
The Interplay between Tumour Microenvironment Components in Malignant Melanoma

 and
and
Abstract
:1. Introduction
2. Clinicopathological and Molecular Hallmarks Update
2.1. General Features
2.2. Histopathological Characteristics
2.3. AJCC Stages and Histological Features in Correlation with Prognosis
2.4. Immunohistochemical Markers of Diagnosis
2.5. Genetics and Specific Markers
2.6. Putative Melanoma Biomarkers
3. Inflammatory Microenvironment in Malignant Melanoma
3.1. Cancer Inflammasome
3.2. Interactions in Melanoma’s Inflammasome
3.3. Melanoma’s Microenvironment Components
4. Melanoma CSCs—The Origin of Heterogeneity, Plasticity, Aggressiveness, and Therapy Resistance
5. Melanoma Cells-Adipocytes “Dialogue”
5.1. Hypodermis Role in Cancer Microenvironment
5.2. Extracellular Vesicles of Tumour Niche
6. Microbiota in Malignant Melanoma
7. Current Melanoma Therapeutic Approaches
7.1. Therapy Targets and Potential Therapeutic Biomarkers
7.2. Therapeutic Strategies Associated to Melanoma Immune Microenvironment
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Richetta, A.G.; Silvestri, V.; Giancristoforo, S.; Rizzolo, P.; D’Epiro, S.; Graziano, V.; Mattozzi, C.; Navazio, A.S.; Falchetti, M.; Calvieri, S.; et al. Mutational profiling in melanocytic tumors: Multiple somatic mutations and clinical implications. Oncology 2014, 86, 104–108. [Google Scholar] [CrossRef]
- de Menezes, F.C.; de Sousa Cabral, L.G.; Petrellis, M.C.; Neto, C.F.; Maria, D.A. Antitumor effect of cell therapy with mesenchymal stem cells on murine melanoma B16-F10. Biomed. Pharmacother. 2020, 128, 110294. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Mazurkiewicz, J.; Simiczyjew, A.; Dratkiewicz, E.; Ziętek, M.; Matkowski, R.; Nowak, D. Stromal cells present in the melanoma niche affect tumor invasiveness and its resistance to therapy. Int. J. Mol. Sci. 2021, 22, 529. [Google Scholar] [CrossRef]
- Belter, B.; Haase-Kohn, C.; Pietzsch, J. Biomarkers in malignant melanoma: Recent trends and critical perspective. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Australia, 2017; ISBN 9780994438140. [Google Scholar]
- Mimeault, M.; Batra, S.K. Novel biomarkers and therapeutic targets for optimizing the therapeutic management of melanomas. World J. Clin. Oncol. 2012, 3, 32–42. [Google Scholar] [CrossRef]
- Han, D.; Han, G.; Morrison, S.; Leong, S.P.; Kashani-Sabet, M.; Vetto, J.; White, R.; Schneebaum, S.; Pockaj, B.; Mozzillo, N.; et al. Factors predicting survival in thick melanoma: Do all thick melanomas have the same prognosis? Surgery 2020, 168, 518–526. [Google Scholar] [CrossRef]
- Gaudy-Marqueste, C.; Macagno, N.; Loundou, A.; Pellegrino, E.; Ouafik, L.; Budden, T.; Mundra, P.; Gremel, G.; Akhras, V.; Lin, L.; et al. Molecular characterization of fast-growing melanomas. J. Am. Acad. Dermatol. 2021, 86, 312–321. [Google Scholar] [CrossRef]
- Vereecken, P.; Cornelis, F.; Van Baren, N.; Vandersleyen, V.; Baurain, J.-F. A synopsis of serum biomarkers in cutaneous melanoma patients. Dermatol. Res. Pract. 2012, 2012, 260643. [Google Scholar] [CrossRef] [Green Version]
- Carr, S.; Smith, C.; Wernberg, J. Epidemiology and risk factors of melanoma. Surg. Clin. N. Am. 2020, 100, 1–12. [Google Scholar] [CrossRef]
- Torres-Cabala, C.; Li-Ning-Tapia, E.; Hwu, W.-J. Pathology-based biomarkers useful for clinical decisions in melanoma. Arch. Med. Res. 2020, 51, 827–838. [Google Scholar] [CrossRef]
- Wilson, M.L. Histopathologic and molecular diagnosis of melanoma. Clin. Plast. Surg. 2021, 48, 587–598. [Google Scholar] [CrossRef]
- Palmer, S.R.; Erickson, L.A.; Ichetovkin, I.; Knauer, D.J.; Markovic, S.N. Circulating serologic and molecular biomarkers in malignant melanoma. Mayo Clin. Proc. 2011, 86, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Alegre, E.; Sammamed, M.; Fernández-Landázuri, S.; Zubiri, L.; González, Á. Circulating biomarkers in malignant melanoma. Adv. Clin. Chem. 2015, 69, 47–89. [Google Scholar] [CrossRef]
- Karagiannis, P.; Fittall, M.; Karagiannis, S.N. Evaluating biomarkers in melanoma. Front. Oncol. 2014, 4, 383. [Google Scholar] [CrossRef] [Green Version]
- Levesque, M.P. Multi-dimensional biomarkers for the personalized treatment of melanoma. Syst. Med. 2021, 2, 361–364. [Google Scholar] [CrossRef]
- Santonocito, C.; Concolino, P.; Lavieri, M.M.; Ameglio, F.; Gentileschi, S.; Capizzi, R.; Rocchetti, S.; Amerio, P.; Castagnola, M.; Zuppi, C.; et al. Comparison between Three Molecular Methods for Detection of Blood Melanoma Tyrosinase MRNA. Correlation with Melanoma Stages and S100B, LDH, NSE Biochemical Markers. Clin. Chim. Acta 2005, 362, 85–93. [Google Scholar] [CrossRef]
- Vendittelli, F.; Santonocito, C.; Paradisi, A.; Romitelli, F.; Concolino, P.; Silveri, S.L.; Sisto, T.; Capizzi, R.; Catricalà, C.; Mulè, A.; et al. A New Standardized Absolute Quantitative RT-PCR Method for Detection of Tyrosinase MRNAs in Melanoma Patients: Technical and Operative Instructions. Clin. Chim. Acta 2009, 409, 100–105. [Google Scholar] [CrossRef]
- Clement, E.; Lazar, I.; Muller, C.; Nieto, L. Obesity and melanoma: Could fat be fueling malignancy? Pigment Cell Melanoma Res. 2017, 30, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, M.J.C.; Seftor, E.A.; Margaryan, N.V.; Seftor, R.E.B. Heterogeneity and plasticity of melanoma: Challenges of current therapies. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Australia, 2017; ISBN 9780994438140. [Google Scholar]
- Mitsuhashi, A.; Okuma, Y. Perspective on immune oncology with liquid biopsy, peripheral blood mononuclear cells, and microbiome with non-invasive biomarkers in cancer patients. Clin. Transl. Oncol. 2018, 20, 966–974. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Oak, A.S.W.; Slominski, R.M.; Brożyna, A.A.; Slominski, A.T. Current molecular markers of melanoma and treatment targets. Int. J. Mol. Sci. 2020, 21, 3535. [Google Scholar] [CrossRef]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dréno, B.; Fargnoli, M.C.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics—Update 2019. Eur. J. Cancer 2020, 126, 141–158. [Google Scholar] [CrossRef] [Green Version]
- Elder, D.E.; Masii, R.; Scolyer, R.A.; Willemze, R. WHO Classification of Skin Tumours, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2018; Volume 4. [Google Scholar]
- Tyrrell, H.; Payne, M. Combatting mucosal melanoma: Recent advances and future perspectives. Melanoma Manag. 2018, 5, MMT11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majem, M.; Manzano, J.L.; Marquez-Rodas, I.; Mujika, K.; Muñoz-Couselo, E.; Pérez-Ruiz, E.; de la Cruz-Merino, L.; Espinosa, E.; Gonzalez-Cao, M.; Berrocal, A. SEOM clinical guideline for the management of cutaneous melanoma (2020). Clin. Transl. Oncol. 2021, 23, 948–960. [Google Scholar] [CrossRef]
- Elder, D.E.; Bastian, B.C.; Cree, I.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization Classification of cutaneous, mucosal, and uveal melanoma: Detailed analysis of 9 distinct subtypes defined by their evolutionary pathway. Arch. Pathol. Lab. Med. 2020, 144, 500–522. [Google Scholar] [CrossRef] [Green Version]
- McGovern, V.J.; Shaw, H.M.; Milton, G.W.; Farago, G.A. Is malignant melanoma arising in a Hutchinson’s melanotic freckle a separate disease entity? Histopathology 1980, 4, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Longo, C.; Pellacani, G. Melanomas. Dermatol. Clin. 2016, 34, 411–419. [Google Scholar] [CrossRef]
- DeWane, M.E.; Kelsey, A.; Oliviero, M.; Rabinovitz, H.; Grant-Kels, J.M. Melanoma on chronically sun-damaged skin: Lentigo maligna and desmoplastic melanoma. J. Am. Acad. Dermatol. 2019, 81, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Goydos, J.S.; Shoen, S.L. Acral lentiginous melanoma. Cancer Treat. Res. 2016, 167, 321–329. [Google Scholar] [CrossRef]
- Patel, K.R.; Chernock, R.; Lewis, J.S.; Raptis, C.A.; Gilani, M.A.; Dehner, L.P. Lipomatous congenital melanocytic nevus presenting as a neck mass in a young adult. Head Neck Pathol. 2013, 7, 404–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccavale, S.; Calabrese, G.; Mattiello, E.; Broganelli, P.; Ramondetta, A.; Pieretti, G.; Alfano, R.; Argenziano, G. Cutaneous melanoma arising in congenital melanocytic nevus: A retrospective observational study. Dermatology 2021, 237, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Chua, R.F.; Pico, J. Dermal melanocytosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Helm, M.F.; Bax, M.J.; Bogner, P.N.; Chung, C.G. Metastatic melanoma with features of blue nevus and tumoral melanosis identified during pembrolizumab therapy. JAAD Case Rep. 2017, 3, 135–137. [Google Scholar] [CrossRef] [Green Version]
- Granter, S.R.; McKee, P.H.; Calonje, E.; Mihm, M.C.; Busam, K. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: A clinicopathologic study of 10 cases on the spectrum of so-called “Malignant blue nevus”. Am. J. Surg. Pathol. 2001, 25, 316–323. [Google Scholar] [CrossRef]
- Lützow-Holm, C.; Gjersvik, P.; Helsing, P. Melanom, føflekk eller talgvorte? Tidsskriftet 2013, 133, 1167–1168. [Google Scholar] [CrossRef] [PubMed]
- Su, A.; Dry, S.M.; Binder, S.W.; Said, J.; Shintaku, P.; Sarantopoulos, G.P. Malignant melanoma with neural differentiation: An exceptional case report and brief review of the pertinent literature. Am. J. Dermatopathol. 2014, 36, e5–e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keung, E.Z.; Gershenwald, J.E. The eighth edition American Joint Committee on Cancer (AJCC) melanoma staging system: Implications for melanoma treatment and care. Expert Rev. Anticancer Ther. 2018, 18, 775–784. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Thompson, J.F.; Long, G.V.; Ross, M.I.; Lazar, A.I.; Atkins, M.B.; Balch, C.M.; Barnhill, R.L.; et al. Melanoma of the skin. In AJCC Cancer Staging Manual, 8th ed.; Amin, M.B., Edge, S.B., Green, F., Byrd, D.R., Brookland, R.K., Washington, M.K., Gershenwald, J.E., Compton, C.C., Hess, K.R., Sullivan, D.C., et al., Eds.; Springer: Cham, Switzerland, 2017; pp. 563–585. ISBN 9783319406176. [Google Scholar]
- Ossio, R.; Roldán-Marín, R.; Martínez-Said, H.; Adams, D.J.; Robles-Espinoza, C.D. Melanoma: A global perspective. Nat. Rev. Cancer 2017, 17, 393–394. [Google Scholar] [CrossRef]
- Lattanzi, M.; Lee, Y.; Simpson, D.; Moran, U.; Darvishian, F.; Kim, R.H.; Hernando, E.; Polsky, D.; Hanniford, D.; Shapiro, R.; et al. Primary melanoma histologic subtype: Impact on survival and response to therapy. J. Natl. Cancer Inst. 2019, 111, 180–188. [Google Scholar] [CrossRef]
- Aung, P.P.; Nagarajan, P.; Prieto, V.G. Regression in primary cutaneous melanoma: Etiopathogenesis and clinical significance. Lab. Investig. 2017, 97, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Chen, N.; Ge, C.; Li, R.; Li, Z.; Zeng, B.; Li, C.; Wang, Y.; Xue, Y.; Song, X.; et al. Prognostic value of tumor-infiltrating lymphocytes in melanoma: A systematic review and meta-analysis. OncoImmunology 2019, 8, e1593806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osella-Abate, S.; Conti, L.; Annaratone, L.; Senetta, R.; Bertero, L.; Licciardello, M.; Caliendo, V.; Picciotto, F.; Quaglino, P.; Cassoni, P.; et al. Phenotypic characterisation of immune cells associated with histological regression in cutaneous melanoma. Pathology 2019, 51, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Berg, J.H.; Heemskerk, B.; van Rooij, N.; Gomez-Eerland, R.; Michels, S.; van Zon, M.; de Boer, R.; Bakker, N.A.M.; Jorritsma-Smit, A.; van Buuren, M.M.; et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: Boosting of neoantigen-specific T cell reactivity and long-term follow-up. J. Immunother. Cancer 2020, 8, e000848. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Buzaid, A.C.; Soong, S.-J.; Atkins, M.B.; Cascinelli, N.; Coit, D.G.; Fleming, I.D.; Gershenwald, J.E.; Houghton, A.; Kirkwood, J.M.; et al. New TNM melanoma staging system: Linking biology and natural history to clinical outcomes. Semin. Surg. Oncol. 2003, 21, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.-J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [Green Version]
- Szumera-Ciećkiewicz, A.; Bosisio, F.; Teterycz, P.; Antoranz, A.; Delogu, F.; Koljenović, S.; van de Wiel, B.A.; Blokx, W.; van Kempen, L.C.; Rutkowski, P.; et al. SOX10 is as specific as S100 protein in detecting metastases of melanoma in lymph nodes and is recommended for sentinel lymph node assessment. Eur. J. Cancer 2020, 137, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Prieto, V.G.; Shea, C.R. Use of immunohistochemistry in melanocytic lesions. J. Cutan. Pathol. 2008, 35 (Suppl. S2), 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Herberth, F.I.; Karamchandani, J.; Kim, J.; Dadras, S.S. SOX10 immunostaining distinguishes desmoplastic melanoma from excision scar. J. Cutan. Pathol. 2010, 37, 944–952. [Google Scholar] [CrossRef]
- Palla, B.; Su, A.; Binder, S.; Dry, S. SOX10 Expression distinguishes desmoplastic melanoma from its histologic mimics. Am. J. Dermatopathol. 2013, 35, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Scatena, C.; Murtas, D.; Tomei, S. Cutaneous melanoma classification: The importance of high-throughput genomic technologies. Front. Oncol. 2021, 11, 635488. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, L.; Lontano, A.; Merli, M.; Dika, E.; Nagore, E.; Quaglino, P.; Puig, S.; Ribero, S. Familial Melanoma and Susceptibility Genes: A Review of the Most Common Clinical and Dermoscopic Phenotypic Aspect, Associated Malignancies and Practical Tips for Management. J. Clin. Med. 2021, 10, 3760. [Google Scholar] [CrossRef] [PubMed]
- Wolf Horrell, E.M.; Boulanger, M.C.; D’Orazio, J.A. Melanocortin 1 receptor: Structure, function, and regulation. Front. Genet. 2016, 7, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simiczyjew, A.; Dratkiewicz, E.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. The influence of tumor microenvironment on immune escape of melanoma. Int. J. Mol. Sci. 2020, 21, 8359. [Google Scholar] [CrossRef] [PubMed]
- Inamdar, G.S.; Madhunapantula, S.V.; Robertson, G.P. Targeting the MAPK Pathway in Melanoma: Why Some Approaches Succeed and Other Fail. Biochem. Pharmacol. 2010, 80, 624–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diefenbach, R.J.; Lee, J.H.; Menzies, A.M.; Carlino, M.S.; Long, G.V.; Saw, R.P.M.; Howle, J.R.; Spillane, A.J.; Scolyer, R.A.; Kefford, R.F.; et al. Design and Testing of a Custom Melanoma Next Generation Sequencing Panel for Analysis of Circulating Tumor DNA. Cancers 2020, 12, 2228. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Chatila, W.K.; Arora, A.; Sanchez-Vega, F.; Kantheti, H.S.; Rojas Zamalloa, J.A.; Krieger, P.; Callahan, M.K.; Betof Warner, A.; Postow, M.A.; et al. Therapeutic Implications of Detecting MAPK-Activating Alterations in Cutaneous and Unknown Primary Melanomas. Clin. Cancer Res. 2021, 27, 2226–2235. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Liu, W.; Huynh, M.V.; Waqas, M.A.; Gillahan, J.E.; Clark, K.S.; Fu, K.; Martin, B.L.; Jeck, W.R.; Souroullas, G.P.; et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov. 2014, 4, 1418–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uguen, A.; Talagas, M.; Costa, S.; Samaison, L.; Paule, L.; Alavi, Z.; De Braekeleer, M.; Le Marechal, C.; Marcorelles, P. NRAS (Q61R), BRAF (V600E) immunohistochemistry: A concomitant tool for mutation screening in melanomas. Diagn. Pathol. 2015, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.T.; Luong, M.C.; Van Hoang, D.; Doucet, A. AI outperformed every dermatologist: Improved dermoscopic melanoma diagnosis through customizing batch logic and loss function in an optimized deep CNN architecture. arXiv 2020, arXiv:2003.02597. [Google Scholar]
- Pilloni, L.; Bianco, P.; DiFelice, E.; Cabras, S.; Castellanos, M.E.; Atzori, L.; Ferreli, C.; Mulas, P.; Nemolato, S.; Faa, G. The usefulness of C-Kit in the immunohistochemical assessment of melanocytic lesions. Eur. J. Histochem. 2011, 55, 20. [Google Scholar] [CrossRef] [Green Version]
- Qadeer, Z.A.; Harcharik, S.; Valle-Garcia, D.; Chen, C.; Birge, M.B.; Vardabasso, C.; Duarte, L.F.; Bernstein, E. Decreased expression of the chromatin remodeler ATRX associates with melanoma progression. J. Investig. Dermatol. 2014, 134, 1768–1772. [Google Scholar] [CrossRef] [Green Version]
- Fukumoto, T.; Lin, J.; Fatkhutdinov, N.; Liu, P.; Somasundaram, R.; Herlyn, M.; Zhang, R.; Nishigori, C. ARID2 deficiency correlates with the response to immune checkpoint blockade in melanoma. J. Investig. Dermatol. 2021, 141, 1564–1572.e4. [Google Scholar] [CrossRef] [PubMed]
- Fahey, C.C.; Davis, I.J. Setting the stage for cancer development: SETD2 and the consequences of lost methylation. Cold Spring Harb. Perspect. Med. 2017, 7, a026468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Shah, C.P.; Weis, E.; Lajous, M.; Shields, J.A.; Shields, C.L. Intermittent and chronic ultraviolet light exposure and uveal melanoma: A meta-analysis. Ophthalmology 2005, 112, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Weis, E.; Shah, C.P.; Lajous, M.; Shields, J.A.; Shields, C.L. The association between host susceptibility factors and uveal melanoma: A meta-analysis. Arch. Ophthalmol. 2006, 124, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaliki, S.; Shields, C.L. Uveal melanoma: Relatively rare but deadly cancer. Eye 2017, 31, 241–257. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Gong, Q.; Wang, Y.; Li, M.; Wang, L.; Ding, H.; Li, P. The biological function and clinical significance of SF3B1 mutations in cancer. Biomark. Res. 2020, 8, 38. [Google Scholar] [CrossRef]
- Cai, H.; Sobue, T.; Kitamura, T.; Sawada, N.; Iwasaki, M.; Shimazu, T.; Tsugane, S. Epidemiology of nonmelanoma skin cancer in Japan: Occupational type, lifestyle, and family history of cancer. Cancer Sci. 2020, 111, 4257–4265. [Google Scholar] [CrossRef]
- Griewank, K.G.; Schilling, B. Next-generation sequencing to guide treatment of advanced melanoma. Am. J. Clin. Dermatol. 2017, 18, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Cirenajwis, H.; Lauss, M.; Ekedahl, H.; Törngren, T.; Kvist, A.; Saal, L.H.; Olsson, H.; Staaf, J.; Carneiro, A.; Ingvar, C.; et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol. Oncol. 2017, 11, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.R.; Look, A.T.; DeClue, J.E.; Valentine, M.B.; Lowy, D.R. Inactivation of the NF1 gene in human melanoma and neuroblastoma cell lines without impaired regulation of GTP.Ras. Proc. Natl. Acad. Sci. USA 1993, 90, 5539–5543. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Bhat, T.; Gutmann, D.H.; Johnson, K.J. Melanoma in individuals with neurofibromatosis type 1: A retrospective study. Dermatol. Online J. 2019, 25, 13030/qt5ck3f722. [Google Scholar] [CrossRef] [PubMed]
- Ranzani, M.; Alifrangis, C.; Perna, D.; Dutton-Regester, K.; Pritchard, A.; Wong, K.; Rashid, M.; Robles-Espinoza, C.D.; Hayward, N.K.; McDermott, U.; et al. BRAF/NRAS wild-type melanoma, NF1 status and sensitivity to trametinib. Pigment Cell Melanoma Res. 2015, 28, 117–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Cancer genome atlas network genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The AACR Project GENIE Consortium; Andre, F.; Arnedos, M.; Baras, A.S.; Baselga, J.; Bedard, P.L.; Berger, M.F.; Bierkens, M.; Calvo, F.; Cerami, E.; et al. AACR Project GENIE: Powering precision medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [Green Version]
- De, P.; Aske, J.C.; Dey, N. RAC1 takes the lead in solid tumors. Cells 2019, 8, 382. [Google Scholar] [CrossRef] [Green Version]
- Ye, T.; Zhang, J.; Liu, X.; Yang, M.; Zhou, Y.; Yuan, S.; Chen, Y.; Gao, C.; Huang, M.; Ye, C.; et al. The predictive value of MAP2K1/2 mutations for efficiency of immunotherapy in melanoma. J. Clin. Oncol. 2021, 39 (Suppl. S15), e21587. [Google Scholar] [CrossRef]
- Tandler, N.; Mosch, B.; Pietzsch, J. Protein and non-protein biomarkers in melanoma: A critical update. Amino Acids 2012, 43, 2203–2230. [Google Scholar] [CrossRef] [PubMed]
- Massi, D.; Landriscina, M.; Piscazzi, A.; Cosci, E.; Kirov, A.; Paglierani, M.; Di Serio, C.; Mourmouras, V.; Fumagalli, S.; Biagioli, M.; et al. S100A13 is a new angiogenic marker in human melanoma. Mod. Pathol. 2010, 23, 804–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.H.; Kim, D.H.; Surh, Y.J. dynamic roles of inflammasomes in inflammatory tumor microenvironment. NPJ Precis. Oncol. 2021, 5, 18. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Machado, J.C.; Pharoah, P.; Sousa, S.; Carvalho, R.; Oliveira, C.; Figueiredo, C.; Amorim, A.; Seruca, R.; Caldas, C.; Carneiro, F.; et al. Interleukin 1B and interleukin 1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology 2001, 121, 823–829. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Z.; Yu, J.; Lei, G.; Wang, S. Three polymorphisms in interleukin-1β gene and risk for breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2010, 124, 821–825. [Google Scholar] [CrossRef]
- da Silva, W.C.; Oshiro, T.M.; de Sá, D.C.; Franco, D.D.G.S.; Festa Neto, C.; Pontillo, A. Genotyping and differential expression analysis of inflammasome genes in sporadic malignant melanoma reveal novel contribution of CARD8, IL1B and IL18 in melanoma susceptibility and progression. Cancer Genet. 2016, 209, 474–480. [Google Scholar] [CrossRef]
- Pandey, A.; Shen, C.; Feng, S.; Man, S.M. Cell biology of inflammasome activation. Trends Cell Biol. 2021, 31, 924–939. [Google Scholar] [CrossRef]
- Man, S.M. Inflammasomes in the gastrointestinal tract: Infection, cancer and gut microbiota homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 721–737. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Rathinam, V.A.K.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Kantono, M.; Guo, B. Inflammasomes and Cancer: The Dynamic Role of the Inflammasome in Tumor Development. Front. Immunol. 2017, 8, 1132. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Bhattacharya, M.; Roy, S.; Tian, Y.; Yin, Q. Immunobiology and structural biology of AIM2 inflammasome. Mol. Asp. Med. 2020, 76, 100869. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.V.; Kim, S.J.; Donovan, E.L.; Chen, M.; Gross, A.C.; Webster Marketon, J.I.; Barsky, S.H.; Glaser, R. Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: Implications for stress-related enhancement of tumor progression. Brain Behav. Immun. 2009, 23, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Burrows, F.J.; Haskard, D.O.; Hart, I.R.; Marshall, J.F.; Selkirk, S.; Poole, S.; Thorpe, P.E. Influence of tumor-derived interleukin 1 on melanoma-endothelial cell interactions in vitro. Cancer Res. 1991, 51, 4768–4775. [Google Scholar]
- Vidal-Vanaclocha, F.; Amézaga, C.; Asumendi, A.; Kaplanski, G.; Dinarello, C.A. Interleukin-1 receptor blockade reduces the number and size of murine B16 melanoma hepatic metastases. Cancer Res. 1994, 54, 2667–2672. [Google Scholar] [PubMed]
- Vidal-Vanaclocha, F.; Alvarez, A.; Asumendi, A.; Urcelay, B.; Tonino, P.; Dinarello, C.A. Interleukin 1 (IL-1)-dependent melanoma hepatic metastasis in vivo; Increased endothelial adherence by IL-1-induced mannose receptors and growth factor production in vitro. J. Natl. Cancer Inst. 1996, 88, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Voronov, E.; Dvorkin, T.; Fima, E.; Cagnano, E.; Benharroch, D.; Shendler, Y.; Bjorkdahl, O.; Segal, S.; Dinarello, C.A.; et al. Differential effects of IL-1 alpha and IL-1 beta on tumorigenicity patterns and invasiveness. J. Immunol. 2003, 171, 6448–6456. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 Is Required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1 beta. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, J.H.; Ellis, L.Z.; Fujita, M. Inflammasomes as molecular mediators of inflammation and cancer: Potential role in melanoma. Cancer Lett. 2012, 314, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Drexler, S.K.; Bonsignore, L.; Masin, M.; Tardivel, A.; Jackstadt, R.; Hermeking, H.; Schneider, P.; Gross, O.; Tschopp, J.; Yazdi, A.S. Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 18384–18389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möller, M.; Wasel, J.; Schmetzer, J.; Weiß, U.; Meissner, M.; Schiffmann, S.; Weigert, A.; Möser, C.V.; Niederberger, E. The specific IKKε/TBK1 inhibitor amlexanox suppresses human melanoma by the inhibition of autophagy, NF-κB and MAP kinase pathways. Int. J. Mol. Sci. 2020, 21, 4721. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Liu, W.; Kaur, M.; Luo, Y.; Domenico, J.; Samson, J.M.; Shellman, Y.G.; Norris, D.A.; Dinarello, C.A.; Spritz, R.A.; et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017, 36, 3820–3830. [Google Scholar] [CrossRef] [Green Version]
- Tengesdal, I.W.; Menon, D.R.; Osborne, D.G.; Neff, C.P.; Powers, N.E.; Gamboni, F.; Mauro, A.G.; D’Alessandro, A.; Stefanoni, D.; Henen, M.A.; et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc. Natl. Acad. Sci. USA 2021, 118, e2000915118. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Muneer, K.M.; Tamimi, I.A.; Chang, M.E.; Ata, M.O.; Yusuf, N. Thymoquinone suppresses metastasis of melanoma cells by inhibition of NLRP3 inflammasome. Toxicol. Appl. Pharmacol. 2013, 270, 70–76. [Google Scholar] [CrossRef]
- Liu, W.; Luo, Y.; Dunn, J.H.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Dual role of apoptosis-associated speck-like protein containing a CARD (ASC) in tumorigenesis of human melanoma. J. Investig. Dermatol. 2013, 133, 518–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, O.; Thomas, C.J.; Guarda, G.; Tschopp, J. The inflammasome: An integrated view. Immunol. Rev. 2011, 243, 136–151. [Google Scholar] [CrossRef]
- Makarenkova, H.P.; Shestopalov, V.I. The role of pannexin hemichannels in inflammation and regeneration. Front. Physiol. 2014, 5, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, D.; Bivik, C.; Farahani, E.; Synnerstad, I.; Fredrikson, M.; Enerbäck, C.; Rosdahl, I.; Söderkvist, P. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res. 2012, 25, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Nikolova, P.N.; Pawelec, G.P.; Mihailova, S.M.; Ivanova, M.I.; Myhailova, A.P.; Baltadjieva, D.N.; Marinova, D.I.; Ivanova, S.S.; Naumova, E.J. Association of cytokine gene polymorphisms with malignant melanoma in Caucasian population. Cancer Immunol. Immunother. 2007, 56, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Howell, W.M.; Turner, S.J.; Theaker, J.M.; Bateman, A.C. Cytokine gene single nucleotide polymorphisms and susceptibility to and prognosis in cutaneous malignant melanoma. Eur. J. Immunogenet. 2003, 30, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Raman, D.; Baugher, P.J.; Thu, Y.M.; Richmond, A. Role of chemokines in tumor growth. Cancer Lett. 2007, 256, 137–165. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Nold-Petry, C.A.; Nold, M.F.; Joosten, L.A.B.; Opitz, B.; van der Meer, J.H.M.; van de Veerdonk, F.L.; Ferwerda, G.; Heinhuis, B.; Devesa, I.; et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009, 113, 2324–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobono, S.; Santini, R.; Gagliardi, S.; Dapporto, F.; Colecchia, D.; Chiariello, M.; Leone, C.; Valoti, M.; Manetti, F.; Petricci, E.; et al. Targeted inhibition of Hedgehog-GLI signaling by novel acylguanidine derivatives inhibits melanoma cell growth by inducing replication stress and mitotic catastrophe. Cell Death Dis. 2018, 9, 142. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Wei, L.; Hui, H.; Dai, Q.; You, Q.-D.; Guo, Q.-L.; Lu, N. Wogonin suppresses melanoma cell B16-F10 invasion and migration by inhibiting Ras-medicated pathways. PLoS ONE 2014, 9, e106458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Gu, T.; Wang, J.-H.; Xiong, H.; Wang, Y.-Q.; Liu, G.-L.; Qu, Y.; Zhang, N. Effects of wogonin on the mechanism of melanin synthesis in A375 cells. Exp. Ther. Med. 2017, 14, 4547–4553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Ji, Y.; Zhang, L.; Cai, H.; Ji, Z.; Gu, L.; Yang, S. Wogonin inhibits the growth of HT144 melanoma via regulating hedgehog signaling-mediated inflammation and glycolysis. Int. Immunopharmacol. 2021, 101, 108222. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Shelton, M.; Anene, C.A.; Nsengimana, J.; Roberts, W.; Newton-Bishop, J.; Boyne, J.R. The role of CAF derived exosomal microRNAs in the tumour microenvironment of melanoma. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188456. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Cesano, A.; Warren, S. Bringing the next generation of immuno-oncology biomarkers to the clinic. Biomedicines 2018, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzagalli, M.; Ebelt, N.D.; Manuel, E.R. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin. Cancer Biol. 2019, 59, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Gan, J.; Long, Z.; Guo, G.; Shi, X.; Wang, C.; Zang, Y.; Ding, Z.; Chen, J.; Zhang, J.; et al. Targeted delivery of let-7b to reprogramme tumor-associated macrophages and tumor infiltrating dendritic cells for tumor rejection. Biomaterials 2016, 90, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef]
- Maria, A.G.; Dillenburg-Pilla, P.; Reis, R.I.; Floriano, E.M.; Tefe-Silva, C.; Ramos, S.G.; Pesquero, J.B.; Nahmias, C.; Costa-Neto, C.M. Host kinin B1 receptor plays a protective role against melanoma progression. Sci. Rep. 2016, 6, 22078. [Google Scholar] [CrossRef]
- Maria, A.G.; Dillemburg-Pilla, P.; Durand, M.T.; Floriano, E.M.; Manfiolli, A.O.; Ramos, S.G.; Pesquero, J.B.; Nahmias, C.; Costa-Neto, C.M. Activation of the kinin B1 receptor by its agonist reduces melanoma metastasis by playing a dual effect on tumor cells and host immune response. Front. Pharmacol. 2019, 10, 1106. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.J.; van den Braber, M.; Rozeman, E.A.; Haanen, J.B.A.G.; Blank, C.U.; et al. Dysfunctional CD8 T Cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 2019, 176, 775–789.e18. [Google Scholar] [CrossRef] [PubMed]
- Marconcini, R.; Spagnolo, F.; Stucci, L.S.; Ribero, S.; Marra, E.; Rosa, F.D.; Picasso, V.; Di Guardo, L.; Cimminiello, C.; Cavalieri, S.; et al. Current status and perspectives in immunotherapy for metastatic melanoma. Oncotarget 2018, 9, 12452–12470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvani, M.; Bruno, G.; Dal Monte, M.; Nassini, R.; Fontani, F.; Casini, A.; Cavallini, L.; Becatti, M.; Bianchini, F.; De Logu, F.; et al. β(3)-Adrenoceptor as a potential immuno-suppressor agent in melanoma. Br. J. Pharmacol. 2019, 176, 2509–2524. [Google Scholar] [CrossRef] [PubMed]
- Ladányi, A.; Kiss, J.; Mohos, A.; Somlai, B.; Liszkay, G.; Gilde, K.; Fejös, Z.; Gaudi, I.; Dobos, J.; Tímár, J. Prognostic impact of B-cell density in cutaneous melanoma. Cancer Immunol. Immunother. 2011, 60, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, R.; Zhang, G.; Fukunaga-Kalabis, M.; Perego, M.; Krepler, C.; Xu, X.; Wagner, C.; Hristova, D.; Zhang, J.; Tian, T.; et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat. Commun. 2017, 8, 607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jonge, K.; Tillé, L.; Lourenco, J.; Maby-El Hajjami, H.; Nassiri, S.; Racle, J.; Gfeller, D.; Delorenzi, M.; Verdeil, G.; Baumgaertner, P.; et al. Inflammatory B cells correlate with failure to checkpoint blockade in melanoma patients. OncoImmunology 2021, 10, 1873585. [Google Scholar] [CrossRef] [PubMed]
- Falleni, M.; Savi, F.; Tosi, D.; Agape, E.; Cerri, A.; Moneghini, L.; Bulfamante, G.P. M1 and M2 macrophages’ clinicopathological significance in cutaneous melanoma. Melanoma Res. 2017, 27, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Di Martino, J.S.; Bowman, R.L.; Campbell, N.R.; Baksh, S.C.; Simon-Vermot, T.; Kim, I.S.; Haldeman, P.; Mondal, C.; Yong-Gonzales, V.; et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. 2018, 8, 1006–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Huang, Y.; Bong, R.; Ding, Y.; Song, N.; Wang, X.; Song, X.; Luo, Y. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin. Cancer Res. 2011, 17, 7230–7239. [Google Scholar] [CrossRef] [Green Version]
- Filippi, L.; Bruno, G.; Domazetovic, V.; Favre, C.; Calvani, M. Current therapies and new targets to fight melanoma: A promising role for the β(3)-adrenoreceptor. Cancers 2020, 12, 1415. [Google Scholar] [CrossRef]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8 T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical role for CD103 + /CD141 + dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- Pietra, G.; Vitale, M.; Moretta, L.; Mingari, M.C. How melanoma cells inactivate NK cells. OncoImmunology 2012, 1, 974–975. [Google Scholar] [CrossRef] [Green Version]
- Papaccio, F.; Kovacs, D.; Bellei, B.; Caputo, S.; Migliano, E.; Cota, C.; Picardo, M. Profiling cancer-associated fibroblasts in melanoma. Int. J. Mol. Sci. 2021, 22, 7255. [Google Scholar] [CrossRef]
- Kharaishvili, G.; Simkova, D.; Bouchalova, K.; Gachechiladze, M.; Narsia, N.; Bouchal, J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell Int. 2014, 14, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, S.; Massi, D.; Farini, V.; Baroni, G.; Parri, M.; Innocenti, S.; Cecchi, R.; Chiarugi, P. Beta-adrenoceptors are upregulated in human melanoma and their activation releases pro-tumorigenic cytokines and metalloproteases in melanoma cell lines. Lab. Investig. 2013, 93, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Yang, K.; Andl, T.; Wickett, R.R.; Zhang, Y. Perspective of Targeting Cancer-Associated Fibroblasts in Melanoma. J. Cancer 2015, 6, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vukman, K.V.; Försönits, A.; Oszvald, Á.; Tóth, E.; Buzás, E.I. Mast cell secretome: Soluble and vesicular components. Semin. Cell Dev. Biol. 2017, 67, 65–73. [Google Scholar] [CrossRef]
- Grimbaldeston, M.A.; Skov, L.; Finlay-Jones, J.J.; Hart, P.H. Increased dermal mast cell prevalence and susceptibility to development of basal cell carcinoma in humans. Methods 2002, 28, 90–96. [Google Scholar] [CrossRef]
- Motti, M.L.; Minopoli, M.; Di Carluccio, G.; Ascierto, P.A.; Carriero, M.V. MicroRNAs as key players in melanoma cell resistance to MAPK and immune checkpoint inhibitors. Int. J. Mol. Sci. 2020, 21, 4544. [Google Scholar] [CrossRef]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials—An ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Sakai, R. Direct interaction between carcinoma cells and cancer associated fibroblasts for the regulation of cancer invasion. Cancers 2015, 7, 2054–2062. [Google Scholar] [CrossRef]
- Boussadia, Z.; Lamberti, J.; Mattei, F.; Pizzi, E.; Puglisi, R.; Zanetti, C.; Pasquini, L.; Fratini, F.; Fantozzi, L.; Felicetti, F.; et al. Acidic microenvironment plays a key role in human melanoma progression through a sustained exosome mediated transfer of clinically relevant metastatic molecules. J. Exp. Clin. Cancer Res. 2018, 37, 245. [Google Scholar] [CrossRef]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.-J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Erra Díaz, F.; Dantas, E.; Geffner, J. Unravelling the interplay between extracellular acidosis and immune cells. Mediat. Inflamm. 2018, 2018, 1218297. [Google Scholar] [CrossRef] [PubMed]
- Böhme, I.; Bosserhoff, A. Extracellular acidosis triggers a senescence-like phenotype in human melanoma cells. Pigment Cell Melanoma Res. 2020, 33, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Calvani, M.; Pelon, F.; Comito, G.; Taddei, M.L.; Moretti, S.; Innocenti, S.; Nassini, R.; Gerlini, G.; Borgognoni, L.; Bambi, F.; et al. Norepinephrine promotes tumor microenvironment reactivity through Β3-Adrenoreceptors during melanoma progression. Oncotarget 2015, 6, 4615–4632. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Cho, D.; Kim, H.J.; Chong, S.J.; Lee, K.H.; Yu, D.S.; Park, C.J.; Lee, J.Y.; Cho, B.K.; Park, H.J. Investigation of the corticotropin-releasing hormone-proopiomelanocortin axis in various skin tumours. Br. J. Dermatol. 2006, 155, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Malek, Z.A.; Knittel, J.; Kadekaro, A.L.; Swope, V.B.; Starner, R. The melanocortin 1 receptor and the UV response of human melanocytes–a shift in paradigm. Photochem. Photobiol. 2008, 84, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Park, H.; Yang, Y.; Kim, T.S.; Bang, S.I.; Cho, D. Enhancement of cell migration by corticotropin-releasing hormone through ERK1/2 pathway in murine melanoma cell line, B16F10. Exp. Dermatol. 2007, 16, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Arnette, C.R.; Roth-Carter, Q.R.; Koetsier, J.L.; Broussard, J.A.; Burks, H.E.; Cheng, K.; Amadi, C.; Gerami, P.; Johnson, J.L.; Green, K.J. Keratinocyte cadherin desmoglein 1 controls melanocyte behavior through paracrine signaling. Pigment Cell Melanoma Res. 2020, 33, 305–317. [Google Scholar] [CrossRef]
- Wu, J.C.; Tsai, H.E.; Liu, G.S.; Wu, C.S.; Tai, M.H. Autophagic cell death participates in POMC-induced melanoma suppression. Cell Death Discov. 2018, 4, 11. [Google Scholar] [CrossRef]
- Zhou, J.; Feng, J.Y.; Wang, Q.; Shang, J. Calcitonin gene-related peptide cooperates with substance P to inhibit melanogenesis and induces apoptosis of B16F10 cells. Cytokine 2015, 74, 137–144. [Google Scholar] [CrossRef]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzam, A.A.H.; McDonald, J.; Lambert, D.G. Hot topics in opioid pharmacology: Mixed and biased opioids. Br. J. Anaesth. 2019, 122, e136–e145. [Google Scholar] [CrossRef]
- Slominski, A.T.; Zmijewski, M.A.; Zbytek, B.; Brozyna, A.A.; Granese, J.; Pisarchik, A.; Szczesniewski, A.; Tobin, D.J. Regulated proenkephalin expression in human skin and cultured skin cells. J. Investig. Dermatol. 2011, 131, 613–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.M.; Jiao, X.; Plotnikoff, N.P.; Griffin, N.; Qi, R.Q.; Gao, X.H.; Shan, F.P. Killing effect of methionine enkephalin on melanoma in vivo and in vitro. Oncol. Rep. 2017, 38, 2132–2140. [Google Scholar] [CrossRef] [Green Version]
- Fell, G.L.; Robinson, K.C.; Mao, J.; Woolf, C.J.; Fisher, D.E. Skin β-endorphin mediates addiction to UV light. Cell 2014, 157, 1527–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coupland, S.E.; Thornton, S.; Kalirai, H. Importance of partial losses of chromosome 3 in uveal melanoma in the BAP1 gene region. JAMA Ophthalmol. 2020, 138, 188. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Zhang, Y. Identification of survival-related genes and a novel gene-based prognostic signature involving the tumor microenvironment of uveal melanoma. Int. Immunopharmacol. 2021, 96, 107816. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.J.; Brouwer, N.J.; Esmaeli, B. The cancer genome atlas project: An integrated molecular view of uveal melanoma. Ophthalmology 2018, 125, 1139–1142. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [Green Version]
- Souri, Z.; Wierenga, A.P.A.; Mulder, A.; Jochemsen, A.G.; Jager, M.J. HLA Expression in Uveal Melanoma: An indicator of malignancy and a modifiable immunological target. Cancers 2019, 11, 1132. [Google Scholar] [CrossRef] [Green Version]
- Bronkhorst, I.H.G.; Jager, M.J. Inflammation in uveal melanoma. Eye 2013, 27, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gezgin, G.; Dogrusöz, M.; van Essen, T.H.; Kroes, W.G.M.; Luyten, G.P.M.; van der Velden, P.A.; Walter, V.; Verdijk, R.M.; van Hall, T.; van der Burg, S.H.; et al. Genetic evolution of uveal melanoma guides the development of an inflammatory microenvironment. Cancer Immunol. Immunother. 2017, 66, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Babchia, N.; Landreville, S.; Clément, B.; Coulouarn, C.; Mouriaux, F. The bidirectional crosstalk between metastatic uveal melanoma cells and hepatic stellate cells engenders an inflammatory microenvironment. Exp. Eye Res. 2019, 181, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Gorain, M.; Kundu, G.; Kundu, G.C. Therapeutic implications of cellular and molecular biology of cancer stem cells in melanoma. Mol. Cancer 2017, 16, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, N.Y.; Schatton, T.; Kim, S.; Zhan, Q.; Wilson, B.J.; Ma, J.; Saab, K.R.; Osherov, V.; Widlund, H.R.; Gasser, M.; et al. VEGFR-1 expressed by malignant melanoma-initiating cells is required for tumor growth. Cancer Res. 2011, 71, 1474–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Yin, J.; Kim, S.-H.; Sohn, Y.-W.; Beck, S.; Lim, Y.C.; Nam, D.-H.; Choi, Y.-J.; Kim, H. EGFR-AKT-Smad signaling promotes formation of glioma stem-like cells and tumor angiogenesis by ID3-driven cytokine induction. Cancer Res. 2011, 71, 7125–7134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Molley, T.G.; Seward, C.H.; Abdeen, A.A.; Zhang, H.; Wang, X.; Gandhi, H.; Yang, J.-L.; Gaus, K.; Kilian, K.A. Geometric regulation of histone state directs melanoma reprogramming. Commun. Biol. 2020, 3, 341. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monzani, E.; Facchetti, F.; Galmozzi, E.; Corsini, E.; Benetti, A.; Cavazzin, C.; Gritti, A.; Piccinini, A.; Porro, D.; Santinami, M.; et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur. J. Cancer 2007, 43, 935–946. [Google Scholar] [CrossRef]
- Keshet, G.I.; Goldstein, I.; Itzhaki, O.; Cesarkas, K.; Shenhav, L.; Yakirevitch, A.; Treves, A.J.; Schachter, J.; Amariglio, N.; Rechavi, G. MDR1 expression identifies human melanoma stem cells. Biochem. Biophys. Res. Commun. 2008, 368, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzagalli, M.; Moretti, R.M.; Messi, E.; Marelli, M.M.; Fontana, F.; Anastasia, A.; Bani, M.R.; Beretta, G.; Limonta, P. Targeting melanoma stem cells with the Vitamin E derivative δ-tocotrienol. Sci. Rep. 2018, 8, 587. [Google Scholar] [CrossRef] [Green Version]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A Temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupas, V.; Weishaupt, C.; Siepmann, D.; Kaserer, M.-L.; Eickelmann, M.; Metze, D.; Luger, T.A.; Beissert, S.; Loser, K. RANK is expressed in metastatic melanoma and highly upregulated on melanoma-initiating cells. J. Investig. Dermatol. 2011, 131, 944–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; et al. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma Initiating Cells. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef] [PubMed]
- Koshio, J.; Kagamu, H.; Nozaki, K.; Saida, Y.; Tanaka, T.; Shoji, S.; Igarashi, N.; Miura, S.; Okajima, M.; Watanabe, S.; et al. DEAD/H (Asp-Glu-Ala-Asp/His) box polypeptide 3, X-linked is an immunogenic target of cancer stem cells. Cancer Immunol. Immunother. 2013, 62, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sharma, P.; Kumar, D.; Chakraborty, G.; Gorain, M.; Kundu, G.C. Functional characterization of stromal osteopontin in melanoma progression and metastasis. PLoS ONE 2013, 8, e69116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampilafkos, P.; Melachrinou, M.; Kefalopoulou, Z.; Lakoumentas, J.; Sotiropoulou-Bonikou, G. Epigenetic modifications in cutaneous malignant melanoma: EZH2, H3K4me2, and H3K27me3 immunohistochemical expression is enhanced at the invasion front of the tumor. Am. J. Dermatopathol. 2015, 37, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Wan, H.; Zhang, X. Requirement of splicing factor hnRNP A2B1 for tumorigenesis of melanoma stem cells. Stem Cell Res. Ther. 2021, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Bruttel, V.S.; Wischhusen, J. Cancer stem cell immunology: Key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.F.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fomeshi, M.R.; Ebrahimi, M.; Mowla, S.J.; Khosravani, P.; Firouzi, J.; Khayatzadeh, H. Evaluation of the expressions pattern of miR-10b, 21, 200c, 373 and 520c to find the correlation between epithelial-to-mesenchymal transition and melanoma stem cell potential in isolated cancer stem cells. Cell. Mol. Biol. Lett. 2015, 20, 448–465. [Google Scholar] [CrossRef]
- Skvortsov, S.; Debbage, P.; Lukas, P.; Skvortsova, I. Crosstalk between DNA repair and cancer stem cell (CSC) associated intracellular pathways. Semin. Cancer Biol. 2015, 31, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.P.; Moideen, K.; George, P.J.; Dolla, C.; Kumaran, P.; Babu, S. Coincident diabetes mellitus modulates Th1-, Th2-, and Th17-cell responses in latent tuberculosis in an IL-10- and TGF-β-dependent manner. Eur. J. Immunol. 2016, 46, 390–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Q.; Shi, X.; Lan, S.; Jin, H.; Wu, D. Effect of melanoma stem cells on melanoma metastasis (Review). Oncol. Lett. 2021, 22, 566. [Google Scholar] [CrossRef]
- Taghizadeh, R.; Noh, M.; Huh, Y.H.; Ciusani, E.; Sigalotti, L.; Maio, M.; Arosio, B.; Nicotra, M.R.; Natali, P.; Sherley, J.L.; et al. CXCR6, a newly defined biomarker of tissue-specific stem cell asymmetric self-renewal, identifies more aggressive human melanoma cancer stem cells. PLoS ONE 2010, 5, e15183. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kumar, S.; Gorain, M.; Tomar, D.; Patil, H.S.; Radharani, N.N.V.; Kumar, T.V.S.; Patil, T.V.; Thulasiram, H.; Kundu, G.C. Notch1-MAPK signaling axis regulates CD133(+) cancer stem cell-mediated melanoma growth and angiogenesis. J. Investig. Dermatol. 2016, 136, 2462–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Coz, V.; Zhu, C.; Devocelle, A.; Vazquez, A.; Boucheix, C.; Azzi, S.; Gallerne, C.; Eid, P.; Lecourt, S.; Giron-Michel, J. IGF-1 contributes to the expansion of melanoma-initiating cells through an epithelial-mesenchymal transition process. Oncotarget 2016, 7, 82511–82527. [Google Scholar] [CrossRef] [PubMed]
- Prasmickaite, L.; Engesaeter, B.Ø.; Skrbo, N.; Hellenes, T.; Kristian, A.; Oliver, N.K.; Suo, Z.; Maelandsmo, G.M. Aldehyde dehydrogenase (ALDH) activity does not select for cells with enhanced aggressive properties in malignant melanoma. PLoS ONE 2010, 5, e10731. [Google Scholar] [CrossRef]
- Ma, I.; Allan, A.L. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. Rep. 2011, 7, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Dallaglio, K.; Chen, Y.; Robinson, W.A.; Robinson, S.E.; McCarter, M.D.; Wang, J.; Gonzalez, R.; Thompson, D.C.; Norris, D.A.; et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells 2012, 30, 2100–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Yang, Z.; Qi, F. Aldehyde dehydrogenase-positive melanoma stem cells in tumorigenesis, drug resistance and anti-neoplastic immunotherapy. Mol. Biol. Rep. 2020, 47, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Ohmura-Kakutani, H.; Akiyama, K.; Maishi, N.; Ohga, N.; Hida, Y.; Kawamoto, T.; Iida, J.; Shindoh, M.; Tsuchiya, K.; Shinohara, N.; et al. Identification of tumor endothelial cells with high aldehyde dehydrogenase activity and a highly angiogenic phenotype. PLoS ONE 2014, 9, e113910. [Google Scholar] [CrossRef]
- Ravindran Menon, D.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Bonyadi Rad, E.; et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene 2015, 34, 4448–4459. [Google Scholar] [CrossRef] [Green Version]
- Murphy, G.F.; Wilson, B.J.; Girouard, S.D.; Frank, N.Y.; Frank, M.H. Stem cells and targeted approaches to melanoma cure. Mol. Asp. Med. 2014, 39, 33–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.-W.; Liu, Y.-Z.; Pan, J.-X. Verteporfin induces apoptosis and eliminates cancer stem-like cells in uveal melanoma in the absence of light activation. Am. J. Cancer Res. 2016, 6, 2816–2830. [Google Scholar] [PubMed]
- Sarvi, S.; Crispin, R.; Lu, Y.; Zeng, L.; Hurley, T.D.; Houston, D.R.; von Kriegsheim, A.; Chen, C.-H.; Mochly-Rosen, D.; Ranzani, M.; et al. ALDH1 bio-activates nifuroxazide to eradicate ALDH high melanoma-initiating cells. Cell Chem. Biol. 2018, 25, 1456–1469.e6. [Google Scholar] [CrossRef] [Green Version]
- Petrachi, T.; Romagnani, A.; Albini, A.; Longo, C.; Argenziano, G.; Grisendi, G.; Dominici, M.; Ciarrocchi, A.; Dallaglio, K. Therapeutic potential of the metabolic modulator phenformin in targeting the stem cell compartment in melanoma. Oncotarget 2017, 8, 6914–6928. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhang, J.; Pan, J. Pristimerin effectively inhibits the malignant phenotypes of uveal melanoma cells by targeting NF-κB pathway. Int. J. Oncol. 2017, 51, 887–898. [Google Scholar] [CrossRef]
- Liu, S.; Gao, X.; Zhang, L.; Qin, S.; Wei, M.; Liu, N.; Zhao, R.; Li, B.; Meng, Y.; Lin, G.; et al. A novel anti-cancer stem cells compound optimized from the natural symplostatin 4 scaffold inhibits Wnt/β-catenin signaling pathway. Eur. J. Med. Chem. 2018, 156, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Dashti, A.; Ebrahimi, M.; Hadjati, J.; Memarnejadian, A.; Moazzeni, S.M. Dendritic cell based immunotherapy using tumor stem cells mediates potent antitumor immune responses. Cancer Lett. 2016, 374, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, L.; Xia, Y.; Chen, X.; Chang, A.E.; Hollingsworth, R.E.; Hurt, E.; Owen, J.; Moyer, J.S.; Prince, M.E.P.; et al. Therapeutic efficacy of cancer stem cell vaccines in the adjuvant setting. Cancer Res. 2016, 76, 4661–4672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badrinath, N.; Yoo, S.Y. Recent advances in cancer stem cell-targeted immunotherapy. Cancers 2019, 11, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, F.; Dang, J.; Zha, H.; Zhang, B.; Lin, M.; Cheng, F. PD-L1 Promotes self-renewal and tumorigenicity of malignant melanoma initiating cells. Biomed Res. Int. 2017, 2017, 1293201. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, N.; Almeida, A.; Partyka, K.A.; Lu, Y.; Schwan, J.V.; Lambert, K.; Rogers, M.; Robinson, W.A.; Robinson, S.E.; Applegate, A.J.; et al. Combining a GSI and BCL-2 inhibitor to overcome melanoma’s resistance to current treatments. Oncotarget 2016, 7, 84594–84607. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, N.; Lu, Y.; Almeida, A.; Lambert, K.; Shiau, C.-W.; Su, J.-C.; Luo, Y.; Fujita, M.; Robinson, W.A.; Robinson, S.E.; et al. Use of a MCL-1 inhibitor alone to de-bulk melanoma and in combination to kill melanoma initiating cells. Oncotarget 2017, 8, 46801–46817. [Google Scholar] [CrossRef]
- Magnoni, C.; Giudice, S.; Pellacani, G.; Bertazzoni, G.; Longo, C.; Veratti, E.; Morini, D.; Benassi, L.; Vaschieri, C.; Azzoni, P.; et al. Stem cell properties in cell cultures from different stage of melanoma progression. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 171–181. [Google Scholar] [CrossRef]
- Booth, A.; Magnuson, A.; Fouts, J.; Foster, M. Adipose tissue, obesity and adipokines: Role in cancer promotion. Horm. Mol. Biol. Clin. Investig. 2015, 21, 57–74. [Google Scholar] [CrossRef]
- Coelho, P.; Almeida, J.; Prudêncio, C.; Fernandes, R.; Soares, R. Effect of adipocyte secretome in melanoma progression and vasculogenic mimicry. J. Cell. Biochem. 2016, 117, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
- Robado de Lope, L.; Alcíbar, O.L.; Amor López, A.; Hergueta-Redondo, M.; Peinado, H. Tumour-adipose tissue crosstalk: Fuelling tumour metastasis by extracellular vesicles. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160485. [Google Scholar] [CrossRef] [PubMed]
- Malvi, P.; Chaube, B.; Pandey, V.; Vijayakumar, M.V.; Boreddy, P.R.; Mohammad, N.; Singh, S.V.; Bhat, M.K. Obesity induced rapid melanoma progression is reversed by orlistat treatment and dietary intervention: Role of adipokines. Mol. Oncol. 2015, 9, 689–703. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; Arabi, S.; Lelliott, E.J.; McArthur, G.A.; Sheppard, K.E. Obesity and the impact on cutaneous melanoma: Friend or foe? Cancers 2020, 12, 1583. [Google Scholar] [CrossRef] [PubMed]
- Oba, J.; Wei, W.; Gershenwald, J.E.; Johnson, M.M.; Wyatt, C.M.; Ellerhorst, J.A.; Grimm, E.A. Elevated serum leptin levels are associated with an increased risk of sentinel lymph node metastasis in cutaneous melanoma. Medicine 2016, 95, e3073. [Google Scholar] [CrossRef] [PubMed]
- Amjadi, F.; Javanmard, S.H.; Zarkesh-Esfahani, H.; Khazaei, M.; Narimani, M. Leptin promotes melanoma tumor growth in mice related to increasing circulating endothelial progenitor cells numbers and plasma NO production. J. Exp. Clin. Cancer Res. 2011, 30, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katira, A.; Tan, P.H. Evolving role of adiponectin in cancer-controversies and update. Cancer Biol. Med. 2016, 13, 101–119. [Google Scholar] [CrossRef] [Green Version]
- Pandey, V.; Vijayakumar, M.V.; Ajay, A.K.; Malvi, P.; Bhat, M.K. Diet-induced obesity increases melanoma progression: Involvement of Cav-1 and FASN. Int. J. Cancer 2012, 130, 497–508. [Google Scholar] [CrossRef]
- Pereira, F.V.; Melo, A.C.L.; Silva, M.B.; de Melo, F.M.; Terra, F.F.; Castro, I.A.; Perandini, L.A.; Miyagi, M.T.; Sato, F.T.; Origassa, C.S.T.; et al. Interleukin-6 and the gut microbiota influence melanoma progression in obese mice. Nutr. Cancer 2021, 73, 642–651. [Google Scholar] [CrossRef]
- Gilbert, C.A.; Slingerland, J.M. Cytokines, obesity, and cancer: New insights on mechanisms linking obesity to cancer risk and progression. Annu. Rev. Med. 2013, 64, 45–57. [Google Scholar] [CrossRef]
- Chen, G.-L.; Luo, Y.; Eriksson, D.; Meng, X.; Qian, C.; Bäuerle, T.; Chen, X.-X.; Schett, G.; Bozec, A. High fat diet increases melanoma cell growth in the bone marrow by inducing osteopontin and interleukin 6. Oncotarget 2016, 7, 26653–26669. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chiang, H.-C.; Sun, X.; Yuan, B.; Mitra, P.; Hu, Y.; Curiel, T.J.; Li, R. Genetic ablation of adipocyte PD-L1 reduces tumor growth but accentuates obesity-associated inflammation. J. Immunother. Cancer 2020, 8, e000964. [Google Scholar] [CrossRef]
- Logozzi, M.; De Milito, A.; Lugini, L.; Borghi, M.; Calabrò, L.; Spada, M.; Perdicchio, M.; Marino, M.L.; Federici, C.; Iessi, E.; et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS ONE 2009, 4, e5219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, M.; Chen, J.; Ye, Y.; Tseng, H.-Y.; Lai, F.; Tay, K.H.; Jin, L.; Guo, S.T.; Jiang, C.C.; Zhang, X.D. Adipocytes contribute to resistance of human melanoma cells to chemotherapy and targeted therapy. Curr. Med. Chem. 2014, 21, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Jiang, L.; Ding, C.; Hu, B.; Loh, X.J.; Li, Z.; Wu, Y.-L. Surfactant free delivery of docetaxel by poly[(R)-3-hydroxybutyrate-(R)-3-hydroxyhexanoate]-based polymeric micelles for effective melanoma treatments. Adv. Healthc. Mater. 2018, 7, e1801221. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Zoico, E.; Darra, E.; Rizzatti, V.; Tebon, M.; Franceschetti, G.; Mazzali, G.; Rossi, A.P.; Fantin, F.; Zamboni, M. Role of Adipose tissue in melanoma cancer microenvironment and progression. Int. J. Obes. 2018, 42, 344–352. [Google Scholar] [CrossRef]
- Hood, J.L. Natural melanoma-derived extracellular vesicles. Semin. Cancer Biol. 2019, 59, 251–265. [Google Scholar] [CrossRef]
- Mannavola, F.; D’Oronzo, S.; Cives, M.; Stucci, L.S.; Ranieri, G.; Silvestris, F.; Tucci, M. Extracellular vesicles and epigenetic modifications are hallmarks of melanoma progression. Int. J. Mol. Sci. 2019, 21, 52. [Google Scholar] [CrossRef] [Green Version]
- Xi, F.-X.; Wei, C.-S.; Xu, Y.-T.; Ma, L.; He, Y.-L.; Shi, X.-E.; Yang, G.-S.; Yu, T.-Y. MicroRNA-214-3p targeting Ctnnb1 promotes 3T3-L1 preadipocyte differentiation by interfering with the Wnt/β-catenin signaling pathway. Int. J. Mol. Sci. 2019, 20, 1816. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Barry, S.; Kmetz, D.; Egger, M.; Pan, J.; Rai, S.N.; Qu, J.; McMasters, K.M.; Hao, H. Melanoma cell-derived exosomes promote epithelial-mesenchymal transition in primary melanocytes through paracrine/autocrine signaling in the tumor microenvironment. Cancer Lett. 2016, 376, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Rüger, R. The multiple roles of exosomes in metastasis. Cancer Genom. Proteom. 2017, 14, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, E.; Lazar, I.; Attané, C.; Carrié, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef] [PubMed]
- Lazar, I.; Clement, E.; Dauvillier, S.; Milhas, D.; Ducoux-Petit, M.; LeGonidec, S.; Moro, C.; Soldan, V.; Dalle, S.; Balor, S.; et al. Adipocyte exosomes promote melanoma aggressiveness through fatty acid axidation: A novel mechanism linking obesity and cancer. Cancer Res. 2016, 76, 4051–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunniff, B.; McKenzie, A.J.; Heintz, N.H.; Howe, A.K. AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol. Biol. Cell 2016, 27, 2662–2674. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Mitochondria on the move: Emerging paradigms of organelle trafficking in tumour plasticity and metastasis. Br. J. Cancer 2017, 117, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Kushiro, K.; Chu, R.A.; Verma, A.; Núñez, N.P. Adipocytes promote B16BL6 melanoma cell invasion and the epithelial-to-mesenchymal transition. Cancer Microenviron. 2012, 5, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Parikh, R.; Jacob, E.; Vaknine, H.; Zemser-Werner, V.; Hershkovitz, D.; Malcov, H.; Leibou, S.; Reichman, H.; Sheinboim, D.; et al. Adipocytes sensitize melanoma cells to environmental TGF-β cues by repressing the expression of MiR-211. Sci. Signal. 2019, 12, eaav6847. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.-H.; Um, J.-Y.; Lee, S.-G.; Yang, W.M.; Sethi, G.; Ahn, K.S. Conditioned media from adipocytes promote proliferation, migration, and invasion in melanoma and colorectal cancer cells. J. Cell. Physiol. 2019, 234, 18249–18261. [Google Scholar] [CrossRef]
- McQuade, J.L.; Ologun, G.O.; Arora, R.; Wargo, J.A. Gut microbiome modulation via fecal microbiota transplant to augment immunotherapy in patients with melanoma or other cancers. Curr. Oncol. Rep. 2020, 22, 74. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [Green Version]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Panebianco, C.; Andriulli, A.; Pazienza, V. Pharmacomicrobiomics: Exploiting the drug-microbiota interactions in anticancer therapies. Microbiome 2018, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Galluzzi, L.; Viaud, S.; Vétizou, M.; Daillère, R.; Merad, M.; Kroemer, G. Cancer and the gut microbiota: An unexpected link. Sci. Transl. Med. 2015, 7, 271ps1. [Google Scholar] [CrossRef] [Green Version]
- Mego, M.; Holec, V.; Drgona, L.; Hainova, K.; Ciernikova, S.; Zajac, V. Probiotic bacteria in cancer patients undergoing chemotherapy and radiation therapy. Complementary Ther. Med. 2013, 21, 712–723. [Google Scholar] [CrossRef]
- Patel, R.M.; Denning, P.W. Therapeutic use of prebiotics, probiotics, and postbiotics to prevent necrotizing enterocolitis: What is the current evidence? Clin. Perinatol. 2013, 40, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.K.; Barbulescu, P.; Coburn, B.; Reguera-Nuñez, E. Therapeutic Interventions and Mechanisms Associated with Gut Microbiota-Mediated Modulation of Immune Checkpoint Inhibitor Responses. Microbes Infect. 2021, 23, 104804. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubin, K.; Callahan, M.K.; Ren, B.; Khanin, R.; Viale, A.; Ling, L.; No, D.; Gobourne, A.; Littmann, E.; Huttenhower, C.; et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat. Commun. 2016, 7, 10391. [Google Scholar] [CrossRef] [Green Version]
- Freitag, T.L.; Hartikainen, A.; Jouhten, H.; Sahl, C.; Meri, S.; Anttila, V.-J.; Mattila, E.; Arkkila, P.; Jalanka, J.; Satokari, R. Minor effect of antibiotic pre-treatment on the engraftment of donor microbiota in fecal transplantation in mice. Front. Microbiol. 2019, 10, 2685. [Google Scholar] [CrossRef]
- Machowinski, A.; Krämer, H.-J.; Hort, W.; Mayser, P. Pityriacitrin—A potent UV filter produced by Malassezia furfur and its effect on human skin microflora. Mycoses 2006, 49, 388–392. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Butcher, A.M.; Trzoss, L.L.; Nam, S.-J.; Shirakawa, K.T.; Zhou, W.; Oh, J.; Otto, M.; Fenical, W.; et al. A commensal strain of Staphylococcus epidermidis protects against skin neoplasia. Sci. Adv. 2018, 4, eaao4502. [Google Scholar] [CrossRef] [Green Version]
- Mrázek, J.; Mekadim, C.; Kučerová, P.; Švejstil, R.; Salmonová, H.; Vlasáková, J.; Tarasová, R.; Čížková, J.; Červinková, M. Melanoma-related changes in skin microbiome. Folia Microbiol. 2019, 64, 435–442. [Google Scholar] [CrossRef]
- Gracia-Cazaña, T.; González, S.; Parrado, C.; Juarranz, Á.; Gilaberte, Y. Influence of the exposome on skin cancer. Actas Dermosifiliogr. (Engl. Ed.) 2020, 111, 460–470. [Google Scholar] [CrossRef]
- Montagut, C.; Settleman, J. Targeting the RAF-MEK-ERK Pathway in Cancer Therapy. Cancer Lett. 2009, 283, 125–134. [Google Scholar] [CrossRef]
- Young, H.L.; Rowling, E.J.; Bugatti, M.; Giurisato, E.; Luheshi, N.; Arozarena, I.; Acosta, J.-C.; Kamarashev, J.; Frederick, D.T.; Cooper, Z.A.; et al. An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. J. Exp. Med. 2017, 214, 1691–1710. [Google Scholar] [CrossRef] [Green Version]
- Lichterman, J.N.; Reddy, S.M. Mast Cells: A new frontier for cancer immunotherapy. Cells 2021, 10, 1270. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Hamid, O.; Gonzalez, R.; Infante, J.R.; Patel, M.R.; Hodi, F.S.; Lewis, K.D.; Tawbi, H.A.; Hernandez, G.; Wongchenko, M.J.; et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat. Med. 2019, 25, 929–935. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Yang, L.; Lei, S.; Tan, W.; Long, J. NEDD4 negatively regulates GITR via ubiquitination in immune microenvironment of melanoma. OncoTargets Ther. 2019, 12, 10629–10637. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.-J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Boudewijns, S.; Koornstra, R.H.T.; Westdorp, H.; Schreibelt, G.; van den Eertwegh, A.J.M.; Geukes Foppen, M.H.; Haanen, J.B.; de Vries, I.J.M.; Figdor, C.G.; Bol, K.F.; et al. Ipilimumab administered to metastatic melanoma patients who progressed after dendritic cell vaccination. OncoImmunology 2016, 5, e1201625. [Google Scholar] [CrossRef]
- Ledford, H. Cancer-fighting viruses win approval. Nature 2015, 526, 622–623. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Diab, A.; Tannir, N.M.; Bentebibel, S.-E.; Hwu, P.; Papadimitrakopoulou, V.; Haymaker, C.; Kluger, H.M.; Gettinger, S.N.; Sznol, M.; Tykodi, S.S.; et al. Bempegaldesleukin (NKTR-214) plus nivolumab in patients with advanced solid tumors: Phase I dose-escalation study of safety, efficacy, and immune activation (PIVOT-02). Cancer Discov. 2020, 10, 1158–1173. [Google Scholar] [CrossRef]
- Everly, J.J.; Lonial, S. Immunomodulatory effects of human recombinant granulocyte-macrophage colony-stimulating factor (RhuGM-CSF): Evidence of antitumour activity. Expert Opin. Biol. Ther. 2005, 5, 293–311. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Haanen, J.B.A.G.; Robert, C. Immune checkpoint inhibitors. Prog. Tumor Res. 2015, 42, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef]
- Tang, T.; Huang, X.; Zhang, G.; Hong, Z.; Bai, X.; Liang, T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 72. [Google Scholar] [CrossRef]



{kind=link}
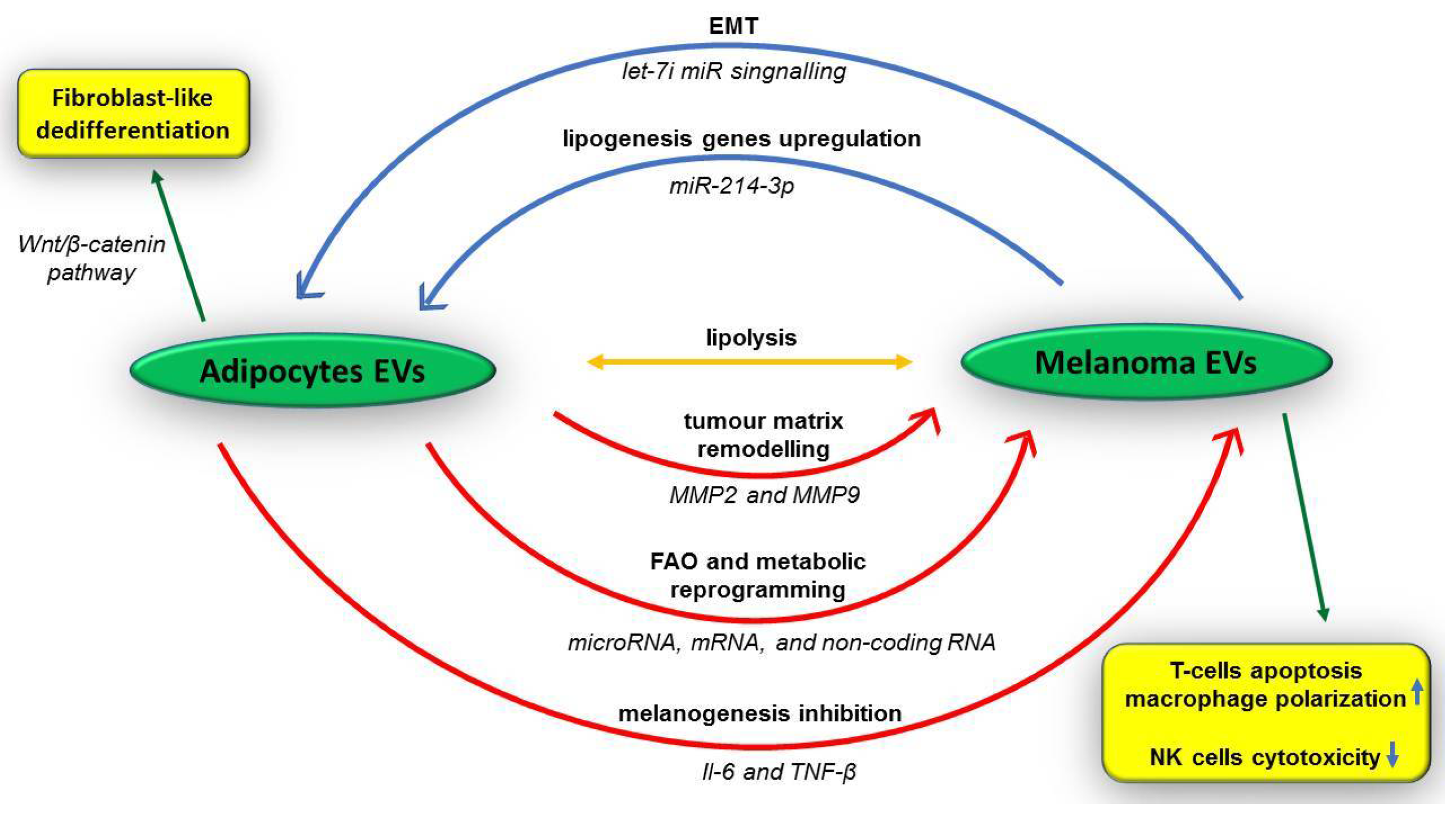{kind=link}
| MM Associated with UV Exposure | MM Not Consistently Associated with UV Exposure | ||||
|---|---|---|---|---|---|
| Low-CDS (intermittent sun exposure) | High-CSD (chronic sun exposure) | mucosal melanoma; melanoma arising in congenital nevi; melanoma arising in blue nevi; Spitz melanoma; acral melanoma; uveal melanoma; nevoid melanoma; nodular melanoma | |||
| superficial spreading melanoma | subset of nodular melanoma | lentigo maligna melanoma | desmoplastic melanoma | subset of nodular melanoma | |
| RGP; young age; precursor lesion (nevi) | VGP; young age; precursor lesion (nevi) | RGP; old age; precursor lesion (MM in situ) | VGP; old age; precursor lesion (nevi); de novo | VGP; young age; precursor lesion (nevi) | VGP and RGP; all ages; precursor lesion (nevi) |
| GENOMIC | |||||
| BRAF (>50%); NRAS (25%); other mutations (rare) | NF1; NRAS; KIT | NF1; NFKBIE; MAPK | NRAS; KIT; BRAF; CDKN2A; TP53; TERT | HRAS; ROS1; NTRK1 and NTRK3; CCND1; TERT; GNAQ; BRAF (10%); KIT (20%); NRAS (15–20%) | |
| Marker/Protein | Locations | IHC Features | Functions |
|---|---|---|---|
| Melan A (MART1) |
|
|
|
| MITF |
|
|
|
| HMB45 |
|
|
|
| SOX10 |
|
|
|
| S-100 |
|
|
|
| Genes | Incidence | Pathway | Actions | MMs Type |
|---|---|---|---|---|
| BRAF (BRAFV600E BRAFV600 BRAFV600K BRAFK601E) | 45% |
|
|
|
| <10% |
| |||
| RAS | 15–30% |
|
|
|
| NRAS | 15% |
|
|
|
| c-KIT (CD117) | <3% |
|
|
|
| 40% |
| |||
| ATRX | 9.11% |
|
|
|
| ARID2 | 13.32% |
|
|
|
| SETD2 | 9.48% |
|
|
|
| GNAQ/GNA11 | 80–90% |
|
|
|
| BAP1 | 6.13% |
|
|
|
| SF3B1 | 33% |
|
|
|
| NF1 | 10–15% |
|
|
|
| RAC1 | 9.2% |
|
|
|
| TERT | 14% |
|
|
|
| KRAS | 2.9% |
|
|
|
| ERBB2/4 | 3.29% |
|
|
|
| CDKN2A | 25–35% |
|
|
|
| TP53 | 15% |
|
|
|
| PTEN | 14% |
|
|
|
| MAP2K1/2 | 10% |
|
|
|
| Type of Molecules | Marker |
|---|---|
| Enzymes |
|
| Soluble proteins and/or antigens |
|
| Melanin-related metabolites |
|
| Circulating cell-free nucleic acids |
|
| Marker/Pathway | Function/Advantage |
|---|---|
| Nodal embryonic signalling |
|
| Nestin |
|
| hTERT |
|
| SOX2 |
|
| SOX10 |
|
| CD20 (MS4A1) |
|
| CD44 |
|
| CD49d/CD29 (α4β1 integrin heterodimer) |
|
| CD49f (Integrin α6) | |
| CD54/ICAM-1 |
|
| CD57/HNK-1 |
|
| CD86/B7-2 |
|
| CD117 (c-KIT) |
|
| CD133 (prominin-1) |
|
| CD144 (vascular-endothelial VE-cadherin) |
|
| CD146/MCAM |
|
| CD166/ALCAM |
|
| CD271/NGFR |
|
| N-cadherin |
|
| miR-10b miR-21 miR200c miR-373 miR-520c |
|
| MDR1 |
|
| L1CAM |
|
| ALDH1A1/A2/A3 |
|
| ABC transporters (ABCB5, ABCG2/BCRP, ABCC1/MRP, ABCC2, ABCC6) |
|
| PD-1 PDL-1 |
|
| CTLA-4 |
|
| RANK |
|
| HIF-1 |
|
| Snail |
|
| Notch4 |
|
| γ-secretase |
|
| Bcl-2 |
|
| GLI (Hh/Glimo) |
|
| DDX3X |
|
| VEGFR-1 |
|
| VEGFR-2 VEGF Ang1/2 Tie2 |
|
| CXCR6 |
|
| JARID1B |
|
| EZH2 |
|
| Histone marks H3K4me2 H3K27me3 H3K9ac |
|
| Hn (heterogeneous ribonucleoproteins) hnRNPs hnRNP A2B1 hnRNP I hnRNP L |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amalinei, C.; Grigoraș, A.; Lozneanu, L.; Căruntu, I.-D.; Giușcă, S.-E.; Balan, R.A. The Interplay between Tumour Microenvironment Components in Malignant Melanoma. Medicina 2022, 58, 365. https://doi.org/10.3390/medicina58030365
Amalinei C, Grigoraș A, Lozneanu L, Căruntu I-D, Giușcă S-E, Balan RA. The Interplay between Tumour Microenvironment Components in Malignant Melanoma. Medicina. 2022; 58(3):365. https://doi.org/10.3390/medicina58030365
Chicago/Turabian StyleAmalinei, Cornelia, Adriana Grigoraș, Ludmila Lozneanu, Irina-Draga Căruntu, Simona-Eliza Giușcă, and Raluca Anca Balan. 2022. "The Interplay between Tumour Microenvironment Components in Malignant Melanoma" Medicina 58, no. 3: 365. https://doi.org/10.3390/medicina58030365