
Identification and Characterization of Novel Mutations in Chronic Kidney Disease (CKD) and Autosomal Dominant Polycystic Kidney Disease (ADPKD) in Saudi Subjects by Whole-Exome Sequencing
 , , ,
, , ,  ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Methods, Materials, and Subjects
2.1. Patients and Family Recruitment
2.2. Ethical Approval and Participants’ Consent
2.3. DNA Extraction
2.4. DNA Library Preparation
2.5. Next-Generation Sequencing (NGS) Technology
2.6. Variant Filtration
2.7. In Silico Analysis
2.8. Bioinformatics
2.9. Statistical Analysis
3. Results
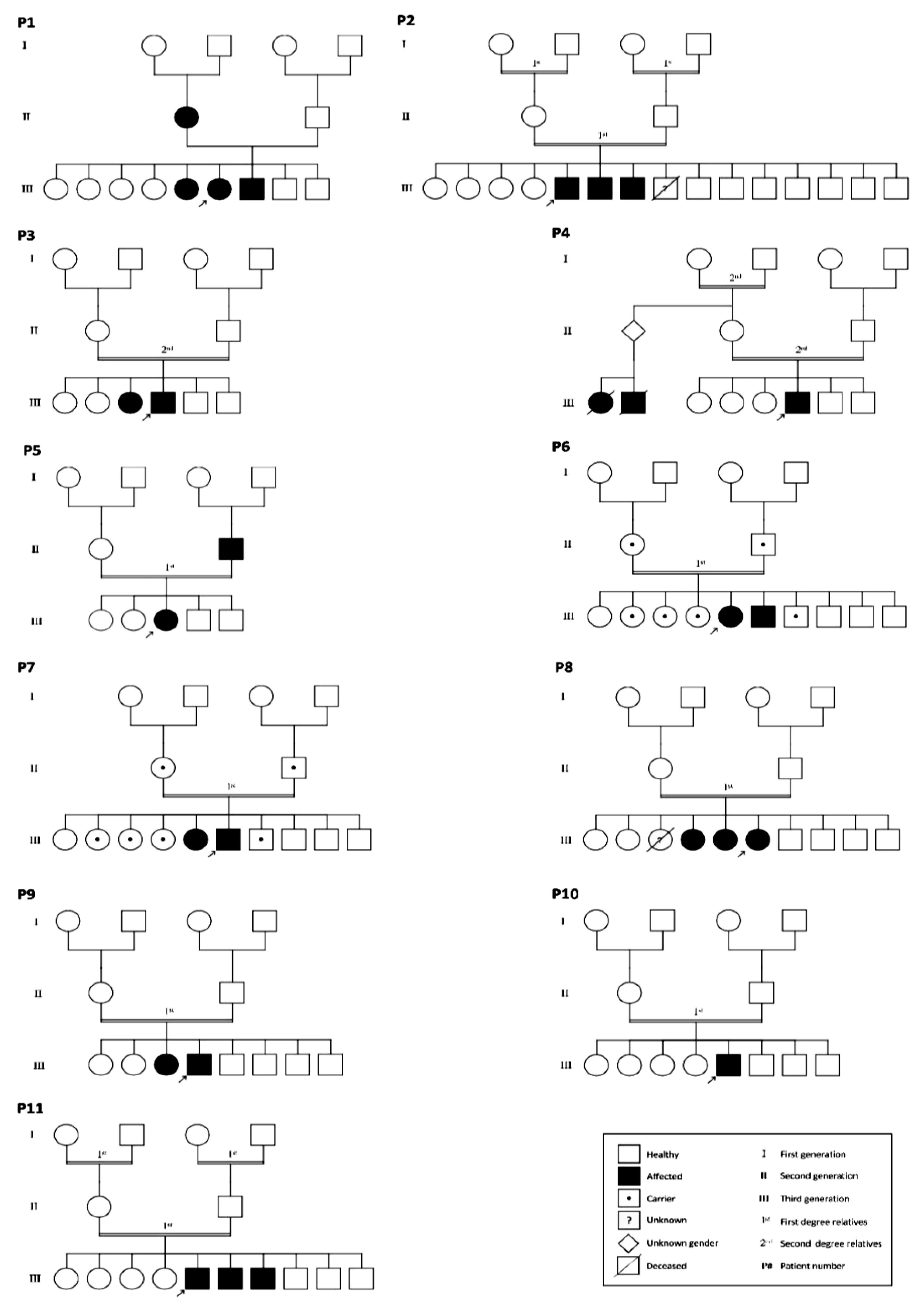
3.1. Basic Clinical Characteristics and Genetic Analysis of the Studied Subjects
3.2. Mutations Identified in Patients with ADPKD
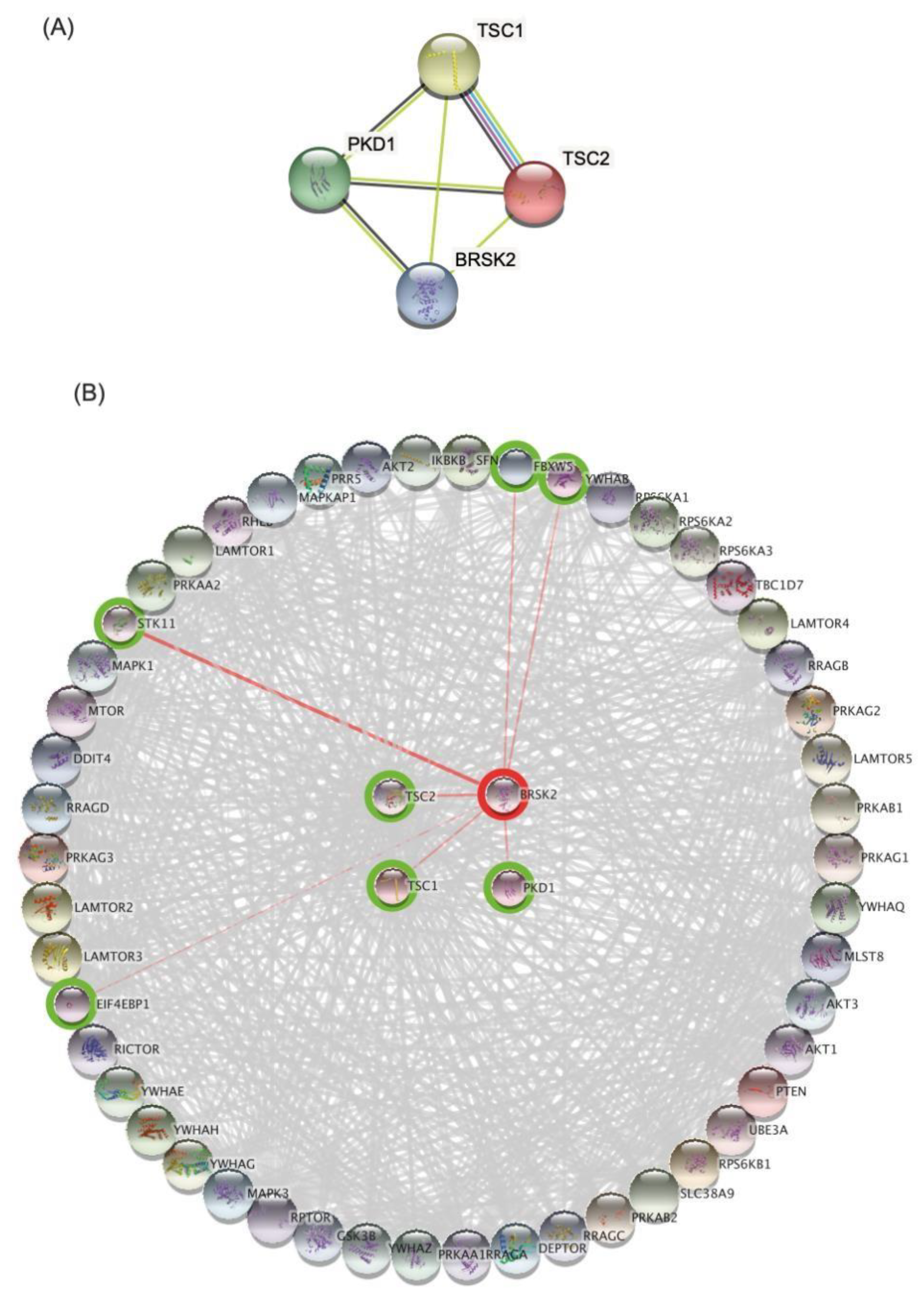
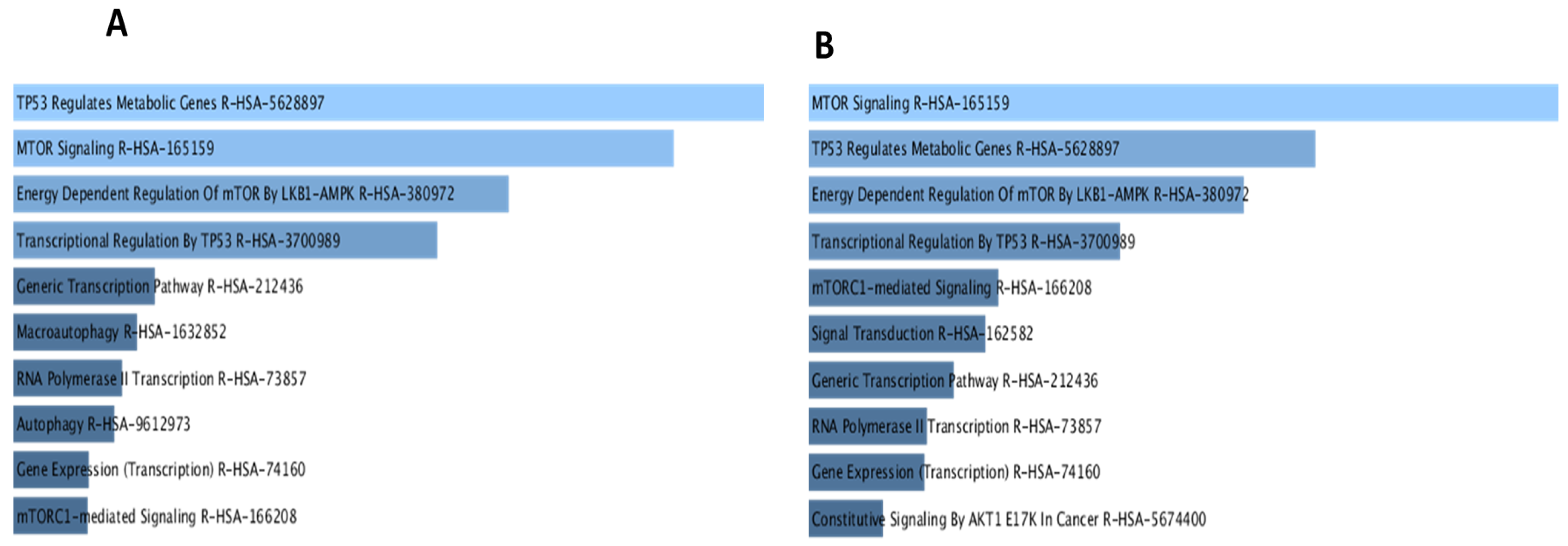
3.3. Bioinformatics Analysis and Data Mining Highlight Potential Proteins Interactions between PKD1 and TSC2 via BRSK2
3.4. Mutation and Allele: Frequency
4. Discussion
4.1. Identification of Novel Variants with a Potential Contribution to the Evolution and Pathogenesis of ADPKD in Saudi Patients
4.2. Potential Damaging Impacts of the Detected Mutations on the Protein Structure and Function
4.3. Prediction of Protein–Protein Interactions Demonstrated a Common Pathogenic Pathway
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Igarashi, P.; Somlo, S. Polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1371–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C. ARPKD and early manifestations of ADPKD: The original polycystic kidney disease and phenocopies. Pediatr. Nephrol. 2015, 30, 15–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahbari-Oskoui, F.; Williams, O.; Chapman, A. Mechanisms and management of hypertension in autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2014, 29, 2194–2201. [Google Scholar] [CrossRef] [Green Version]
- Levey, A.S.; Atkins, R.; Coresh, J.; Cohen, E.P.; Collins, A.J.; Eckardt, K.U.; Nahas, M.E.; Jaber, B.L.; Jadoul, M.; Levin, A.; et al. Chronic kidney disease as a global public health problem: Approaches and initiatives—A position statement from Kidney Disease Improving Global Outcomes. Kidney Int. 2007, 72, 247–259. [Google Scholar] [CrossRef] [Green Version]
- Lanktree, M.B.; Haghighi, A.; Guiard, E.; Iliuta, I.-A.; Song, X.; Harris, P.C.; Paterson, A.D.; Pei, Y. Prevalence estimates of polycystic kidney and liver disease by population sequencing. J. Am. Soc. Nephrol. 2018, 29, 2593–2600. [Google Scholar] [CrossRef] [Green Version]
- Hopp, K.; Ward, C.J.; Hommerding, C.J.; Nasr, S.H.; Tuan, H.-F.; Gainullin, V.G.; Rossetti, S.; Torres, V.E.; Harris, P.C. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Investig. 2012, 122, 4257–4273. [Google Scholar] [CrossRef] [Green Version]
- Lanktree, M.B.; Haghighi, A.; di Bari, I.; Song, X.; Pei, Y. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin. J. Am. Soc. Nephrol. 2021, 16, 790–799. [Google Scholar] [CrossRef]
- Harris, P.C.; Torres, V.E. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Investig. 2014, 124, 2315–2324. [Google Scholar] [CrossRef] [Green Version]
- Rossetti, S.; Consugar, M.B.; Chapman, A.B.; Torres, V.E.; Guay-Woodford, L.M.; Grantham, J.J.; Bennett, W.M.; Meyers, C.M.; Walker, D.L.; Bae, K.; et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 2143–2160. [Google Scholar] [CrossRef] [Green Version]
- Grieben, M.; Pike, A.C.; Shintre, C.A.; Venturi, E.; El-Ajouz, S.; Tessitore, A.; Shrestha, L.; Mukhopadhyay, S.; Mahajan, P.; Chalk, R.; et al. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2). Nat. Struct. Mol. Biol. 2017, 24, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Audrezet, M.P.; Cornec-Le Gall, E.; Chen, J.M.; Redon, S.; Quere, I.; Creff, J.; Benech, C.; Maestri, S.; Le Meur, Y.; Ferec, C. Autosomal dominant polycystic kidney disease: Comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum. Mutat. 2012, 33, 1239–1250. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Blais, J.D.; Irazabal, M.V.; Devuyst, O.; Gansevoort, R.T.; Perrone, R.D.; Chapman, A.B.; Czerwiec, F.S.; Ouyang, J.; Heyer, C.M. Can we further enrich autosomal dominant polycystic kidney disease clinical trials for rapidly progressive patients? Application of the PROPKD score in the TEMPO trial. Nephrol. Dial. Transplant. 2018, 33, 645–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimpel, C.; Bergmann, C.; Bockenhauer, D.; Breysem, L.; Cadnapaphornchai, M.A.; Cetiner, M.; Dudley, J.; Emma, F.; Konrad, M.; Harris, T. International consensus statement on the diagnosis and management of autosomal dominant polycystic kidney disease in children and young people. Nat. Rev. Nephrol. 2019, 15, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.-H.; Barua, M.; McNaught, A.; Khalili, K.; Pei, Y. Imaging-Based Diagnosis of Autosomal Dominant Polycystic Kidney Disease. In Polycystic Kidney Disease; Springer: Berlin/Heidelberg, Germany, 2018; pp. 133–142. [Google Scholar]
- Iliuta, I.-A.; Kalatharan, V.; Wang, K.; Cornec-Le Gall, E.; Conklin, J.; Pourafkari, M.; Ting, R.; Chen, C.; Borgo, A.C.; He, N. Polycystic kidney disease without an apparent family history. J. Am. Soc. Nephrol. 2017, 28, 2768–2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chebib, F.T.; Torres, V.E. Recent advances in the management of autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 1765–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanktree, M.B.; Chapman, A.B. New treatment paradigms for ADPKD: Moving towards precision medicine. Nat. Rev. Nephrol. 2017, 13, 750–768. [Google Scholar] [CrossRef]
- Bullich, G.; Domingo-Gallego, A.; Vargas, I.; Ruiz, P.; Lorente-Grandoso, L.; Furlano, M.; Fraga, G.; Madrid, Á.; Ariceta, G.; Borregán, M. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 2018, 94, 363–371. [Google Scholar] [CrossRef]
- Bondagji, N.S. Antenatal diagnosis, prevalence and outcome of congenital anomalies of the kidney and urinary tract in Saudi Arabia. Urol. Ann. 2014, 6, 36. [Google Scholar] [CrossRef]
- El-Hazmi, M.; Al-Swailem, A.; Warsy, A.; Al-Swailem, A.; Sulaimani, R.; Al-Meshari, A. Consanguinity among the Saudi Arabian population. J. Med. Genet. 1995, 32, 623–626. [Google Scholar] [CrossRef] [Green Version]
- Alkuraya, F.S. Autozygome decoded. Genet. Med. 2010, 12, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, T.K. Genetically transmitted renal diseases in children: A saudi perspective. Saudi J. Kidney Dis. Transplant. 1998, 9, 105. [Google Scholar]
- Al-Hamed, M.H.; Alsahan, N.; Rice, S.J.; Edwards, N.; Nooreddeen, E.; Alotaibi, M.; Kurdi, W.; Alnemer, M.; Altaleb, N.; Ali, W. Bialleleic PKD1 mutations underlie early-onset autosomal dominant polycystic kidney disease in Saudi Arabian families. Pediatr. Nephrol. 2019, 34, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Al-Muhanna, F.A.; Al-Rubaish, A.M.; Vatte, C.; Mohiuddin, S.S.; Cyrus, C.; Ahmad, A.; Shakil Akhtar, M.; Albezra, M.A.; Alali, R.A.; Almuhanna, A.F. Exome sequencing of Saudi Arabian patients with ADPKD. Renal Fail. 2019, 41, 842–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Muhanna, F.A.; Malhotra, K.K.; Saeed, I.; Al-Mueilo, S. Autosomal dominant polycystic kidney disease: Observations from a university hospital in Saudi Arabia. Saudi J. Kidney Dis. Transplant. 1995, 6, 28. [Google Scholar]
- Al-Amer, O.M.; Oyouni, A.A.A.; Alshehri, M.A.; Alasmari, A.; Alzahrani, O.R.; Aljohani, S.A.S.; Alasmael, N.; Theyab, A.; Algahtani, M.; Al Sadoun, H.; et al. Association of SNPs within TMPRSS6 and BMP2 genes with iron deficiency status in Saudi Arabia. PLoS ONE 2021, 16, e0257895. [Google Scholar] [CrossRef]
- Alanazi, I.O.; Shaik, J.P.; Parine, N.R.; Azzam, N.A.; Alharbi, O.; Hawsawi, Y.M.; Oyouni, A.A.A.; Al-Amer, O.M.; Alzahrani, F.; Almadi, M.A.; et al. Association of HER1 and HER2 Gene Variants in the Predisposition of Colorectal Cancer. J. Oncol. 2021, 2021, 6180337. [Google Scholar] [CrossRef]
- Alrefaei, A.F.; Hawsawi, Y.M.; Almaleki, D.; Alafif, T.; Alzahrani, F.A.; Bakhrebah, M.A. Genetic data sharing and artificial intelligence in the era of personalized medicine based on a cross-sectional analysis of the Saudi human genome program. Sci. Rep. 2022, 12, 1405. [Google Scholar] [CrossRef]
- Alsohime, F.; Almaghamsi, T.; Basha, T.A.; Alardati, H.; Alghamdi, M.; Hawsawi, Y.M. Unusual Prominent Pulmonary Involvement in a Homozygous PRF1 Gene Variant in a Female Patient. J. Clin. Immunol. 2021, 41, 217–220. [Google Scholar] [CrossRef]
- Rath, S.; Hawsawi, Y.M.; Alzahrani, F.; Khan, M.I. Epigenetic regulation of inflammation: The metabolomics connection. Semin. Cell Dev. Biol. 2022. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protocols 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, R.; Elfaki, I.; Duhier, F.M.A.; Alotaibi, M.A.; AlAlawy, A.I.; Barnawi, J.; Babakr, A.T.; Mir, M.M.; Mirghani, H.; Hamadi, A.; et al. Molecular Determination of mirRNA-126 rs4636297, Phosphoinositide-3-Kinase Regulatory Subunit 1-Gene Variability rs7713645, rs706713 (Tyr73Tyr), rs3730089 (Met326Ile) and Their Association with Susceptibility to T2D. J. Pers. Med. 2021, 11, 861. [Google Scholar] [CrossRef]
- Saiyin, H.; Na, N.; Han, X.; Fang, Y.; Wu, Y.; Lou, W.; Yang, X. BRSK2 induced by nutrient deprivation promotes Akt activity in pancreatic cancer via downregulation of mTOR activity. Oncotarget 2017, 8, 44669–44681. [Google Scholar] [CrossRef] [Green Version]
- Margaria, J.P.; Campa, C.C.; De Santis, M.C.; Hirsch, E.; Franco, I. The PI3K/Akt/mTOR pathway in polycystic kidney disease: A complex interaction with polycystins and primary cilium. Cell Signal. 2020, 66, 109468. [Google Scholar] [CrossRef]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [Green Version]
- Alzahrani, F.A.; Ahmed, F.; Sharma, M.; Rehan, M.; Mahfuz, M.; Baeshen, M.N.; Hawsawi, Y.; Almatrafi, A.; Alsagaby, S.A.; Kamal, M.A.; et al. Investigating the pathogenic SNPs in BLM helicase and their biological consequences by computational approach. Sci. Rep. 2020, 10, 12377. [Google Scholar] [CrossRef]
- Alzahrani, F.A.; Hawsawi, Y.M.; Altayeb, H.N.; Alsiwiehri, N.O.; Alzahrani, O.R.; Alatwi, H.E.; Al-Amer, O.M.; Alomar, S.; Mansour, L. In silico modeling of the interaction between TEX19 and LIRE1, and analysis of TEX19 gene missense SNPs. Mol. Genet. Genom. Med. 2021, 9, e1707. [Google Scholar] [CrossRef] [PubMed]
- El-Kattawy, A.M.; Algezawy, O.; Alfaifi, M.Y.; Noseer, E.A.; Hawsawi, Y.M.; Alzahrani, O.R.; Algarni, A.; Kahilo, K.A.; El-Magd, M.A. Therapeutic potential of camel milk exosomes against HepaRG cells with potent apoptotic, anti-inflammatory, and anti-angiogenesis effects for colostrum exosomes. Biomed. Pharmacother. 2021, 143, 112220. [Google Scholar] [CrossRef] [PubMed]
- Semlali, A.; Almutairi, M.; Rouabhia, M.; Reddy Parine, N.; Al Amri, A.; Al-Numair, N.S.; Hawsawi, Y.M.; Saud Alanazi, M. Novel sequence variants in the TLR6 gene associated with advanced breast cancer risk in the Saudi Arabian population. PLoS ONE 2018, 13, e0203376. [Google Scholar] [CrossRef] [PubMed]
- Semlali, A.; Parine, N.R.; Al-Numair, N.S.; Almutairi, M.; Hawsawi, Y.M.; Amri, A.A.; Aljebreen, A.M.; Arafah, M.; Almadi, M.A.; Azzam, N.A.; et al. Potential role of Toll-like receptor 2 expression and polymorphisms in colon cancer susceptibility in the Saudi Arabian population. Onco Targets Ther. 2018, 11, 8127–8141. [Google Scholar] [CrossRef] [Green Version]
- Elisakova, V.; Merta, M.; Reiterova, J.; Baxova, A.; Kotlas, J.; Hirschfeldova, K.; Obeidova, L.; Tesar, V.; Stekrova, J. Bilineal inheritance of pathogenic PKD1 and PKD2 variants in a Czech family with autosomal dominant polycystic kidney disease—A case report. BMC Nephrol. 2018, 19, 163. [Google Scholar] [CrossRef]
- Fick, G.M.; Johnson, A.M.; Strain, J.D.; Kimberling, W.J.; Kumar, S.; Manco-Johnson, M.L.; Duley, I.T.; Gabow, P.A. Characteristics of very early onset autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1993, 3, 1863–1870. [Google Scholar] [CrossRef]
- Chang, M.-Y.; Chen, H.-M.; Jenq, C.-C.; Lee, S.-Y.; Chen, Y.-M.; Tian, Y.-C.; Chen, Y.-C.; Hung, C.-C.; Fang, J.-T.; Yang, C.-W.; et al. Novel PKD1 and PKD2 mutations in Taiwanese patients with autosomal dominant polycystic kidney disease. J. Hum. Genet. 2013, 58, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Chen, S.C.; Yang, Y.M.; Yan, K.; Qian, Y.Q.; Zhang, J.Y.; Hu, Y.T.; Dong, M.Y.; Jin, F.; Huang, H.F.; et al. Identification of novel PKD1 and PKD2 mutations in a Chinese population with autosomal dominant polycystic kidney disease. Sci. Rep. 2015, 5, 17468. [Google Scholar] [CrossRef] [Green Version]
- Boucher, C.; Sandford, R. Autosomal dominant polycystic kidney disease (ADPKD, MIM 173900, PKD1 and PKD2 genes, protein products known as polycystin-1 and polycystin-2). Eur. J. Hum. Genet. 2004, 12, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Vouk, K.; Strmecki, L.; Stekrova, J.; Reiterova, J.; Bidovec, M.; Hudler, P.; Kenig, A.; Jereb, S.; Zupanic-Pajnic, I.; Balazic, J.; et al. PKD1 and PKD2 mutations in Slovenian families with autosomal dominant polycystic kidney disease. BMC Med. Genet. 2006, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Ali, H.; Hussain, N.; Naim, M.; Zayed, M.; Al-Mulla, F.; Kehinde, E.O.; Seaburg, L.M.; Sundsbak, J.L.; Harris, P.C. A novel PKD1 variant demonstrates a disease-modifying role in trans with a truncating PKD1 mutation in patients with autosomal dominant polycystic kidney disease. BMC Nephrol. 2015, 16, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornec-Le Gall, E.; Audrézet, M.-P.; Chen, J.-M.; Hourmant, M.; Morin, M.-P.; Perrichot, R.; Charasse, C.; Whebe, B.; Renaudineau, E.; Jousset, P. Type of PKD1 mutation influences renal outcome in ADPKD. J. Am. Soc. Nephrol. 2013, 24, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Audrézet, M.-P.; Rousseau, A.; Hourmant, M.; Renaudineau, E.; Charasse, C.; Morin, M.-P.; Moal, M.-C.; Dantal, J.; Wehbe, B. The PROPKD score: A new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 942–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hateboer, N.; Dijk, M.A.; Bogdanova, N.; Coto, E.; Saggar-Malik, A.K.; San Millan, J.L.; Torra, R.; Breuning, M.; Ravine, D.; Group, E.P.-P.S. Comparison of phenotypes of polycystic kidney disease types 1 and 2. Lancet 1999, 353, 103–107. [Google Scholar] [CrossRef]
- Moszyńska, A.; Gebert, M.; Collawn, J.F.; Bartoszewski, R. SNPs in microRNA target sites and their potential role in human disease. Open Biol. 2017, 7, 170019. [Google Scholar] [CrossRef]
- Consugar, M.B.; Wong, W.C.; Lundquist, P.A.; Rossetti, S.; Kubly, V.J.; Walker, D.L.; Rangel, L.J.; Aspinwall, R.; Niaudet, W.P.; Özen, S. Characterization of large rearrangements in autosomal dominant polycystic kidney disease and the PKD1/TSC2 contiguous gene syndrome. Kidney Int. 2008, 74, 1468–1479. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.R.; Maheshwar, M.M.; Aspinwall, R.; Thompson, P.; Cheadle, J.P.; Ravine, D.; Roy, S.; Haan, E.; Bernstein, J.; Harris, P.C. Renal cystic disease in tuberous sclerosis: Role of the polycystic kidney disease 1 gene. Am. J. Hum. Genet. 1997, 61, 843–851. [Google Scholar] [CrossRef] [Green Version]
- Rosset, C.; Netto, C.B.O.; Ashton-Prolla, P. TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: A review. Genet. Mol. Biol. 2017, 40, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Brook-Carter, P.T.; Peral, B.; Ward, C.J.; Thompson, P.; Hughes, J.; Maheshwar, M.M.; Nellist, M.; Gamble, V.; Harris, P.C.; Sampson, J.R. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease—A contiguous gene syndrome. Nat. Genet. 1994, 8, 328–332. [Google Scholar] [CrossRef]
- Cabrera-Lopez, C.; Bullich, G.; Marti, T.; Catala, V.; Ballarin, J.; Bissler, J.J.; Harris, P.C.; Ars, E.; Torra, R. Insight into response to mTOR inhibition when PKD1 and TSC2 are mutated. BMC Med. Genet. 2015, 16, 39. [Google Scholar] [CrossRef] [Green Version]
- Cornec-Le Gall, E.; Audrézet, M.-P.; Renaudineau, E.; Hourmant, M.; Charasse, C.; Michez, E.; Frouget, T.; Vigneau, C.; Dantal, J.; Siohan, P. PKD2-related autosomal dominant polycystic kidney disease: Prevalence, clinical presentation, mutation spectrum, and prognosis. Am. J. Kidney Dis. 2017, 70, 476–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Adelsberg, J. Peptides from the PKD repeats of polycystin, the PKD1 gene product, modulate pattern formation in the developing kidney. Dev. Genet. 1999, 24, 299–308. [Google Scholar] [CrossRef]
- Bycroft, M.; Bateman, A.; Clarke, J.; Hamill, S.J.; Sandford, R.; Thomas, R.L.; Chothia, C. The structure of a PKD domain from polycystin-1: Implications for polycystic kidney disease. EMBO J. 1999, 18, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Weston, B.S.; Malhas, A.N.; Price, R.G. Structure–function relationships of the extracellular domain of the autosomal dominant polycystic kidney disease-associated protein, polycystin-1. FEBS Lett. 2003, 538, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Ibraghimov-Beskrovnaya, O.; Bukanov, N.O.; Donohue, L.C.; Dackowski, W.R.; Klinger, K.W.; Landes, G.M. Strong homophilic interactions of the Ig-like domains of polycystin-1, the protein product of an autosomal dominant polycystic kidney disease gene, PKD1. Hum. Mol. Genet. 2000, 9, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Streets, A.; Ong, A. Post-translational modifications of the polycystin proteins. Cell Signal. 2020, 72, 109644. [Google Scholar] [CrossRef]
- Drickamer, K.; Taylor, M.E. Recent insights into structures and functions of C-type lectins in the immune system. Curr. Opin. Struct. Biol. 2015, 34, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Cummings, R.D.; McEver, R.P. C-Type Lectins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 435–452. [Google Scholar]
- Ward, M.; Hughes, C.F.; Strain, J.J.; Reilly, R.; Cunningham, C.; Molloy, A.M.; Horigan, G.; Casey, M.; McCarroll, K.; O’Kane, M.; et al. Impact of the common MTHFR 677C-->T polymorphism on blood pressure in adulthood and role of riboflavin in modifying the genetic risk of hypertension: Evidence from the JINGO project. BMC Med. 2020, 18, 318. [Google Scholar] [CrossRef]
- Xu, M.; Ma, L.; Bujalowski, P.J.; Qian, F.; Sutton, R.B.; Oberhauser, A.F. Analysis of the REJ Module of Polycystin-1 Using Molecular Modeling and Force-Spectroscopy Techniques. J. Biophys. 2013, 2013, 525231. [Google Scholar] [CrossRef] [Green Version]
- Qian, F.; Boletta, A.; Bhunia, A.K.; Xu, H.; Liu, L.; Ahrabi, A.K.; Watnick, T.J.; Zhou, F.; Germino, G.G. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc. Natl. Acad. Sci. USA 2002, 99, 16981–16986. [Google Scholar] [CrossRef] [Green Version]
- European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993, 75, 1305–1315. [Google Scholar] [CrossRef]
- Dere, R.; Wilson, P.D.; Sandford, R.N.; Walker, C.L. Carboxy terminal tail of polycystin-1 regulates localization of TSC2 to repress mTOR. PLoS ONE 2010, 5, e9239. [Google Scholar] [CrossRef] [PubMed]
- El-Hashemite, N.; Zhang, H.; Henske, E.P.; Kwiatkowski, D.J. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet 2003, 361, 1348–1349. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostov, K.E. mTOR is out of control in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5247–5248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Pejchinovski, M.; Wang, X.; Fu, X.; Castelletti, D.; Watnick, T.J.; Arcaro, A.; Siwy, J.; Mullen, W.; Mischak, H.; et al. Dual mTOR/PI3K inhibition limits PI3K-dependent pathways activated upon mTOR inhibition in autosomal dominant polycystic kidney disease. Sci. Rep. 2018, 8, 5584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiatt, S.M.; Thompson, M.L.; Prokop, J.W.; Lawlor, J.M.J.; Gray, D.E.; Bebin, E.M.; Rinne, T.; Kempers, M.; Pfundt, R.; van Bon, B.W.; et al. Deleterious Variation in BRSK2 Associates with a Neurodevelopmental Disorder. Am. J. Hum. Genet. 2019, 104, 701–708. [Google Scholar] [CrossRef] [Green Version]
- Nie, J.; Han, X.; Shi, Y. SAD-A and AMPK kinases: The “yin and yang” regulators of mTORC1 signaling in pancreatic beta cells. Cell Cycle 2013, 12, 3366–3369. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wan, B.; Li, D.; Zhou, J.; Li, R.; Bai, M.; Chen, F.; Yu, L. BRSK2 is regulated by ER stress in protein level and involved in ER stress-induced apoptosis. Biochem. Biophys. Res. Commun. 2012, 423, 813–818. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein-protein interactions modulators: Mechanisms and clinical trials. Signal. Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Age | Sex | Nationality | Ethnicity | Hypertension | eGFR (mL/min/1.73m2) | CKD stage | Affected Family Members | Other Diseases |
|---|---|---|---|---|---|---|---|---|---|
| P1 | 46 | F | SA | Arab | Yes | NA | G4/A3 | 3 | PKD |
| P2 | 37 | M | SA | Arab | No | 60–89 mL/min/1.73 | G2/A1 | 2 | Hereditary nephritis and CKD |
| P3 | 19 | M | SA | Arab | No | 15 mL/min/1.73 | G5/A3 | 1 | CKD |
| P4 | 18 | M | SA | Arab | Yes | 15 | G5/A3 | 2 | Hereditary nephritis CKD |
| P5 | 40 | F | SA | Arab | Yes | 60 | G2/A3 | 1 | CKD |
| P6 | 33 | F | SA | Arab | Yes | 5 | G5/A3 | 1 and 6 carrier | CKD ESRD |
| P7 | 30 | M | SA | Arab | Yes | 60 | G2/A2 | 1 and 6 carrier | CKD |
| P8 | 30 | F | SA | Arab | Yes | 60–89 | G2/A3 | 2 | CKD |
| P9 | 26 | M | SA | Arab | No | 49 | G3a/A2 | 1 | CKD |
| P10 | 26 | M | SA | Arab | Yes | NA | G4/A3 | 0 | CKD |
| P11 | 31 | M | SA | Arab | No | NA | G5/A3 | 2 | CKD ESRD |
| Patients | Gene | cDNA Change | Protein Change | Mutation Type | Exome No. | Zygosity | Pathogenicity | SNP ID |
|---|---|---|---|---|---|---|---|---|
| P1 | PKD1L1 | c.3813_3814delinsTG | Non-frameshift substitution | 24 | Hetero | Uncertain significance | ||
| P2 | PKD1 | c.4264G > A | p.A1422T | Missense Variant | 15 | Hetero | Uncertain significance | rs140980374 |
| P3 | PKD2 | c.1445T > G | p.F482C | Missense Variant | 6 | Homo | Pathogenic | rs75762896 |
| P4 | PKD1 | c.1758A > C | p.E586D | Missense Variant | 9 | Hetero | Pathogenic | |
| P5 | PKD1L2 | c.404C > T | p.P135L | Missense Variant | 2 | Hetero | Uncertain significance | rs201455881 |
| P6 | PKD2L2 | c.1364A > T | p.N455I | Missense Variant | 9 | Hetero | Uncertain significance | |
| P7 | TSC2 | c.5038C > T | p.R1680C | N/A | 39 | Exonic | Likely pathogenic | rs45517423 |
| P8 | PKD1 | c.4264G > A | p.A1422T | Missense Variant | 15 | Homo | Uncertain significance | rs140980374 |
| P9 | PKD1 | c.9774T > G | p.F3258L | Missense Variant | 29 | Hetero | Pathogenic | N/A |
| P10 | None | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| P11 | PKD1 | c.4264G > A | p.A1422T | Missense Variant | 15 | Hetero | Uncertain significance | rs140980374 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzahrani, O.R.; Alatwi, H.E.; Alharbi, A.A.; Alessa, A.H.; Al-Amer, O.M.; Alanazi, A.F.R.; Shams, A.M.; Alomari, E.; Naser, A.Y.; Alzahrani, F.a.; et al. Identification and Characterization of Novel Mutations in Chronic Kidney Disease (CKD) and Autosomal Dominant Polycystic Kidney Disease (ADPKD) in Saudi Subjects by Whole-Exome Sequencing. Medicina 2022, 58, 1657. https://doi.org/10.3390/medicina58111657
Alzahrani OR, Alatwi HE, Alharbi AA, Alessa AH, Al-Amer OM, Alanazi AFR, Shams AM, Alomari E, Naser AY, Alzahrani Fa, et al. Identification and Characterization of Novel Mutations in Chronic Kidney Disease (CKD) and Autosomal Dominant Polycystic Kidney Disease (ADPKD) in Saudi Subjects by Whole-Exome Sequencing. Medicina. 2022; 58(11):1657. https://doi.org/10.3390/medicina58111657
Chicago/Turabian StyleAlzahrani, Othman R., Hanan E. Alatwi, Amnah A. Alharbi, Abdulrahman H. Alessa, Osama M. Al-Amer, Abeer F. R. Alanazi, Anwar M. Shams, Esra’a Alomari, Abdallah Y. Naser, Faisal a. Alzahrani, and et al. 2022. "Identification and Characterization of Novel Mutations in Chronic Kidney Disease (CKD) and Autosomal Dominant Polycystic Kidney Disease (ADPKD) in Saudi Subjects by Whole-Exome Sequencing" Medicina 58, no. 11: 1657. https://doi.org/10.3390/medicina58111657