangiectasia/atrio-venous malformation.
3.5. Endotheliopathy-Associated Vascular Microthrombotic Disease
3.5.1. Examples of “Suspected” or Proven Endotheliopathy-Associated Microthrombotic Syndromes
Focal, Multifocal, Local, or Regional EA-VMTD [11,12]
- Hereditary disease in early life
- -
- Hereditary hemorrhagic telangiectasia/Osler–Weber–Rendu syndrome;
- -
- Capillary arterio-venous malformation syndrome;
- -
- Hereditary neurocutaneous hemangiomatosis;
- -
- Hereditary endotheliopathy, retinopathy, nephropathy and stroke (HERNS) syndrome;
- -
- Kasabach–Merritt syndrome with kaposiform hemangioendothelioma;
- -
- Fabry disease with endothelial dysfunction due to α-galactosidase A deficiency;
- Acquired disease in later life
- -
- Susac syndrome with encephalopathy, retinopathy, and audiopathy;
- -
- -
- Vascular dementia due to multifocal endotheliopathy associated with cognitive dysfunction;
- -
- Retinal microaneurysm in diabetes;
- -
- Degos disease associated with skin papules [75];
- -
- Henoch–Shoenlein purpura with subcutaneous hemorrhage and renal failure [76];
- -
- Heyde’s syndrome associated with aortic stenosis and intestinal angiodysplasia [77];
- -
- -
- Critical-illness-associated endotheliopathy;
- Sepsis-associated endotheliopathy (e.g., pathogens, including bacteria, viruses, fungi, rickettsia, and parasites) [6];
- Chemical-/toxin-associated endotheliopathy (e.g., drug, toxin, vaccine, poison, and venom);
- Pregnancy-associated endotheliopathy (e.g., gestational thrombocytopenia; hemolysis, elevated liver enzymes, low platelets [HELLP] syndrome; preeclampsia; abruptio placenta; placenta previa; amniotic fluid embolism; dead fetus syndrome);
- Cancer-associated endotheliopathy (e.g., breasts, stomach, and lungs);
- Polytrauma-associated endotheliopathy (e.g., car accident);
- Transplant-associated endotheliopathy (e.g., kidneys, heart, bone marrow, and stem cells) [83];
- Autoimmune disease-associated endotheliopathy (e.g., systemic lupus erythematosus (SLE) [84];
- Diabetic endotheliopathy due to plasma glycated CD59.
Organ/Multiorgan Dysfunction Syndromes
- Reye’s syndrome with possible hepatic encephalopathy with MODS [85];
- Transfusion-related acute lung injury (TRALI) (possible consumptive thrombocytopenia-associated pulmonary artery endotheliopathy causing ARDS) [86];
- Subacute bacterial endocarditis (SBE) (consistent with inflammatory syndrome and microthrombosis due to endotheliopathy) [87];
- Veno-occlusive disease (VOD)/sinusoidal obstruction syndrome (SOS) consistent with hepatic venous endotheliopathy [88];
- Microvascular myocardial infarction (MVMI) due to diffuse microvascular microthrombosis [89];
- Acute necrotizing pancreatitis, often with arterial endotheliopathy [92];
- Waterhouse–Friderichsen syndrome due to adrenal microvascular microthrombosis and hemorrhage [93];
- Acute renal failure (ARF)/hemolytic-uremic syndrome (HUS) [94];
- Goodpasture syndrome with pulmonary renal syndrome with ADAMTS13 insufficiency [95];
- Idiopathic pulmonary hypertension with pulmonary arterial endotheliopathy and microtubule dysfunction [98];
- Primary biliary cirrhosis with endothelial transformation;
- Felty syndrome with the overexpression of VWF (?);
- EA-VMTD with hepatic coagulopathy (old term: “acute DIC”) [8];
- Paroxysmal nocturnal hemoglobinuria;
- Combined organ syndromes with recognized terms (e.g., hepatorenal syndrome, hepatic encephalopathy, cardio-pulmonary syndrome, pulmonary encephalopathy, pulmonary-renal syndrome, and cardio-renal syndrome);
- MODS [6].
Systemic Syndromes
- TTP-like syndrome (consistent with arterial EA-VMTD) [5];
- Systemic lupus erythematosus (SLE) (consistent with inflammatory cutaneous endotheliopathy and MODS associated with EA-VMTD) [101];
- Kawasaki disease (consistent with inflammatory and lymphocutaneous syndrome in vaccine-induced venous endotheliopathy, and rarely with arterial endotheliopathy) [103];
- Malignant hypertension with hypertensive encephalopathy [104];
Combined Micro–Macrothrombotic Syndromes [6,10,13,14]
- Peripheral gangrene syndromes associated with arterial endotheliopathy and additional in-hospital arterial vascular injury (e.g., symmetrical peripheral gangrene (SPG), Fournier’s disease, Buerger’s disease, gas gangrene, diabetic gangrene, acute necrotizing fasciitis, Raynaud’s phenomenon, “coumadin”-associated gangrene syndrome, and envenomation syndrome);
3.5.2. Pathogenesis of Combined Micro–Macrothrombotic Syndromes [10,13,14]
3.5.3. What Should We Have to Know about the Pathogenesis of EA-VMTD?
- EA-VMTD is a hemostatic disease caused by the activation of ULVWF path of hemostasis;
- It is characterized by increased ULVWF/VWF/VWF antigen expression and increased FVIII activity due to the release from damaged ECs;
- It is associated with platelet consumption and the formation of microthrombi strings, and it may trigger microvascular hemolysis due to the increased shear-stress effect on the red cells, and especially in the arterial system (i.e., MAHA);
- It is often associated with the insufficiency of the ULVWF-cleaving enzyme ADAMTS13 secondary to its gene mutation or polymorphism, or excessive exocytosis of ULVWF over the cleaving capacity of ADAMTS13 in the endothelial damage;
- EA-VMTD orchestrates MODS, such as encephalopathy, ARDS, MVMI, acute necrotizing pancreatitis, rhabdomyolysis, hepatic dysfunction, acute renal failure, and sometimes multifocal microvascular microthrombosis with clinically significant phenotypes, such syndromes as retinal microaneurysm, TIA, Heyde’s syndrome, HERNS syndrome, and Susac syndrome.
3.6. Epiphenomenon
3.6.1. Positive Anti-PF4 Antibodies and Anti-PL Antibodies
3.6.2. Endothelial Epiphenomenon
4. Practical Diagnostic Criteria and Diagnostic Perspective for Endotheliopathy
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hattori, R.; Hamilton, K.K.; McEver, R.P.; Sims, P.J. Complement proteins C5b-9 induce secretion of high molecular weight mul-timers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J. Biol. Chem. 1989, 264, 9053–9060. [Google Scholar] [CrossRef]
- Brunn, G.J.; Saadi, S.; Platt, J.L. Differential Regulation of Endothelial Cell Activation by Complement and Interleukin 1α. Circ. Res. 2006, 98, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Kerr, H.; Richards, A. Complement-mediated injury and protection of endothelium: Lessons from atypical haemolytic uraemic syndrome. Immunobiology 2012, 217, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Thrombocytopenia in critically ill patients due to vascular microthrombotic disease: Pathogenesis based on “two activation theory of the endothelium”. Vasc. Dis. Ther. 2017, 2, 2–7. Available online: https://www.oatext.com/pdf/VDT-2-132.pdf (accessed on 13 September 2022). [CrossRef]
- Chang, J.C. TTP-like syndrome: Novel concept and molecular pathogenesis of endotheliopathy-associated vascular micro-thrombotic disease. Thromb. J. 2018, 16, 20. [Google Scholar] [CrossRef]
- Chang, J.C. Sepsis and septic shock: Endothelial molecular pathogenesis associated with vascular microthrombotic disease. Thromb. J. 2019, 17, 10. [Google Scholar] [CrossRef]
- Chang, J.C. Acute Respiratory Distress Syndrome as an Organ Phenotype of Vascular Microthrombotic Disease: Based on Hemostatic Theory and Endothelial Molecular Pathogenesis. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029619887437. [Google Scholar] [CrossRef]
- Chang, J.C. Disseminated intravascular coagulation: New identity as endotheliopathy-associated vascular microthrombotic disease based on in vivo hemostasis and endothelial molecular pathogenesis. Thromb. J. 2020, 18, 25. [Google Scholar] [CrossRef]
- Chang, J. Pathogenesis of Ebola Viral Haemorrhagic Fever: TTP-like Syndrome Associated with Hepatic Coagulopathy based on “Two Activation Theory of the Endothelium”. J. Prev. Infect. Control 2017, 3, 1–4. [Google Scholar] [CrossRef]
- Chang, J.C. COVID-19 Sepsis: Pathogenesis and Endothelial Molecular Mechanisms Based on “Two-Path Unifying Theory” of Hemostasis and Endotheliopathy-Associated Vascular Microthrombotic Disease, and Proposed Therapeutic Approach with Antimicrothrombotic Therapy. Vasc. Health Risk Manag. 2021, 17, 273–298. [Google Scholar] [CrossRef]
- Chang, J.C. Hemostasis based on a novel ‘two-path unifying theory’ and classification of hemostatic disorders. Blood Coagul. Fibrinolysis 2018, 29, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Thrombogenesis and thrombotic disorders based on “two-path unifying theory of hemostasis: Philosophical, physiological and phenotypical interpretation. Blood Coagul. Fibrinolysis 2018, 29, 585–595. [Google Scholar] [CrossRef]
- Chang, J.C.; Hawley, H.B. Vaccine-Associated Thrombocytopenia and Thrombosis: Venous Endotheliopathy Leading to Venous Combined Micro-Macrothrombosis. Medicina 2021, 57, 1163. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Pathogenesis of Two Faces of DVT: New Identity of Venous Thromboembolism as Combined Micro-Macrothrombosis via Unifying Mechanism Based on “Two-Path Unifying Theory” of Hemostasis and “Two-Activation Theory of the Endothelium”. Life 2022, 12, 220. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. Crosstalk between inflammation and thrombosis. Maturitas 2004, 47, 305–314. [Google Scholar] [CrossRef]
- Aksu, K.; Donmez, A.; Keser, G. Inflammation-induced thrombosis: Mechanisms, disease associations and management. Curr. Pharm. Des. 2012, 18, 1478–1493. [Google Scholar]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Allam, R.; Kumar, S.V.; Darisipudi, M.N.; Anders, H.-J. Extracellular histones in tissue injury and inflammation. J. Mol. Med. 2014, 92, 465–472. [Google Scholar] [CrossRef]
- Palankar, R.; Greinacher, A. Challenging the concept of immunothrombosis. Blood 2019, 133, 508–509. [Google Scholar] [CrossRef]
- Chang, J.C. Disseminated intravascular coagulation (DIC): Is it fact or fancy? Blood Coagul. Fibrinolysis 2018, 29, 330–337. [Google Scholar] [CrossRef]
- Turner, N.A.; Moake, J. Assembly and activation of alternative complement components on endothelial cell-anchored ultra-large von Willebrand factor links complement and hemostasis-thrombosis. PLoS ONE 2013, 8, e59372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amari Chinchilla, K.; Vijayan, M.; Taveras Garcia, B.; Jim, B. Complement-Mediated Disorders in Pregnancy. Adv. Chronic Kidney Dis. 2020, 27, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Sahoo, R.; Vaidya, A.; Chorev, M.; Halperin, J.A. Role of Complement and Complement Regulatory Proteins in the Complications of Diabetes. Endocr. Rev. 2015, 36, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.B.; Jane-Wit, D.; Pober, J.S. Complement Membrane Attack Complex: New Roles, Mechanisms of Action, and Therapeutic Targets. Am. J. Pathol. 2020, 190, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Abagyan, R.; Dong, S.; Gilbert, A.; Nussenzweig, V.; Tomlinson, S. Mapping the Active Site of CD59. J. Exp. Med. 1997, 185, 745–754. [Google Scholar] [CrossRef]
- Wu, G.; Hu, W.; Shahsafaei, A.; Song, W.; Dobarro, M.; Sukhova, G.K.; Bronson, R.R.; Shi, G.-P.; Rother, R.P.; Halperin, J.A.; et al. Complement Regulator CD59 Protects Against Atherosclerosis by Restricting the Formation of Complement Membrane Attack Complex. Circ. Res. 2009, 104, 550–558. [Google Scholar] [CrossRef]
- Blann, A.D. How a damaged blood vessel wall contibutes to thrombosis and hypertension. Pathophysiol. Haemost. Thromb. 2003, 33, 445–448. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.; Rosen, H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef]
- Adam, E.; Zacharowski, K.; Miesbach, W. A comprehensive assessment of the coagulation profile in critically ill COVID-19 patients. Thromb. Res. 2020, 194, 42–44. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Henry, B.M.; Lippi, G. Increased VWF and Decreased ADAMTS-13 in COVID-19: Creating a Milieu for (Micro) Thrombosis. Semin. Thromb. Hemost. 2021, 47, 400–418. [Google Scholar] [CrossRef]
- Bagot, C.N.; Arya, R. Virchow and his triad: A question of attribution. Br. J. Haematol. 2008, 143, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.K.; Goerge, T.; Schneider, S.W.; Wagner, D.D. Formation of platelet strings and microthrombi in the presence of ADAMTS-13 inhibitor does not require P-selectin or beta3 integrin. J. Thromb. Haemost. 2007, 5, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-M. Pathophysiology of thrombotic thrombocytopenic purpura. Int. J. Hematol. 2010, 91, 1–19. [Google Scholar] [CrossRef]
- Nguyen, T.C.; Liu, A.; Liu, L.; Ball, C.; Choi, H.; May, W.S.; Aboulfatova, K.; Bergeron, A.L.; Dong, J.-F. Acquired ADAMTS-13 deficiency in pediatric patients with severe sepsis. Haematologica 2007, 92, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Karim, F.; Adil, S.N.; Afaq, B.; Haq, A.U. Deficiency of ADAMTS-13 in pediatric patients with severe sepsis and impact on in-hospital mortality. BMC Pediatr. 2013, 13, 44. [Google Scholar] [CrossRef]
- Fukushima, H.; Nishio, K.; Asai, H.; Watanabe, T.; Seki, T.; Matsui, H.; Sugimoto, M.; Matsumoto, M.; Fujimura, Y.; Okuchi, K. Ratio of von Willebrand Factor Propeptide to ADAMTS13 Is Associated with Severity of Sepsis. Shock 2013, 39, 409–414. [Google Scholar] [CrossRef]
- Mendolicchio, G.L.; Ruggeri, Z.M. Interaction of von Willebrand factor with platelets and the vessel wall. Hämostaseologie 2015, 35, 211–224. [Google Scholar] [CrossRef]
- Shim, C.Y.; Ni Liu, Y.; Atkinson, T.; Xie, A.; Foster, T.; Davidson, B.P.; Treible, M.; Qi, Y.; López, J.A.; Munday, A.; et al. Molecular Imaging of Platelet–Endothelial Interactions and Endothelial von Willebrand Factor in Early and Mid-Stage Atherosclerosis. Circ. Cardiovasc. Imaging 2015, 8, e002765. [Google Scholar] [CrossRef]
- Stein, E.; McMahon, B.; Kwaan, H.; Altman, J.K.; Frankfurt, O.; Tallman, M.S. The coagulopathy of acute promyelocytic leukaemia revisited. Best Pract. Res. Clin. Haematol. 2009, 22, 153–163. [Google Scholar] [CrossRef]
- Chang, J.C. Stroke Classification: Critical Role of Unusually Large von Willebrand Factor Multimers and Tissue Factor on Clinical Phenotypes Based on Novel “Two-Path Unifying Theory” of Hemostasis. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620913634. [Google Scholar] [CrossRef]
- Thachil, J.; Warkentin, T.E. How do we approach thrombocytopenia in critically ill patients? Br. J. Haematol. 2017, 177, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Bomhof, G.; Mutsaers, P.G.; Leebeek, F.W.; Te Boekhorst, P.A.; Hofland, J.; Croles, F.N.; Jansen, A.G. COVID-19-associated immune thrombocytopenia. Br. J. Haematol. 2020, 190, e61–e64. [Google Scholar] [CrossRef] [PubMed]
- Boehlen, F. Thrombocytopenia during pregnancy. Importance, diagnosis and management. Hamostaseologie 2006, 26, 72–78. [Google Scholar]
- Kwaan, H.C. Thrombotic microangiopathy. Adv. Exp. Med. Biol. 1990, 281, 367–375. [Google Scholar]
- Peerschke, E.I.; Yin, W.; Ghebrehiwet, B. Complement activation on platelets: Implications for vascular inflammation and thrombosis. Mol. Immunol. 2010, 47, 2170–2175. [Google Scholar] [CrossRef] [PubMed]
- Castelli, R.; Delilliers, G.L.; Gidaro, A.; Cicardi, M.; Bergamaschini, L. Complement activation in patients with immune thrombocytopenic purpura according to phases of disease course. Clin. Exp. Immunol. 2020, 201, 258–265. [Google Scholar] [CrossRef]
- Portuguese, A.J.; Sunga, C.; Kruse-Jarres, R.; Gernsheimer, T.; Abkowitz, J. Autoimmune- and complement-mediated hematologic condition recrudescence following SARS-CoV-2 vaccination. Blood Adv. 2021, 5, 2794–2798. [Google Scholar] [CrossRef]
- Godfrey, C.L.; Terrinoni, I.; Laffan, M.; Crawley, J.; Cooper, N. Elevated Plasma Von Willebrand Factor and Decreased ADAMTS13 Antigen Levels in Patients with Immune Thrombocytopenia (ITP). Blood 2012, 120, 1096. [Google Scholar] [CrossRef]
- Moake, J.L.; Rudy, C.K.; Troll, J.H.; Weinstein, M.J.; Colannino, N.M.; Azocar, J.; Seder, R.H.; Hong, S.L.; Deykin, D. Unusually Large Plasma Factor VIII: Von Willebrand Factor Multimers in Chronic Relapsing Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 1982, 307, 1432–1435. [Google Scholar] [CrossRef]
- Farkas, P.; Csuka, D.; Mikes, B.; Sinkovits, G.; Réti, M.; Németh, E.; Rácz, K.; Madách, K.; Gergely, M.; Demeter, J.; et al. Complement activation, inflammation and relative ADAMTS13 deficiency in secondary thrombotic microangiopathies. Immunobiology 2017, 222, 119–127. [Google Scholar] [CrossRef]
- Kalambokis, G.N.; Oikonomou, A.; Christou, L.; Kolaitis, N.I.; Tsianos, E.V.; Christodoulou, D.; Baltayiannis, G. von Willebrand factor and procoagulant imbalance predict outcome in patients with cirrhosis and thrombocytopenia. J. Hepatol. 2016, 65, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Casonato, A.; Fabris, F.; Boscaro, M.; Girolami, A. Increased factor VIII/vWf levels in patients with reduced platelet number. Blut 1987, 54, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Rodeghiero, F. ITP and thrombosis: An intriguing association. Blood Adv. 2017, 1, 2280. [Google Scholar] [CrossRef]
- Swan, D.; Newland, A.; Rodeghiero, F.; Thachil, J. Thrombosis in immune thrombocytopenia—Current status and future per-spectives. Br. J. Haematol. 2021, 194, 822–834. [Google Scholar] [CrossRef]
- Mei, H.; Luo, L.; Hu, Y. Thrombocytopenia and thrombosis in hospitalized patients with COVID-19. J. Hematol. Oncol. 2020, 13, 161. [Google Scholar] [CrossRef]
- Wong, R.S.M.; Bakshi, K.; Brainsky, A. Thrombophilia in patients with chronic immune thrombocytopenia. Scand. J. Clin. Lab. Investig. 2015, 75, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Rock, G.A.; Shumak, K.H.; Buskard, N.A.; Blanchette, V.S.; Kelton, J.G.; Nair, R.C.; Spasoff, R.A.; Canadian Apheresis Study Group. Comparison of Plasma Exchange with Plasma Infusion in the Treatment of Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 1991, 325, 393–397. [Google Scholar] [CrossRef]
- Blanchette, V.; Hogan, V.; McCombie, N.; Drouin, J.; Bormanis, J.; Taylor, R.; Rock, G. Intensive plasma exchange therapy in ten patients with idiopathic thrombocytopenic purpura. Transfusion 1984, 24, 388–394. [Google Scholar] [CrossRef]
- Hansen, R.J.; Balthasar, J.P. Mechanisms of IVIG action in immune thrombocytopenic purpura. Clin. Lab. 2004, 50, 133–140. [Google Scholar]
- Debourdeau, P.; Zammit, C.; Souleau, B.; Védrine, L.; Farge-Bancel, D. Traitement des microangiopathies thrombotiques [Treatment of thrombotic microangiopathies]. Ann. Med. Interne 1999, 150, 374–387. [Google Scholar]
- Raniele, D.P.; Opsahl, J.A.; Kjellstrand, C.M. Should Intravenous Immunoglobulin G Be First-Line Treatment for Acute Thrombotic Thrombocytopenic Purpura? Case Report and Review of the Literature. Am. J. Kidney Dis. 1991, 18, 264–268. [Google Scholar] [CrossRef]
- Lucchini, E.; Zaja, F.; Bussel, J. Rituximab in the treatment of immune thrombocytopenia: What is the role of this agent in 2019? Haematologica 2019, 104, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- George, J.N.; Woodson, R.D.; Kiss, J.E.; Kojouri, K.; Vesely, S. Rituximab therapy for thrombotic thrombocytopenic purpura: A proposed study of the Transfusion Medicine/Hemostasis Clinical Trials Network with a systematic review of rituximab therapy for immune-mediated disorders. J. Clin. Apher. 2006, 21, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fu, A.; Wang, J.; Wu, T.; Li, Z.; Tang, J.; Shen, H.; Zhu, J.; Li, J.; Zhu, Q.; et al. Rituximab as first-line treatment for acquired thrombotic thrombocytopenic purpura. J. Int. Med. Res. 2017, 45, 1253–1260. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Radia, T.; Williams, N.; Agrawal, P.; Harman, K.; Weale, J.; Cook, J.; Gupta, A. Multi-system inflammatory syndrome in children & adolescents (MIS-C): A sys-tematic review of clinical features and presentation. Paediatr. Respir. Rev. 2021, 38, 51–57. [Google Scholar]
- Vogel, T.P.; Top, K.A.; Karatzios, C.; Hilmers, D.C.; Tapia, L.I.; Moceri, P.; Giovannini-Chami, L.; Wood, N.; Chandler, R.E.; Klein, N.P.; et al. Multisystem inflammatory syndrome in children and adults (MIS-C/A): Case definition & guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 2021, 39, 3037–3049. [Google Scholar]
- Sharma, C.; Ganigara, M.; Galeotti, C.; Burns, J.; Berganza, F.M.; Hayes, D.A.; Singh-Grewal, D.; Bharath, S.; Sajjan, S.; Bayry, J. Multisystem inflammatory syndrome in children and Kawasaki disease: A critical comparison. Nat. Rev. Rheumatol. 2021, 17, 731–748. [Google Scholar] [CrossRef]
- Colmegna, I.; Maldonado-Cocco, J.A. Polyarteritis nodosa revisited. Curr. Rheumatol. Rep. 2005, 7, 288–296. [Google Scholar] [CrossRef]
- Lancerotto, L.; Tocco, I.; Salmaso, R.; Vindigni, V.; Bassetto, F. Necrotizing fasciitis: Classification, diagnosis, and management. J. Trauma Acute Care Surg. 2012, 72, 560–566. [Google Scholar] [CrossRef]
- Olin, J.W. Thromboangiitis obliterans (Buerger’s disease). N. Engl. J. Med. 2000, 343, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Vecchi, A.; Dejana, E.; Sozzani, S.; Introna, M. Cytokine Regulation of Endothelial Cell Function. FASEB J. 1992, 6, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Moake, J.L. Factor VIII Is Synthesized in Human Endothelial Cells, Packaged in Weibel-Palade Bodies and Secreted Bound to ULVWF Strings. PLoS ONE 2015, 10, e0140740. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M.; Arena, R. Endothelial dysfunction and pathophysiological correlates in atrial fibrillation. Heart 2009, 95, 102–106. [Google Scholar] [CrossRef]
- Magro, C.M.; Poe, J.C.; Kim, C.; Shapiro, L.; Nuovo, G.; Crow, M.K.; Crow, Y.J. Degos disease: A C5b-9/interferon-α-mediated endotheliopathy syndrome. Am. J. Clin. Pathol. 2011, 135, 599–610. [Google Scholar] [CrossRef]
- Muslu, A.; Islek, I.; Gök, F.; Aliyazicioglu, Y.; Dagdemir, A.; Dundaröz, R.; Kucukoduk, S.; Sakarcan, A.; Daǧdemir, A. Endothelin levels in Henoch-Schonlein purpura. Pediatr. Nephrol. 2002, 17, 920–925. [Google Scholar] [CrossRef]
- Lourdusamy, D.; Mupparaju, V.K.; Sharif, N.F.; Ibebuogu, U.N. Aortic stenosis and Heyde’s syndrome: A comprehensive review. World J. Clin. Cases 2021, 9, 7319–7329. [Google Scholar] [CrossRef]
- de la Torre, J.C.; Stefano, G.B. Evidence that Alzheimer’s disease is a microvascular disorder: The role of constitutive nitric oxide. Brain Res. Brain Res. Rev. 2000, 34, 119–136. [Google Scholar] [CrossRef]
- Matsuyama, S.S.; Jarvik, L.F. Hypothesis: Microtubules, a key to Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 8152–8156. [Google Scholar] [CrossRef]
- Chang, J.C.; Shipstone, A.; Llenado-Lee, M.A. Postoperative thrombotic thrombocytopenic purpura following cardiovascular surgeries. Am. J. Hematol. 1996, 53, 11–17. [Google Scholar] [CrossRef]
- Saltzman, D.J.; Chang, J.C.; Jimenez, J.C.; Carson, J.G.; Abolhoda, A.; Newman, R.S.; Milliken, J.C. Postoperative Thrombotic Thrombocytopenic Purpura after Open Heart Operations. Ann. Thorac. Surg. 2010, 89, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, T.; Baumann, M.; Chang, J. Post-operative thrombotic thrombocytopenic purpura: A review. Int. J. Clin. Pract. 2004, 58, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Young, J.A.; Pallas, C.R.; Knovich, M.A. Transplant-associated thrombotic microangiopathy: Theoretical considerations and a practical approach to an unrefined diagnosis. Bone Marrow Transpl. 2021, 56, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Bijl, M. Endothelial activation, endothelial dysfunction and premature atherosclerosis in systemic autoimmune diseases. Neth. J. Med. 2003, 61, 273–277. [Google Scholar]
- Ferretti, S.; Gatto, A.; Curatola, A.; Pansini, V.; Graglia, B.; Chiaretti, A. Atypical Reye syndrome: Three cases of a problem that pediatricians should consider and remember. Acta Bio-Med. Atenei Parm. 2021, 92, e2021110. [Google Scholar] [CrossRef]
- Zeeuw van der Laan, E.A.N.; van der Velden, S.; Porcelijn, L.; Semple, J.W.; van der Schoot, C.E.; Kapur, R. Evaluation of Platelet Responses in Transfusion-Related Acute Lung Injury (TRALI). Transf. Med. Rev. 2020, 34, 227–233. [Google Scholar] [CrossRef]
- Bayer, A.S.; Theofilopoulos, A.N.; Eisenberg, R.; Friedman, S.G.; Guze, L.B. Thrombotic Thrombocytopenic Purpura-like Syndrome Associated with Infective Endocarditis. JAMA 1977, 238, 408–410. [Google Scholar] [CrossRef]
- Richardson, P.; Ho, V.; Cutler, C.; Glotzbecker, B.; Antin, J.; Soiffer, R. Hepatic Veno-Occlusive Disease after Hematopoietic Stem Cell Transplantation: Novel Insights to Pathogenesis, Current Status of Treatment, and Future Directions. Biol. Blood Marrow Transplant. 2013, 19, S88–S90. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, D.; Kawakami, R.; Guagliumi, G.; Sakamoto, A.; Kawai, K.; Gianatti, A.; Nasr, A.; Kutys, R.; Guo, L.; Cornelissen, A.; et al. Microthrombi as a Major Cause of Cardiac Injury in COVID-19: A Pathologic Study. Circulation 2021, 143, 1031–1042. [Google Scholar] [CrossRef]
- Takaya, H.; Yoshiji, H.; Kawaratani, H.; Sakai, K.; Matsumoto, M.; Fujimura, Y.; Fukui, H. Decreased activity of plasma ADAMTS13 are related to enhanced cytokinemia and endotoxemia in patients with acute liver failure. Biomed. Rep. 2017, 7, 277–285. [Google Scholar] [CrossRef]
- Murakami, J.; Shimizu, Y. Hepatic Manifestations in Hematological Disorders. Int. J. Hepatol. 2013, 2013, 484903. [Google Scholar] [CrossRef] [PubMed]
- Thachil, J. Lessons from acute pancreatitis-induced thrombotic thrombocytopenic purpura. Eur. J. Intern. Med. 2009, 20, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.; Harris, M.D.; Foweraker, J.; Gresham, G.A. Waterhouse-Friderichsen syndrome as a result of non-meningococcal infection. J. Clin. Pathol. 2004, 57, 208–209. [Google Scholar] [CrossRef] [PubMed]
- Torok, N.; Niazi, M.; Al Ahwel, Y.; Taleb, M.; Taji, J.; Assaly, R. Thrombotic thrombocytopenic purpura associated with anti-glomerular basement membrane disease. Nephrol. Dial. Transplant. 2010, 25, 3446–3449. [Google Scholar] [CrossRef]
- Vega-Cabrera, C.; Del Peso, G.; Bajo, A.; Picazo, M.-L.; Rivas-Becerra, B.; Benitez, A.-L.; Ara, J.M.; Olea, T.; Selgas, R. Goodpasture’s syndrome associated with thrombotic thrombocytopenic purpura secondary to an ADAMTS-13 deficit. Int. Urol. Nephrol. 2013, 45, 1785–1789. [Google Scholar] [CrossRef]
- Qahtani, S.A. Acute renal failure and severe rhabdomyolysis in a patient with resistant thrombotic thrombocytopenic purpura. Int. J. Gen. Med. 2011, 4, 687–689. [Google Scholar] [CrossRef]
- Taxbro, K.; Kahlow, H.; Wulcan, H.; Fornarve, A. Rhabdomyolysis and acute kidney injury in severe COVID-19 infection. BMJ Case Rep. 2020, 13, e237616. [Google Scholar] [CrossRef]
- Karki, P.; Birukova, A.A. Microtubules as Major Regulators of Endothelial Function: Implication for Lung Injury. Front. Physiol. 2021, 12, 758313. [Google Scholar] [CrossRef]
- Keeler, E.; Fioravanti, G.; Samuel, B.; Longo, S. Scleroderma Renal Crisis or Thrombotic Thrombocytopenic Purpura: Seeing Through the Masquerade. Lab. Med. 2015, 46, e39–e44. [Google Scholar] [CrossRef]
- Manadan, A.M.; Harris, C.; Block, J. Thrombotic thrombocytopenic purpura in the setting of systemic sclerosis. Semin. Arthritis Rheum. 2005, 34, 683–688. [Google Scholar] [CrossRef]
- Goulielmos, G.N.; Zervou, M.I.; Vazgiourakis, V.M.; Ghodke-Puranik, Y.; Garyfallos, A.; Niewold, T.B. The genetics and molecular pathogenesis of systemic lupus erythematosus (SLE) in populations of different ancestry. Gene 2018, 668, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Mezhov, V.; Segan, J.D.; Tran, H.A.; Cicuttini, F.M. Antiphospholipid syndrome: A clinical review. Med. J. Aust. 2019, 211, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, C.; Bayry, J.; Kone-Paut, I.; Kaveri, S.V. Kawasaki disease: Aetiopathogenesis and therapeutic utility of intravenous immunoglobulin. Autoimmun. Rev. 2010, 9, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Shibagaki, Y.; Fujita, T. Thrombotic microangiopathy in malignant hypertension and hemolytic uremic syndrome (HUS)/thrombotic thrombocytopenic purpura (TTP): Can we differentiate one from the other? Hypertens. Res. 2005, 28, 89–95. [Google Scholar] [CrossRef]
- Colling, M.E.; Bendapudi, P.K. Purpura Fulminans: Mechanism and Management of Dysregulated Hemostasis. Transfus. Med. Rev. 2018, 32, 69–76. [Google Scholar] [CrossRef]
- Biedermann, B.C. Vascular endothelium and graft-versus-host disease. Best Pract. Res. Clin. Haematol. 2008, 21, 129–138. [Google Scholar] [CrossRef]
- Elliott, M.A.; Nichols, W.L.; Plumhoff, E.A.; Ansell, S.M.; Dispenzieri, A.; Gastineau, D.A.; Gertz, M.A.; Inwards, D.J.; Lacy, M.Q.; Micallef, I.N.; et al. Posttransplantation Thrombotic Thrombocytopenic Purpura: A Single-Center Experience and a Contemporary Review. Mayo Clin. Proc. 2003, 78, 421–430. [Google Scholar] [CrossRef]
- Temprano, K.K. A Review of Raynaud’s Disease. Mo. Med. 2016, 113, 123–126. [Google Scholar]
- Saghazadeh, A.; Hafizi, S.; Rezaei, N. Inflammation in venous thromboembolism: Cause or consequence? Int. Immunopharmacol. 2015, 28, 655–665. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Pasalic, L.; Lippi, G. Antibodies against Platelet Factor 4 and Their Associated Pathologies: From HIT/HITT to Spontaneous HIT-Like Syndrome, to COVID-19, to VITT/TTS. Antibodies 2022, 11, 7. [Google Scholar] [CrossRef]
- Cines, D.B.; Bussel, J.B. SARS-CoV-2 Vaccine-Induced Immune Thrombotic Thrombocytopenia. N. Engl. J. Med. 2021, 384, 2254–2256. [Google Scholar] [CrossRef] [PubMed]
- Poudel, D.R.; Ghimire, S.; Dhital, R.; Forman, D.A.; Warkentin, T.E. Spontaneous HIT syndrome post-knee replacement surgery with delayed recovery of thrombocytopenia: A case report and literature review. Platelets 2017, 28, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, E.; Bramanti, A.; Ciurleo, R.; Tchorbanov, A.I.; Giordano, A.; Fagone, P.; Belizna, C.; Bramanti, P.; Shoenfeld, Y.; Nicoletti, F. Entangling COVID-19 associated thrombosis into a secondary antiphospholipid antibody syndrome: Diagnostic and therapeutic perspectives (Review). Int. J. Mol. Med. 2020, 46, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gkrouzman, E.; Andrade, D.C.O.; Andreoli, L.; Barbhaiya, M.; Belmont, H.M.; Branch, D.W.; de Jesús, G.R.; Efthymiou, M.; Ríos-Garcés, R.; et al. COVID-19 and antiphospholipid antibodies: A position statement and management guidance from AntiPhospholipid Syndrome Alliance for Clinical Trials and International Networking (APS ACTION). Lupus 2021, 30, 2276–2285. [Google Scholar] [CrossRef]
- Uzun, G.; Althaus, K.; Bakchoul, T. No Correlation between Anti-PF4 and Anti-SARS-CoV-2 Antibodies after ChAdOx1 nCoV-19 Vaccination. N. Engl. J. Med. 2021, 385, 1334–1336. [Google Scholar] [CrossRef] [PubMed]
- Nazi, I.; Arnold, D.M.; Warkentin, T.E.; Smith, J.W.; Staibano, P.; Kelton, J.G. Distinguishing between anti-platelet factor 4/heparin antibodies that can and cannot cause heparin-induced thrombocytopenia. J. Thromb. Haemost. 2015, 13, 1900–1907. [Google Scholar] [CrossRef]
- Terpos, E.; Politou, M.; Ntanasis-Stathopoulos, I.; Karalis, V.; Merkouri, E.; Fotiou, D.; Gavriatopoulou, M.; Malandrakis, P.; Kastritis, E.; Trougakos, I.; et al. High Prevalence of Anti-PF4 Antibodies Following ChAdOx1 nCov-19 (AZD1222) Vaccination Even in the Absence of Thrombotic Events. Vaccines 2021, 9, 712. [Google Scholar] [CrossRef]
- Pascreau, T.; Ballester, M.C.; Van Dreden, P.; Zia-Chahabi, S.; Zuber, B.; Choucair, J.; Bironien, R.; Farfour, E.; Vasse, M. The high frequency of anti-PF4/heparin antibodies in patients with COVID-19 is neither related to heparin treatment or to an increased incidence of thrombosis. Clin. Chem. Lab. Med. 2021, 59, e405–e408. [Google Scholar] [CrossRef]
- Warkentin, T.E. Clinical picture of heparin-induced thrombocytopenia (HIT) and its differentiation from non-HIT thrombocytopenia. Thromb. Haemost. 2016, 116, 813–822. [Google Scholar] [CrossRef]
- Castillo-Martínez, D.; Torres, Z.; Amezcua-Guerra, L.M.; Pineda, C. Are antiphospholipid antibodies just a common epi-phenomenon or are they causative of immune-mediated coagulopathy in COVID-19? Clin. Rheumatol. 2021, 40, 3015–3019. [Google Scholar] [CrossRef]
- Chauhan, A.K. Degradation of platelet-von Willebrand factor complexes by plasmin: An alternative/backup mechanism to ADAMTS13. Circulation 2014, 129, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Lord, M.S.; Cheng, B.; Farrugia, B.L.; McCarthy, S.; Whitelock, J.M. Platelet Factor 4 Binds to Vascular Proteoglycans and Controls Both Growth Factor Activities and Platelet Activation. J. Biol. Chem. 2017, 292, 4054–4063. [Google Scholar] [CrossRef]
- Opdenakker, G.; Proost, P.; Van Damme, J. Microbiomic and Posttranslational Modifications as Preludes to Autoimmune Diseases. Trends Mol. Med. 2016, 22, 746–757. [Google Scholar] [CrossRef]
- Zavala-Cerna, M.G.; Martínez-García, E.A.; Bugarin, O.T.; Rubio-Jurado, B.; Riebeling, C.; Nava, A. The Clinical Significance of Posttranslational Modification of Autoantigens. Clin. Rev. Allergy Immunol. 2014, 47, 73–90. [Google Scholar] [CrossRef]
- Shaw, P.X.; Goodyear, C.S.; Chang, M.-K.; Witztum, J.L.; Silverman, G.J. The Autoreactivity of Anti-Phosphorylcholine Antibodies for Atherosclerosis-Associated Neo-Antigens and Apoptotic Cells. J. Immunol. 2003, 170, 6151–6157. [Google Scholar] [CrossRef]
- Eggleton, P.; Haigh, R.; Winyard, P. Consequence of neo-antigenicity of the ‘altered self’. Rheumatology 2008, 47, 567–571. [Google Scholar] [CrossRef]
- Ankri, A.; Bonmarchand, M.; Coutellier, A.; Herson, S.; Karmochkine, M. Antiphospholipid antibodies are an epiphenomenon in HIV-infected patients. AIDS 1999, 13, 1282. [Google Scholar] [CrossRef]
- Singer, H.S.; Krumholz, A.; Giuliano, J.; Kiessling, L.S. Antiphospholipid antibodies: An epiphenomenon in tourette syndrome. Mov. Disord. 1997, 12, 738–742. [Google Scholar] [CrossRef]
- Martinuzzo, M.E.; Forastiero, R.R.; Adamczuk, Y.; Pombo, G.; Carreras, L.O. Antiplatelet Factor 4—Heparin Antibodies in Patients with Antiphospholipid Antibodies. Thromb. Res. 1999, 95, 271–279. [Google Scholar] [CrossRef]
- Chang, J. White clot syndrome associated with heparin-induced thrombocytopenia: A review of 23 cases. Heart Lung 1987, 16, 403–407. [Google Scholar]
- Williams, R.T.; Damaraju, L.V.; Mascelli, M.A.; Barnathan, E.S.; Califf, R.M.; Simoons, M.L.; Deliargyris, E.N.; Sane, D.C. Anti-platelet factor 4/heparin antibodies: An independent predictor of 30-day myocardial infarction after acute coronary ischemic syndromes. Circulation 2003, 107, 2307–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hemostasis | Coagulation | Thrombosis | |
|---|---|---|---|
| Term concept | Philosophical | Physiological | Structural |
| Implied meaning | Natural process in vivo | Artificial process in vitro Physiologic process at bleeding site | Pathological process in vivo |
| Involved site | Blood-vessel wall | Test tube Extravascular trauma to vessel wall | Intravascular lumen |
| Products | Hemostatic plug | Fibrin mesh/fibrin clot * | Microthrombi/macrothrombi |
| Critical role | Vascular-wall injury | Coagulation test Hemorrhage | Physiologic thrombogenesis |
| Components | Endothelium SET EVT Blood in circulation | TF/thromboplastin Coagulation proteins/serine proteases | ULVWF/FVIII from ECs Platelets+ from circulation Coagulation proteins/serine proteases TF from SET/EVT |
| Phenotypes | Determined by the level of vascular damage | Determined by participating coagulation factors | Determined by ULVWF, platelets, TF, hemostatic factors, and unifying mechanism |
| Inciting example | Endotheliopathy Vascular injury | Coagulation tests for PT and aPTT | VMTD/MODS due to microthrombosis EA-VMTD: TTP-like syndrome: “DIC” Arterial macrothrombosis VTE Combined micro–macrothrombosis |
| (1) Hemostatic Principles | ||
| ||
| (2) Major Participating Components | ||
| Components | Origin | Mechanism |
| (1) ECs/SET/EVT | Blood-vessel wall/EVT | Protective barrier |
| (2) ULVWF | ECs | Endothelial exocytosis/anchoring and microthrombogenesis |
| (3) Platelets | Circulation | Adhesion to ULVWF strings/assembling and microthrombogenesis |
| (4) TF | SET and EVT | Release from tissue due to vascular injury/leading and fibrinogenesis |
| (5) Coagulation factors | Circulation | Activation of coagulation factors/forming and macrothrombogenesis |
| (3) Vascular Injury and Hemostatic Phenotypes | ||
| Injury-Induced Damage | Involved Hemostatic Path | Level of Vascular Injury and Examples |
| (1) Endothelium | ULVWF | Level 1 damage—microthrombosis (e.g., TIA [focal]; Heyde’s syndrome [local); EA-VMTD [disseminated]) |
| (2) Endothelium/SET | ULVWF + sTF | Level 2 damage—macrothrombosis (e.g., AIS; DVT; PTE; AA) |
| (3) Endothelium/SET/EVT | ULVWF + eTF | Level 3 damage—macrothrombosis with hemorrhage (e.g., THS; THMI) |
| (4) EVT alone | eTF | Level e damage—fibrin clot disease (e.g., AHS (e.g., SDH; EDH); ICH; organ/tissue hematoma) |
| Hemostatic Phenotypes | Causes | Genesis |
| (1) Hemorrhage | External bodily injury | Trauma-induced external bleeding (e.g., accident; assault; self-inflicted injury) |
| (2) Hematoma | Internal EVT injury | Obtuse trauma-induced bleeding (e.g., tissue and cavitary hematoma; hemarthrosis) |
| (3) Thrombosis | Intravascular injury | Intravascular injury (e.g., atherosclerosis; diabetes; indwelling venous catheter; surgery; vascular access) |
| Thrombosis Mechanism | Microthrombogenesis | Fibrinogenesis | Macrothrombogenesis |
|---|---|---|---|
| Utilizing hemostatic path | ULVWF path | TF path | Combined ULVWF and TF path |
| Examples | Sepsis (bacterial, viral, fungal, rickettsial, parasitic) Polytrauma, surgery, transplant Toxin, drug, venom, vaccine Pregnancy Diseases (autoimmune, cancer, diabetes) | APL SDH EDH | DVT (distal) VTE (central DVT/PTE) Arterial thrombosis Peripheral gangrene |
| Thrombosis character | Microthrombi strings | Fibrin clots | Macrothrombus Combined micro–macrothrombi |
| Vascular lesion and phenotype example | Focal—retinal microaneurysm Local—hepatic VOD Multifocal—HERNS, Susac syndrome Disseminated—EA-VMTD/MODS | Disseminated—true DIC | Local—distal DVT, arterial thrombosis, AIS |
| Complex vascular/ hematologic phenotypes | Venous—venous microthrombi (ITP-like syndrome) Arterial—arterial microthrombi (TTP-like syndrome, MODS) | Venous/arterial—DIC (fibrin clot disease) | Combined micro–macrothrombosis Venous—VTE, PTE, CVST Arterial—SPG, limb gangrene |
| Clinical Phenotype | Arterial Endotheliopathy | Venous Endotheliopathy |
|---|---|---|
| Underlying pathology | aEA-VMTD | vEA-VMTD |
| Physiological/hemodynamic difference | Efferent circulation from the heart (oxygen delivery) High-pressure flow High shear stress Capillary and arteriolar microvascular events | Afferent circulation into the heart (CO2 disposal) Low-pressure flow Low shear stress Venous and pulmonary microvascular events |
| Primary cause | ||
| Vascular injury (ECs) | Sepsis-induced arterial microvascular endotheliopathy | Sepsis-induced venous endotheliopathy Vaccine-induced venous endotheliopathy |
| Vascular pathology site | Disseminated aEA-VMTD at the microvasculature | Transient or “silent” vEA-VMTD at the venous system |
| Activated hemostatic path | ULVWF path | ULVWF path |
| Thrombosis component | Microthrombi strings in the microvasculature | Microthrombi strings in the venous system |
| Microthrombotic event | Disseminated VMTD | Silent microthrombosis with efficient (?) microthrombolysis |
| Clinical phenotypes | TTP-like syndrome
| ITP-like syndrome
|
Clinical Features
|
Laboratory and Molecular Features
|
Diagnostic Blood Tests
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.C. Molecular Pathogenesis of Endotheliopathy and Endotheliopathic Syndromes, Leading to Inflammation and Microthrombosis, and Various Hemostatic Clinical Phenotypes Based on “Two-Activation Theory of the Endothelium” and “Two-Path Unifying Theory” of Hemostasis. Medicina 2022, 58, 1311. https://doi.org/10.3390/medicina58091311
Chang JC. Molecular Pathogenesis of Endotheliopathy and Endotheliopathic Syndromes, Leading to Inflammation and Microthrombosis, and Various Hemostatic Clinical Phenotypes Based on “Two-Activation Theory of the Endothelium” and “Two-Path Unifying Theory” of Hemostasis. Medicina. 2022; 58(9):1311. https://doi.org/10.3390/medicina58091311
Chicago/Turabian StyleChang, Jae C. 2022. "Molecular Pathogenesis of Endotheliopathy and Endotheliopathic Syndromes, Leading to Inflammation and Microthrombosis, and Various Hemostatic Clinical Phenotypes Based on “Two-Activation Theory of the Endothelium” and “Two-Path Unifying Theory” of Hemostasis" Medicina 58, no. 9: 1311. https://doi.org/10.3390/medicina58091311