
Association between Microbiome-Related Human Genetic Variants and Fasting Plasma Glucose in a High-Cardiovascular-Risk Mediterranean Population

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants and Study Design
2.2. Baseline Anthropometric, Clinical, Biochemical, and Lifestyle Variables
2.3. DNA Isolation, Genotyping and GWAS in This Mediterranean Population
2.4. Selection of the Mi-RSNPs from Previously Published mbGWAS and Statistical Analysis
2.5. Two-Sample Mendelian Randomization Analysis for Microbiome Effects on Fasting Glucose
3. Results
3.1. General Characteristics of the Participants
3.2. Association between the Microbiome and Fasting Plasma Glucose
3.3. Association between the Microbiome and Type 2 Diabetes Prevalence
3.4. Interactions between the Mi-RSNPs and Adherence to the Mediterranean Diet in Determining Fasting Plasma Glucose
3.5. Interactions between the Mi-RSNPs and Sex in Determining Fasting Plasma Glucose
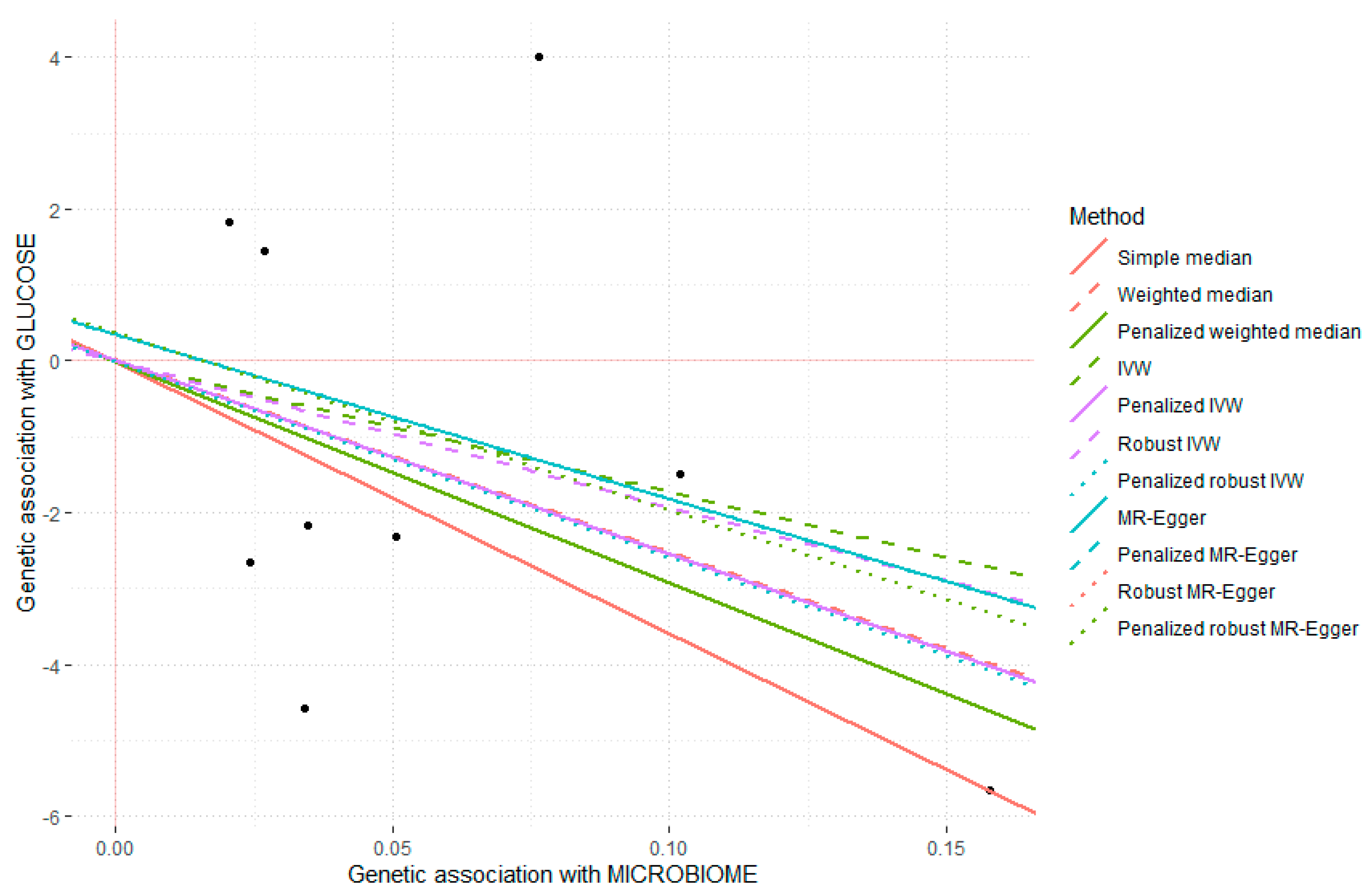
3.6. Mendelian Randomization Sub-Study including Multiple Mi-RSNPs in Determining Plasma Fasting Glucose
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shi, X.; Ma, T.; Sakandar, H.A.; Menghe, B.; Sun, Z. Gut Microbiome and Aging Nexus and Underlying Mechanism. Appl. Microbiol. Biotechnol. 2022, 106, 5349–5358. [Google Scholar] [CrossRef]
- Rahman, M.M.; Islam, F.; -Or-Rashid, M.H.; Mamun, A.A.; Rahaman, M.S.; Islam, M.M.; Meem, A.F.K.; Sutradhar, P.R.; Mitra, S.; Mimi, A.A.; et al. The Gut Microbiota (Microbiome) in Cardiovascular Disease and Its Therapeutic Regulation. Front. Cell. Infect. Microbiol. 2022, 12, 903570. [Google Scholar] [CrossRef]
- Liu, C.-F.; Tang, W.H.W. Gut Microbiota in Sarcopenia and Heart Failure. J. Cardiovasc. Aging 2022, 2, 35. [Google Scholar] [CrossRef]
- Huang, R. Gut Microbiota: A Key Regulator in the Effects of Environmental Hazards on Modulates Insulin Resistance. Front. Cell. Infect. Microbiol. 2021, 11, 800432. [Google Scholar] [CrossRef]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef] [PubMed]
- Tokarek, J.; Gadzinowska, J.; Młynarska, E.; Franczyk, B.; Rysz, J. What Is the Role of Gut Microbiota in Obesity Prevalence? A Few Words about Gut Microbiota and Its Association with Obesity and Related Diseases. Microorganisms 2021, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Jian, C.; Silvestre, M.P.; Middleton, D.; Korpela, K.; Jalo, E.; Broderick, D.; de Vos, W.M.; Fogelholm, M.; Taylor, M.W.; Raben, A.; et al. Gut Microbiota Predicts Body Fat Change Following a Low-Energy Diet: A PREVIEW Intervention Study. Genome Med. 2022, 14, 54. [Google Scholar] [CrossRef]
- Amabebe, E.; Robert, F.O.; Agbalalah, T.; Orubu, E.S.F. Microbial Dysbiosis-Induced Obesity: Role of Gut Microbiota in Homoeostasis of Energy Metabolism. Br. J. Nutr. 2020, 123, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Verhaar, B.J.H.; Hendriksen, H.M.A.; de Leeuw, F.A.; Doorduijn, A.S.; van Leeuwenstijn, M.; Teunissen, C.E.; Barkhof, F.; Scheltens, P.; Kraaij, R.; van Duijn, C.M.; et al. Gut Microbiota Composition Is Related to AD Pathology. Front. Immunol. 2021, 12, 794519. [Google Scholar] [CrossRef]
- Connell, E.; Le Gall, G.; Pontifex, M.G.; Sami, S.; Cryan, J.F.; Clarke, G.; Müller, M.; Vauzour, D. Microbial-Derived Metabolites as a Risk Factor of Age-Related Cognitive Decline and Dementia. Mol. Neurodegener. 2022, 17, 43. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, T.; Cao, M.; Yuan, C.; Reiter, R.J.; Zhao, Z.; Zhao, Y.; Chen, L.; Fan, W.; Wang, X.; et al. Gut Microbiota Dysbiosis Induced by Decreasing Endogenous Melatonin Mediates the Pathogenesis of Alzheimer’s Disease and Obesity. Front. Immunol. 2022, 13, 900132. [Google Scholar] [CrossRef]
- Hashimoto, K. Gut-Microbiota-Brain Axis by Bile Acids in Depression. Psychiatry Clin. Neurosci. 2022, 76, 281. [Google Scholar] [CrossRef] [PubMed]
- Knuesel, T.; Mohajeri, M.H. The Role of the Gut Microbiota in the Development and Progression of Major Depressive and Bipolar Disorder. Nutrients 2021, 14, 37. [Google Scholar] [CrossRef]
- Chadchan, S.B.; Singh, V.; Kommagani, R. Female Reproductive Dysfunctions and the Gut Microbiota. J. Mol. Endocrinol. 2022, 69, R81–R94. [Google Scholar] [CrossRef]
- Aghamajidi, A.; Maleki Vareki, S. The Effect of the Gut Microbiota on Systemic and Anti-Tumor Immunity and Response to Systemic Therapy against Cancer. Cancers 2022, 14, 3563. [Google Scholar] [CrossRef]
- Gubatan, J.; Boye, T.L.; Temby, M.; Sojwal, R.S.; Holman, D.R.; Sinha, S.R.; Rogalla, S.R.; Nielsen, O.H. Gut Microbiome in Inflammatory Bowel Disease: Role in Pathogenesis, Dietary Modulation, and Colitis-Associated Colon Cancer. Microorganisms 2022, 10, 1371. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Cai, L.; Guo, W. New Advances in Improving Bone Health Based on Specific Gut Microbiota. Front. Cell. Infect. Microbiol. 2022, 12, 821429. [Google Scholar] [CrossRef]
- Lepage, P.; Leclerc, M.C.; Joossens, M.; Mondot, S.; Blottière, H.M.; Raes, J.; Ehrlich, D.; Doré, J. A Metagenomic Insight into Our Gut’s Microbiome. Gut 2013, 62, 146–158. [Google Scholar] [CrossRef]
- Maccaferri, S.; Biagi, E.; Brigidi, P. Metagenomics: Key to Human Gut Microbiota. Dig. Dis. 2011, 29, 525–530. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the Gut Microbiota in Nutrition and Health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the Human Gut Microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, J. Role of Intestinal Microbiota and Metabolites on Gut Homeostasis and Human Diseases. BMC Immunol. 2017, 18, 2. [Google Scholar] [CrossRef]
- Di Segni, A.; Braun, T.; BenShoshan, M.; Farage Barhom, S.; Glick Saar, E.; Cesarkas, K.; Squires, J.E.; Keller, N.; Haberman, Y. Guided Protocol for Fecal Microbial Characterization by 16S RRNA-Amplicon Sequencing. J. Vis. Exp. 2018, 133, e56845. [Google Scholar] [CrossRef]
- Durazzi, F.; Sala, C.; Castellani, G.; Manfreda, G.; Remondini, D.; De Cesare, A. Comparison between 16S RRNA and Shotgun Sequencing Data for the Taxonomic Characterization of the Gut Microbiota. Sci. Rep. 2021, 11, 3030. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An Integrated Catalog of Reference Genes in the Human Gut Microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Hugon, P.; Dufour, J.-C.; Colson, P.; Fournier, P.-E.; Sallah, K.; Raoult, D. A Comprehensive Repertoire of Prokaryotic Species Identified in Human Beings. Lancet Infect. Dis. 2015, 15, 1211–1219. [Google Scholar] [CrossRef]
- Jeske, J.T.; Gallert, C. Microbiome Analysis via OTU and ASV-Based Pipelines-A Comparative Interpretation of Ecological Data in WWTP Systems. Bioengineering 2022, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S RRNA Gene Sequencing. Microb. Ecol. 2021, 6, e01202–e01220. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A Metagenome-Wide Association Study of Gut Microbiota in Type 2 Diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, X.; Xia, Y. Further Analysis Reveals New Gut Microbiome Markers of Type 2 Diabetes Mellitus. Antonie Van Leeuwenhoek 2017, 110, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Brunkwall, L.; Orho-Melander, M. The Gut Microbiome as a Target for Prevention and Treatment of Hyperglycaemia in Type 2 Diabetes: From Current Human Evidence to Future Possibilities. Diabetologia 2017, 60, 943–951. [Google Scholar] [CrossRef]
- Zeng, Q.; Zhao, M.; Wang, F.; Li, Y.; Li, H.; Zheng, J.; Chen, X.; Zhao, X.; Ji, L.; Gao, X.; et al. Integrating Choline and Specific Intestinal Microbiota to Classify Type 2 Diabetes in Adults: A Machine Learning Based Metagenomics Study. Front. Endocrinol. 2022, 13, 906310. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling Type 2 Diabetes and Metformin Treatment Signatures in the Human Gut Microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.J.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut Microbiota in Human Adults with Type 2 Diabetes Differs from Non-Diabetic Adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, D.; Fang, Z.; Jie, Z.; Qiu, X.; Zhang, C.; Chen, Y.; Ji, L. Human Gut Microbiota Changes Reveal the Progression of Glucose Intolerance. PLoS ONE 2013, 8, e71108. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut Metagenome in European Women with Normal, Impaired and Diabetic Glucose Control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Davenport, E.R.; Waters, J.L.; Clark, A.G.; Ley, R.E. Cross-Species Comparisons of Host Genetic Associations with the Microbiome. Science 2016, 352, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Doms, S.; Fokt, H.; Rühlemann, M.C.; Chung, C.J.; Kuenstner, A.; Ibrahim, S.M.; Franke, A.; Turner, L.M.; Baines, J.F. Key Features of the Genetic Architecture and Evolution of Host-Microbe Interactions Revealed by High-Resolution Genetic Mapping of the Mucosa-Associated Gut Microbiome in Hybrid Mice. Elife 2022, 11, e75419. [Google Scholar] [CrossRef] [PubMed]
- Awany, D.; Allali, I.; Dalvie, S.; Hemmings, S.; Mwaikono, K.S.; Thomford, N.E.; Gomez, A.; Mulder, N.; Chimusa, E.R. Host and Microbiome Genome-Wide Association Studies: Current State and Challenges. Front. Genet. 2018, 9, 637. [Google Scholar] [CrossRef]
- Bonder, M.J.; Kurilshikov, A.; Tigchelaar, E.F.; Mujagic, Z.; Imhann, F.; Vila, A.V.; Deelen, P.; Vatanen, T.; Schirmer, M.; Smeekens, S.P.; et al. The Effect of Host Genetics on the Gut Microbiome. Nat. Genet. 2016, 48, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.R.; Cusanovich, D.A.; Michelini, K.; Barreiro, L.B.; Ober, C.; Gilad, Y. Genome-Wide Association Studies of the Human Gut Microbiota. PLoS ONE 2015, 10, e0140301. [Google Scholar] [CrossRef] [PubMed]
- Groot, H.E.; van de Vegte, Y.J.; Verweij, N.; Lipsic, E.; Karper, J.C.; van der Harst, P. Human Genetic Determinants of the Gut Microbiome and Their Associations with Health and Disease: A Phenome-Wide Association Study. Sci. Rep. 2020, 10, 14771. [Google Scholar] [CrossRef]
- Hughes, D.A.; Bacigalupe, R.; Wang, J.; Rühlemann, M.C.; Tito, R.Y.; Falony, G.; Joossens, M.; Vieira-Silva, S.; Henckaerts, L.; Rymenans, L.; et al. Genome-Wide Associations of Human Gut Microbiome Variation and Implications for Causal Inference Analyses. Nat. Microbiol. 2020, 5, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Kurilshikov, A.; Medina-Gomez, C.; Bacigalupe, R.; Radjabzadeh, D.; Wang, J.; Demirkan, A.; Le Roy, C.I.; Garay, J.A.R.; Finnicum, C.T.; Liu, X.; et al. Large-Scale Association Analyses Identify Host Factors Influencing Human Gut Microbiome Composition. Nat. Genet. 2021, 53, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Kato, K.; Tanaka, M.; Odamaki, T.; Kubo, R.; Mitsuyama, E.; Xiao, J.-Z.; Yamaguchi, R.; Uematsu, S.; Imoto, S.; et al. Genome-Wide Association Studies and Heritability Analysis Reveal the Involvement of Host Genetics in the Japanese Gut Microbiota. Commun. Biol. 2020, 3, 686. [Google Scholar] [CrossRef] [PubMed]
- Lopera-Maya, E.A.; Kurilshikov, A.; van der Graaf, A.; Hu, S.; Andreu-Sánchez, S.; Chen, L.; Vila, A.V.; Gacesa, R.; Sinha, T.; Collij, V.; et al. Effect of Host Genetics on the Gut Microbiome in 7,738 Participants of the Dutch Microbiome Project. Nat. Genet. 2022, 54, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Rühlemann, M.C.; Hermes, B.M.; Bang, C.; Doms, S.; Moitinho-Silva, L.; Thingholm, L.B.; Frost, F.; Degenhardt, F.; Wittig, M.; Kässens, J.; et al. Genome-Wide Association Study in 8,956 German Individuals Identifies Influence of ABO Histo-Blood Groups on Gut Microbiome. Nat. Genet. 2021, 53, 147–155. [Google Scholar] [CrossRef]
- Wang, J.; Kurilshikov, A.; Radjabzadeh, D.; Turpin, W.; Croitoru, K.; Bonder, M.J.; Jackson, M.A.; Medina-Gomez, C.; Frost, F.; Homuth, G.; et al. Meta-Analysis of Human Genome-Microbiome Association Studies: The MiBioGen Consortium Initiative. Microbiome 2018, 6, 101. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Havulinna, A.S.; Liu, Y.; Jousilahti, P.; Ritchie, S.C.; Tokolyi, A.; Sanders, J.G.; Valsta, L.; Brożyńska, M.; Zhu, Q.; et al. Combined Effects of Host Genetics and Diet on Human Gut Microbiota and Incident Disease in a Single Population Cohort. Nat. Genet. 2022, 54, 134–142. [Google Scholar] [CrossRef]
- Smith, G.D.; Ebrahim, S. “Mendelian Randomization”: Can Genetic Epidemiology Contribute to Understanding Environmental Determinants of Disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, P.M.A.; Grant, S.F.A. Mendelian Randomization in the Era of Genomewide Association Studies. Clin. Chem. 2010, 56, 723–728. [Google Scholar] [CrossRef]
- Holmes, M.V.; Ala-Korpela, M.; Smith, G.D. Mendelian Randomization in Cardiometabolic Disease: Challenges in Evaluating Causality. Nat. Rev. Cardiol. 2017, 14, 577–590. [Google Scholar] [CrossRef]
- Lv, B.-M.; Quan, Y.; Zhang, H.-Y. Causal Inference in Microbiome Medicine: Principles and Applications. Trends Microbiol. 2021, 29, 736–746. [Google Scholar] [CrossRef]
- Zheng, J.; Baird, D.; Borges, M.-C.; Bowden, J.; Hemani, G.; Haycock, P.; Evans, D.M.; Smith, G.D. Recent Developments in Mendelian Randomization Studies. Curr. Epidemiol. Rep. 2017, 4, 330–345. [Google Scholar] [CrossRef]
- Mukamal, K.J.; Stampfer, M.J.; Rimm, E.B. Genetic Instrumental Variable Analysis: Time to Call Mendelian Randomization What It Is. The Example of Alcohol and Cardiovascular Disease. Eur. J. Epidemiol. 2020, 35, 93–97. [Google Scholar] [CrossRef]
- Davies, N.M.; Holmes, M.V.; Smith, G.D. Reading Mendelian Randomisation Studies: A Guide, Glossary, and Checklist for Clinicians. BMJ 2018, 362, k601. [Google Scholar] [CrossRef]
- Ortega-Azorín, C.; Coltell, O.; Asensio, E.M.; Sorlí, J.V.; González, J.I.; Portolés, O.; Saiz, C.; Estruch, R.; Ramírez-Sabio, J.B.; Pérez-Fidalgo, A.; et al. Candidate Gene and Genome-Wide Association Studies for Circulating Leptin Levels Reveal Population and Sex-Specific Associations in High Cardiovascular Risk Mediterranean Subjects. Nutrients 2019, 11, 2751. [Google Scholar] [CrossRef]
- Coltell, O.; Ortega-Azorín, C.; Sorlí, J.V.; Portolés, O.; Asensio, E.M.; Saiz, C.; Barragán, R.; Estruch, R.; Corella, D. Circulating Adiponectin and Its Association with Metabolic Traits and Type 2 Diabetes: Gene-Diet Interactions Focusing on Selected Gene Variants and at the Genome-Wide Level in High-Cardiovascular Risk Mediterranean Subjects. Nutrients 2021, 13, 541. [Google Scholar] [CrossRef]
- Barragán, R.; Coltell, O.; Asensio, E.M.; Francés, F.; Sorlí, J.V.; Estruch, R.; Salas-Huetos, A.; Ordovas, J.M.; Corella, D. MicroRNAs and Drinking: Association between the Pre-MiR-27a Rs895819 Polymorphism and Alcohol Consumption in a Mediterranean Population. Int. J. Mol. Sci. 2016, 17, 1338. [Google Scholar] [CrossRef]
- Schröder, H.; Fitó, M.; Estruch, R.; Martínez-González, M.A.; Corella, D.; Salas-Salvadó, J.; Lamuela-Raventós, R.; Ros, E.; Salaverría, I.; Fiol, M.; et al. A Short Screener Is Valid for Assessing Mediterranean Diet Adherence among Older Spanish Men and Women. J. Nutr. 2011, 141, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Turner, S.D. Qqman: An R Package for Visualizing GWAS Results Using Q-Q and Manhattan Plots. J. Open Source Softw. 2014, 3, 731. [Google Scholar] [CrossRef]
- Yang, Q.; Lin, S.L.; Kwok, M.K.; Leung, G.M.; Schooling, C.M. The Roles of 27 Genera of Human Gut Microbiota in Ischemic Heart Disease, Type 2 Diabetes Mellitus, and Their Risk Factors: A Mendelian Randomization Study. Am. J. Epidemiol. 2018, 187, 1916–1922. [Google Scholar] [CrossRef]
- Hartwig, F.P.; Davies, N.M.; Hemani, G.; Smith, G.D. Two-Sample Mendelian Randomization: Avoiding the Downsides of a Powerful, Widely Applicable but Potentially Fallible Technique. Int. J. Epidemiol. 2016, 45, 1717–1726. [Google Scholar] [CrossRef]
- Haycock, P.C.; Burgess, S.; Wade, K.H.; Bowden, J.; Relton, C.; Smith, G.D. Best (but Oft-Forgotten) Practices: The Design, Analysis, and Interpretation of Mendelian Randomization Studies. Am. J. Clin. Nutr. 2016, 103, 965–978. [Google Scholar] [CrossRef]
- Bowden, J.; Smith, G.D.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Boef, A.G.C.; Dekkers, O.M.; le Cessie, S. Mendelian Randomization Studies: A Review of the Approaches Used and the Quality of Reporting. Int. J. Epidemiol. 2015, 44, 496–511. [Google Scholar] [CrossRef]
- Bowden, J.; Smith, G.D.; Burgess, S. Mendelian Randomization with Invalid Instruments: Effect Estimation and Bias Detection through Egger Regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Rees, J.M.B.; Wood, A.M.; Burgess, S. Extending the MR-Egger Method for Multivariable Mendelian Randomization to Correct for Both Measured and Unmeasured Pleiotropy. Stat. Med. 2017, 36, 4705–4718. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.M.B.; Wood, A.M.; Dudbridge, F.; Burgess, S. Robust Methods in Mendelian Randomization via Penalization of Heterogeneous Causal Estimates. PLoS ONE 2019, 14, e0222362. [Google Scholar] [CrossRef] [PubMed]
- Yavorska, O.O.; Burgess, S. MendelianRandomization: An R Package for Performing Mendelian Randomization Analyses Using Summarized Data. Int. J. Epidemiol. 2017, 46, 1734–1739. [Google Scholar] [CrossRef]
- Broadbent, J.R.; Foley, C.N.; Grant, A.J.; Mason, A.M.; Staley, J.R.; Burgess, S. MendelianRandomization v0.5.0: Updates to an R Package for Performing Mendelian Randomization Analyses Using Summarized Data. Wellcome Open Res. 2020, 5, 252. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, Y.; Xia, F.; Abudukerimu, B.; Zhang, W.; Guo, Y.; Wang, N.; Lu, Y. A Glucagon-Like Peptide-1 Receptor Agonist Lowers Weight by Modulating the Structure of Gut Microbiota. Front. Endocrinol. 2018, 9, 233. [Google Scholar] [CrossRef]
- Fu, J.; Wang, Y.; Tan, S.; Wang, J. Effects of Banana Resistant Starch on the Biochemical Indexes and Intestinal Flora of Obese Rats Induced by a High-Fat Diet and Their Correlation Analysis. Front. Bioeng. Biotechnol. 2021, 9, 575724. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut Microbiota in Human Metabolic Health and Disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Ryu, E.P.; Davenport, E.R. Host Genetic Determinants of the Microbiome Across Animals: From Caenorhabditis Elegans to Cattle. Annu. Rev. Anim. Biosci. 2022, 10, 203–226. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R.; Franzosa, E.A.; Rahnavard, G.; Hall, A.B.; Vlamakis, H.; Stevens, C.; Daly, M.J.; Xavier, R.J.; Huttenhower, C. Host Genetic Variation and Its Microbiome Interactions within the Human Microbiome Project. Genome Med. 2018, 10, 6. [Google Scholar] [CrossRef]
- Markowitz, R.H.G.; LaBella, A.L.; Shi, M.; Rokas, A.; Capra, J.A.; Ferguson, J.F.; Mosley, J.D.; Bordenstein, S.R. Microbiome-Associated Human Genetic Variants Impact Phenome-Wide Disease Risk. Proc. Natl. Acad. Sci. USA 2022, 119, e2200551119. [Google Scholar] [CrossRef]
- García-Santisteban, I.; Cilleros-Portet, A.; Moyua-Ormazabal, E.; Kurilshikov, A.; Zhernakova, A.; Garcia-Etxebarria, K.; Fernandez-Jimenez, N.; Bilbao, J.R. A Two-Sample Mendelian Randomization Analysis Investigates Associations Between Gut Microbiota and Celiac Disease. Nutrients 2020, 12, 1420. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Huang, S.-Y.; Chen, S.-D.; Zhang, Y.-R.; Huang, Y.-Y.; Yu, J.-T. Investigating Casual Associations Among Gut Microbiota, Metabolites, and Neurodegenerative Diseases: A Mendelian Randomization Study. J. Alzheimers Dis. 2022, 87, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Xiang, K.; Zhang, J.-J.; Xu, Y.-Y.; Zhong, X.; Ni, J.; Pan, H.-F. Genetically Predicted Causality of 28 Gut Microbiome Families and Type 2 Diabetes Mellitus Risk. Front. Endocrinol. 2022, 13, 780133. [Google Scholar] [CrossRef] [PubMed]
- Xiang, K.; Wang, P.; Xu, Z.; Hu, Y.-Q.; He, Y.-S.; Chen, Y.; Feng, Y.-T.; Yin, K.-J.; Huang, J.-X.; Wang, J.; et al. Causal Effects of Gut Microbiome on Systemic Lupus Erythematosus: A Two-Sample Mendelian Randomization Study. Front. Immunol. 2021, 12, 667097. [Google Scholar] [CrossRef]
- Zhang, L.; Zhuang, Z.; Zhang, G.; Huang, T.; Fan, D. Assessment of Bidirectional Relationships between 98 Genera of the Human Gut Microbiota and Amyotrophic Lateral Sclerosis: A 2-Sample Mendelian Randomization Study. BMC Neurol. 2022, 22, 8. [Google Scholar] [CrossRef] [PubMed]
- Mathieson, I.; McVean, G. Differential Confounding of Rare and Common Variants in Spatially Structured Populations. Nat. Genet. 2012, 44, 243–246. [Google Scholar] [CrossRef]
- Du, Y.; Neng, Q.; Li, Y.; Kang, Y.; Guo, L.; Huang, X.; Chen, M.; Yang, F.; Hong, J.; Zhou, S.; et al. Gastrointestinal Autonomic Neuropathy Exacerbates Gut Microbiota Dysbiosis in Adult Patients With Type 2 Diabetes Mellitus. Front. Cell. Infect. Microbiol. 2021, 11, 804733. [Google Scholar] [CrossRef]
- Camargo, A.; Vals-Delgado, C.; Alcala-Diaz, J.F.; Villasanta-Gonzalez, A.; Gomez-Delgado, F.; Haro, C.; Leon-Acuña, A.; Cardelo, M.P.; Torres-Peña, J.D.; Guler, I.; et al. A Diet-Dependent Microbiota Profile Associated with Incident Type 2 Diabetes: From the CORDIOPREV Study. Mol. Nutr. Food Res. 2020, 64, e2000730. [Google Scholar] [CrossRef] [PubMed]
- Barlow, G.M.; Yu, A.; Mathur, R. Role of the Gut Microbiome in Obesity and Diabetes Mellitus. Nutr. Clin. Pract. 2015, 30, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yi, G.; Peng, H.; Li, Z.; Chen, S.; Zhong, H.; Chen, Y.; Wang, Z.; Deng, Q.; Fu, M. How Ocular Surface Microbiota Debuts in Type 2 Diabetes Mellitus. Front. Cell. Infect. Microbiol. 2019, 9, 202. [Google Scholar] [CrossRef] [PubMed]
- Rogall, E.T.; Jacob, S.; Triebskorn, R.; Schwartz, T. The Impact of the Anti-Diabetic Drug Metformin on the Intestinal Microbiome of Larval Brown Trout (Salmo trutta f. Fario). Environ. Sci. Eur. 2020, 32, 65. [Google Scholar] [CrossRef]
- diCenzo, G.C.; Muhammed, Z.; Østerås, M.; O’Brien, S.A.P.; Finan, T.M. A Key Regulator of the Glycolytic and Gluconeogenic Central Metabolic Pathways in Sinorhizobium meliloti. Genetics 2017, 207, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Unno, T.; Kim, B.Y.; Park, M.S. Sex Differences in Gut Microbiota. World J. Men’s Health 2020, 38, 48–60. [Google Scholar] [CrossRef]
- Ruuskanen, M.O.; Erawijantari, P.P.; Havulinna, A.S.; Liu, Y.; Méric, G.; Tuomilehto, J.; Inouye, M.; Jousilahti, P.; Salomaa, V.; Jain, M.; et al. Gut Microbiome Composition Is Predictive of Incident Type 2 Diabetes in a Population Cohort of 5,572 Finnish Adults. Diabetes Care 2022, 45, 811–818. [Google Scholar] [CrossRef]
- Wang, H.; Gou, W.; Su, C.; Du, W.; Zhang, J.; Miao, Z.; Xiao, C.; Jiang, Z.; Wang, Z.; Fu, Y.; et al. Association of Gut Microbiota with Glycaemic Traits and Incident Type 2 Diabetes, and Modulation by Habitual Diet: A Population-Based Longitudinal Cohort Study in Chinese Adults. Diabetologia 2022, 65, 1145–1156. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (n = 1020) | Men (n = 371) | Women (n = 649) | p | |
|---|---|---|---|---|
| Age (years) | 66.9 ± 0.2 | 66.4 ± 0.3 | 67.2 ± 0.2 | 0.054 |
| Weight (kg) | 77.1 ± 0.4 | 81.8 ± 0.6 | 74.4 ± 0.5 | <0.001 |
| BMI (kg/m2) | 30.6 ± 0.2 | 29.6 ± 0.2 | 31.2 ± 0.2 | <0.001 |
| SBP (mmHg) | 147.2 ± 0.7 | 148.5 ± 1.1 | 146.4 ± 0.9 | 0.127 |
| DBP (mmHg) | 81.8 ± 0.3 | 82.5 ± 0.6 | 81.4 ± 0.4 | 0.106 |
| Total cholesterol (mg/dL) | 208.3 ± 1.2 | 200.5 ± 1.9 | 212.7 ± 1.6 | <0.001 |
| LDL-C (mg/dL) | 129.5 ± 1.1 | 125.2 ± 1.8 | 131.9 ± 1.4 | 0.004 |
| HDL-C (mg/dL) | 52.7 ± 0.4 | 48.3 ± 0.6 | 55.2 ± 0.5 | <0.001 |
| Triglycerides 1 (mg/dL) | 131.3 ± 2.2 | 135.1 ± 3.6 | 129.2 ± 2.7 | 0.191 |
| Fasting glucose (mg/dL) | 120.1 ± 1.2 | 126.4 ± 2.1 | 116.5 ± 1.5 | <0.001 |
| Total energy intake (kcal/day) | 2203.6 ± 19.7 | 2336.9 ± 33.9 | 2127.3 ± 23.7 | <0.001 |
| Physical Activity (MET-min/day) | 170.0 ± 5.4 | 228.6 ± 11.5 | 136.5 ± 5.0 | <0.001 |
| Adherence to MedDiet (P14) 2 | 8.5 ± 0.1 | 8.6 ± 0.1 | 8.4 ± 0.1 | 0.147 |
| High Adherence MedDiet 3 (≥9) | 505 (49.5) | 187 (50.4) | 318 (49.0) | 0.666 |
| Current smokers: n, % | 128 (12.5) | 99 (26.7) | 29 (4.5) | <0.001 |
| Type 2 diabetes: n, % | 474 (46.5) | 199 (53.6) | 275 (42.4) | 0.001 |
| Chr | SNP | Beta | p | MAF | Gene | Microbial Trait |
|---|---|---|---|---|---|---|
| 13 | rs9569095 | −7.917 | 0.00099 | 0.144 | LOC105370213 | C_Gammaproteobacteria_HB |
| 10 | rs17103336 | 4.563 | 0.00593 | 0.219 | BTBD16 | order_Rhizobiales |
| 4 | rs11940694 | −3.685 | 0.00918 | 0.499 | KLB | genus.Ruminococcustorquesgroup.id.14377 |
| 14 | rs10498633 | 4.845 | 0.01025 | 0.153 | SLC24A4 | genus.Ruminococcustorquesgroup.id.14377 |
| 1 | rs10752747 | 3.258 | 0.03628 | 0.497 | MMEL1 | genus.Tyzzerella3.id.11335 |
| 17 | rs2716882 | 3.737 | 0.05294 | 0.131 | Intergenic | genus_Sporacetigenium |
| 20 | rs6108958 | 2.758 | 0.05605 | 0.359 | LOC105372529 | genus_Abiotrophia |
| 20 | rs6030140 | −4.860 | 0.05747 | 0.139 | PTPRT | G_Holdemanella_HB |
| 2 | rs1004787 | 2.674 | 0.06220 | 0.197 | LOC107985879 | genus.Allisonella.id.2174 |
| 4 | rs2736990 | −2.748 | 0.06539 | 0.393 | SNCA | phylum.Actinobacteria.id.400 |
| 10 | rs860912 | 3.173 | 0.07632 | 0.276 | LOC105378531 | G_Subdoligranulum_HB |
| 1 | rs867426 | −2.812 | 0.07730 | 0.476 | Intergenic | G_unclassified_P_Proteobacteria_HB |
| 2 | rs7580217 | 4.193 | 0.09186 | 0.242 | KCNK12 | g__Parabacteroides |
| 9 | rs1014306 | −2.481 | 0.09196 | 0.184 | DAPK1 | O_Rhodospirillales_RNT |
| 12 | rs987019 | −3.049 | 0.09574 | 0.193 | PPM1H | s__Romboutsia ilealis |
| 2 | rs182549 | −2.375 | 0.09772 | 0.163 | MCM6 | s__Bifidobacterium adolescentis |
| 14 | rs1951597 | 2.368 | 0.10150 | 0.480 | LOC105370413 | G_Odoribacter_HB |
| 3 | rs6766789 | −3.874 | 0.10630 | 0.306 | FHIT | g__CAG-448 |
| 6 | rs3010562 | 2.291 | 0.12010 | 0.334 | Intergenic | genus_Anaerofilum |
| 6 | rs2854275 | −3.225 | 0.12090 | 0.065 | HLA-DQB1 | genus.Streptococcus.id.1853 |
| Chr | SNP | OR | p | MAF | Gene | Microbial Trait |
|---|---|---|---|---|---|---|
| 10 | rs17551124 | 0.775 | 0.00906 | 0.255 | Intergenic | order_Rhizobiales |
| 18 | rs7235005 | 1.249 | 0.01406 | 0.443 | LOC105372112 | G_unclassified_F_Erysipelotrichaceae_HB |
| 4 | rs17063777 | 0.640 | 0.01939 | 0.162 | Intergenic | F_Desulfovibrionaceae_HB |
| 8 | rs10090365 | 1.228 | 0.02043 | 0.403 | Intergenic | genus.CandidatusSoleaferrea.id.11350 |
| 8 | rs10091895 | 1.289 | 0.02241 | 0.428 | CSMD1 | genus.Romboutsia.id.11347 |
| 13 | rs9569095 | 1.364 | 0.04048 | 0.144 | LOC105370213 | C_Gammaproteobacteria_HB |
| 9 | rs10780691 | 0.835 | 0.04640 | 0.345 | NTRK2 | family.Oxalobacteraceae.id.2966 |
| 4 | rs11947461 | 1.200 | 0.04968 | 0.336 | Intergenic | o__Chloroflexales |
| 10 | rs10994397 | 0.645 | 0.05116 | 0.160 | ANK3 | order.Gastranaerophilales.id.1591 |
| 5 | rs10064431 | 1.191 | 0.05420 | 0.411 | FAM172A | genus.Romboutsia.id.11347 |
| 20 | rs1035177 | 0.818 | 0.05659 | 0.315 | MACROD2 | g__Holdemania |
| 4 | rs7654391 | 0.687 | 0.06058 | 0.096 | Intergenic | F_Desulfovibrionaceae_HB |
| 2 | rs1507705 | 1.244 | 0.06471 | 0.380 | DTNB | genus_Desulfovibrio |
| 12 | rs10777875 | 0.819 | 0.06689 | 0.472 | RMST | genus.Ruminococcus1.id.11373 |
| 8 | rs6468721 | 0.827 | 0.06831 | 0.152 | Intergenic | c__Syntrophorhabdia |
| 17 | rs228770 | 0.691 | 0.07381 | 0.053 | NAGS | G_Parasutterella_HB |
| 20 | rs6108958 | 1.176 | 0.07503 | 0.359 | LOC105372529 | genus_Abiotrophia |
| Chr | SNP | Beta 1 | SE 1 | Beta 2 | SE 2 | p_GenexDiet | MAF | Gene | Microbial Trait |
|---|---|---|---|---|---|---|---|---|---|
| 17 | rs2716882 | 12.440 | 3.485 | −0.451 | 3.142 | 0.0060 | 0.1312 | Intergenic | genus_Sporacetigenium |
| 10 | rs10823909 | −4.936 | 3.900 | 7.916 | 3.563 | 0.0150 | 0.0379 | ANAPC16 | g__CAG-776 |
| 1 | rs16833405 | −5.816 | 3.839 | 5.812 | 3.202 | 0.0200 | 0.2568 | Intergenic | O_Rhodospirillales_HB |
| 2 | rs7580217 | 7.420 | 4.211 | −6.183 | 4.368 | 0.0250 | 0.2422 | KCNK12 | g__Parabacteroides |
| 2 | rs6717477 | −3.776 | 2.605 | 3.563 | 2.299 | 0.0347 | 0.3391 | Intergenic | genus_Acinetobacter |
| 2 | rs6546314 | 6.379 | 3.711 | −4.390 | 3.577 | 0.0367 | 0.2274 | DTNB | genus_Desulfovibrio |
| 11 | rs7104796 | −6.851 | 8.103 | 15.920 | 7.374 | 0.0376 | 0.0986 | LDLRAD3 | F_Sutterellaceae_HB |
| 4 | rs10520163 | 3.063 | 2.722 | −4.335 | 2.337 | 0.0392 | 0.4655 | CLCN3 | genus.Allisonella.id.2174 |
| 6 | rs7773795 | −10.310 | 4.327 | 1.553 | 3.979 | 0.0436 | 0.0962 | RTN4IP1 | g__Achromobacter |
| 9 | rs11141878 | 4.587 | 2.693 | −2.560 | 2.419 | 0.0483 | 0.4209 | DAPK1 | O_Rhodospirillales_RNT |
| 16 | rs12597384 | 13.820 | 6.666 | −2.754 | 5.129 | 0.0488 | 0.0519 | Intergenic | G_Odoribacter_HB |
| 13 | rs9569095 | 1.127 | 4.270 | −10.350 | 4.022 | 0.0504 | 0.1442 | LOC105370213 | C_Gammaproteobacteria_HB |
| Method | Estimate | SE | 95%_CI | p-Value | |
|---|---|---|---|---|---|
| Simple_median | −35.841 | 19.400 | −73.863 | 2.182 | 0.065 |
| Weighted_median | −25.292 | 15.332 | −55.342 | 4.759 | 0.099 |
| Penalized_weighted_median | −29.185 | 15.782 | −60.117 | 1.747 | 0.064 |
| IVW | −17.198 | 14.415 | −45.451 | 11.055 | 0.233 |
| Penalized_IVW | −25.517 | 12.300 | −49.626 | −1.409 | 0.038 |
| Robust_IVW | −19.300 | 13.537 | −45.832 | 7.232 | 0.154 |
| Penalized_robust_IVW | −25.811 | 8.198 | −41.879 | −9.743 | 0.002 |
| MR-Egger | −21.692 | 26.192 | −73.027 | 29.644 | 0.408 |
| (intercept) | 0.351 | 1.657 | −2.896 | 3.598 | 0.832 |
| Penalized_MR-Egger | −21.692 | 26.192 | −73.027 | 29.644 | 0.408 |
| (intercept) | 0.351 | 1.657 | −2.896 | 3.598 | 0.832 |
| Robust_MR-Egger | −23.403 | 18.326 | −59.321 | 12.515 | 0.202 |
| (intercept) | 0.363 | 1.287 | −2.159 | 2.885 | 0.778 |
| Penalized_robust_MR-Egger | −23.403 | 18.326 | −59.321 | 12.515 | 0.202 |
| (intercept) | 0.363 | 1.287 | −2.159 | 2.885 | 0.778 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asensio, E.M.; Ortega-Azorín, C.; Barragán, R.; Alvarez-Sala, A.; Sorlí, J.V.; Pascual, E.C.; Fernández-Carrión, R.; Villamil, L.V.; Corella, D.; Coltell, O. Association between Microbiome-Related Human Genetic Variants and Fasting Plasma Glucose in a High-Cardiovascular-Risk Mediterranean Population. Medicina 2022, 58, 1238. https://doi.org/10.3390/medicina58091238
Asensio EM, Ortega-Azorín C, Barragán R, Alvarez-Sala A, Sorlí JV, Pascual EC, Fernández-Carrión R, Villamil LV, Corella D, Coltell O. Association between Microbiome-Related Human Genetic Variants and Fasting Plasma Glucose in a High-Cardiovascular-Risk Mediterranean Population. Medicina. 2022; 58(9):1238. https://doi.org/10.3390/medicina58091238
Chicago/Turabian StyleAsensio, Eva M., Carolina Ortega-Azorín, Rocío Barragán, Andrea Alvarez-Sala, José V. Sorlí, Eva C. Pascual, Rebeca Fernández-Carrión, Laura V. Villamil, Dolores Corella, and Oscar Coltell. 2022. "Association between Microbiome-Related Human Genetic Variants and Fasting Plasma Glucose in a High-Cardiovascular-Risk Mediterranean Population" Medicina 58, no. 9: 1238. https://doi.org/10.3390/medicina58091238