Anti-Trypanosomal and Antimalarial Properties of Tetralone Derivatives and Structurally Related Benzocycloalkanones

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. In Vitro Anti-Trypanosomal Assay
2.2. In Vitro Cytotoxicity Assay
2.3. In Vitro Antiplasmodial Assay
3. Results
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brun, R.; Schumacher, R.; Schmid, C.; Kunz, C.; Burri, C. The phenomenon of treatment failures in human African trypanosomiasis. Trop. Med. Int. Health 2001, 6, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.; Burchmore, R.; Stich, A.; Lazzari, J.; Frasch, A.; Cazzulo, J.; Krishna, S. The trypanosomiases. Lancet 2003, 362, 1469–1480. [Google Scholar] [CrossRef]
- Beteck, R.M.; Legoabe, L.J.; Isaacs, M.; Hoppe, H.C. In vitro Anti-trypanosomal activities of indanone-based chalcones. Drug Res. 2018, 68. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Bray, M.; Cal, M.; Trunz, B.; Torreele, E.; Brun, R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef]
- MacLean, L.; Reiber, H.; Kennedy, P.; Sternberg, J. Stage progression and neurological symptoms in trypanosoma brucei rhodesience sleeping sickness: Role of the CNS inflammatory response. PLoS Negl. Trop. Dis. 2012, 6, e1857. [Google Scholar] [CrossRef]
- Kennedy, P.G. Human African trypanosomiasis of the CNS: Current issues and challenges. J. Clin. Investig. 2004, 113, 496–504. [Google Scholar] [CrossRef]
- Stich, A.; Abel, P.; Krishna, S. Human African trypanosomiasis. BMJ 2002, 325, 203–206. [Google Scholar] [CrossRef]
- Checchi, F.; Filipe, J.; Haydon, D.; Chandramohan, D.; Chappuis, F. Estimates of the duration of the early and late stage of gambiense sleeping sickness. Infect. Dis. 2008, 8, 16. [Google Scholar] [CrossRef]
- Global Health Observatory Data. Available online: http://www.who.int/gho/neglected_diseases/human_african_trypanosomiasis/en/ (accessed on 28 December 2018).
- Kuepfer, I.; Hhary, E.; Allan, M.; Edielu, A.; Burri, C.; Blum, J. Clinical presentation of T.b. rhodesiense sleeping sickness in second stage patients from Tanzania and Uganda. PLoS. Negl. Trop. Dis. 2011, 5, e968. [Google Scholar] [CrossRef]
- Simarro, P.; Cecchi, G.; Franco, J.; Paone, M.; Diarra, A.; Ruiz-Postigo, J.; Fe’vre, E.; Mattioli, R.; Jannin, G. Estimating and mapping the population at risk of sleeping sickness. PLoS. Negl. Trop. Dis. 2012, 6, e1859. [Google Scholar] [CrossRef]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Babokhov, P.; Sanyaolu, A.; Oyibo, W.; Fagbenro-Beyioku, A.; Iriemenam, N. A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis. Pathog. Glob. Health 2013, 107, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Baker, N.; Rothert, M.; Henke, B.; Jeacock, L.; Horn, D.; Beitz, E. Pentamidine is not a permeant but a nanomolar inhibitor of the Trypanosoma brucei aquaglyceroporin-2. PLoS Pathog. 2016, 12, e1005436. [Google Scholar] [CrossRef] [PubMed]
- Alirol, E.; Schrumpf, D.; Amici Heradi, J.; Riedel, A.; de Patoul, C.; Quere, M.; Chappuis, F. Nifurtimox-eflornithine combination therapy for second-stage gambiense human African trypanosomiasis: Médecins San Frontières experience in the Democratic Republic of Congo. Clin. Infect. Dis. 2013, 56, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. The development of drugs for treatment of sleeping sickness: A historical review. Parasit. Vectors 2010, 3, 15. [Google Scholar] [CrossRef]
- Beteck, R.M.; Isaacs, M.; Hoppe, H.C.; Khanye, S.D. Synthesis, in vitro cytotoxicity and trypanocidal evaluation of novel 1,3,6-substituted non-fluoroquinolones. S. Afr. J. Chem. 2018, 71, 188–195. [Google Scholar] [CrossRef]
- Berninger, M.; Schmidt, I.; Ponte-Sucre, A.; Holzgrabe, U. Novel lead compounds in pre-clinical development against African sleeping sickness. Med. Chem. Commun. 2017, 8, 1872–1890. [Google Scholar] [CrossRef]
- Troeberg, L.; Chen, X.; Flaherty, T.; Morty, R.; Cheng, M.; Hua, H.; Springer, C.; McKerrow, J.; Kenyon, G.; Lonsdale-Eccles, J.; et al. Chalcone, acyl hydrazide, and related amides kill cultured Trypanosoma brucei brucei. Mol. Med. 2000, 6, 660–669. [Google Scholar] [CrossRef]
- Connor, R.J. The impact of nagana. Onderstepoort. J. Vet. Res. 1994, 61, 379–383. [Google Scholar] [PubMed]
- Giordani, F.; Morrison, L.; Rowan, T.; de Koning, H.; Barrett, M. The animal trypanosomiases and their chemotherapy: A review. Parasitology 2016, 143, 1862–1889. [Google Scholar] [CrossRef]
- Veale, C.G.; Hoppe, H.C. Screening of the Pathogen Box reveals new starting points for anti-trypanosomal drug discovery. Med. Chem. Commun. 2018, 9, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- WHO. World Malaria Report 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Meyrowitsch, D.; Pedersen, E.; Alifrangis, M.; Scheike, M.; Malecela, M.; Magesa, S.; Derua, Y.; Rwegoshora, R.; Michael, E.; Simonsen, P. Is the current decline in malaria burden in sub-Saharan Africa due to a decrease in vector population? Mal. J. 2011, 10, 188. [Google Scholar] [CrossRef]
- Roberts, L. Drug-resistant malaria advances in Mekong. Science 2017, 358, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E. Prophylaxis of Malaria. Mediterr. J. Hematol. Infect. Dis. 2012, 4, e201245. [Google Scholar] [CrossRef]
- Na-Bangchang, K.; Karbwang, J. Current status of malaria chemotherapy and the role of pharmacology in antimalarial drug research and development. Fundam. Clin. Pharmacol. 2009, 23, 387–409. [Google Scholar] [CrossRef]
- Enato, E.F.; Okhamafe, A.O. Plasmodium falciparum malaria and antimalarial interventions in sub-Saharan Africa: Challenges and opportunities. Afr. J. Biotechnol. 2005, 4, 1598–1605. [Google Scholar] [CrossRef]
- Verlinden, B.K.; Louw, A.; Birkholtz, L.M. Resisting resistance: Is there a solution for malaria? Expert Opin. Drug Discov. 2016, 11, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Devine, W.; Woodring, J.L.; Swaminathan, U.; Amata, E.; Patel, G.; Erath, J.; Roncal, N.E.; Lee, P.J.; Leed, S.E.; Rodriguez, A.; et al. Protozoan parasite growth inhibitors discovered by cross-screening yield potent scaffolds for lead discovery. J. Med. Chem. 2015, 58, 5522–5537. [Google Scholar] [CrossRef]
- Spangenberg, T.; Burrows, J.N.; Kowalczyk, P.; McDonald, S.; Wells, T.N.; Willis, P. The open access malaria box: A drug discovery catalyst for neglected diseases. PLoS ONE 2013, 8, e62906. [Google Scholar] [CrossRef]
- Van Voorhis, W.C.; Adams, J.H.; Adelfio, R.; Ahyong, V.; Akabas, M.; Alano, P.; Alday, A.; Resto, Y.; Alsibaee, A.; Alzualde, A.; et al. Open source drug discovery with the malaria box compound collection for neglected diseases and beyond. PLoS Pathog. 2016, 12, e1005763. [Google Scholar] [CrossRef]
- Monti, L.; Wang, S.; Oukoloff, K.; Smith, A., III; Brunden, K.; Caffrey, C.; Ballatore, C. Brain-penetrant, triazolopyrimidine and phenylpyrimidine microtubule-stabilizers as potential leads to treat Human African Trypanosomiasis. ChemMedChem 2018, 13, 1751–1754. [Google Scholar] [CrossRef] [PubMed]
- Amakali, K.T.; Legoabe, L.J.; Petzer, A.; Petzer, J.P. Synthesis and in vitro evaluation of 2-heteroarylidene-1-tetralone derivatives as monoamine oxidase inhibitors. Drug Res. (Stuttg.) 2018, 68, 687–695. [Google Scholar] [CrossRef]
- Amakali, K.T.; Legoabe, L.J.; Petzer, A.; Petzer, J.P. Synthesis and valuation of 2-benzylidene-1-tetralone derivatives for monoamine oxidase inhibitory activity. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Gumbo, M.; Beteck, R.M.; Mandizvo, T.; Seldon, R.; Warner, D.F.; Hoppe, H.C.; Isaacs, M.; Laming, D.; Tam, C.C.; Cheng, L.W.; et al. Cinnamoyl-Oxaborole Amides: Synthesis and Their in Vitro Biological Activity. Molecules 2018, 23, 2038. [Google Scholar] [CrossRef]
- Darrell, O.T.; Hulushe, S.T.; Mtshare, T.E.; Beteck, R.M.; Isaacs, M.; Laming, D.; Hoppe, H.C.; Krause, R.W.M.; Khanye, S.D. Synthesis, antiplasmodial and antitrypanosomal evaluation of a series of novel 2-oxoquinoline-based thiosemicarbazone derivatives. S. Afr. J. Chem. 2018, 71, 174–181. [Google Scholar] [CrossRef]
- Makler, M.T.; Ries, J.M.; Williams, J.A.; Bancroft, J.E.; Piper, R.C.; Gibbins, B.L.; Hinrichs, D.J. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am. J. Trop. Med. Hyg. 1993, 48, 739–741. [Google Scholar] [CrossRef] [PubMed]




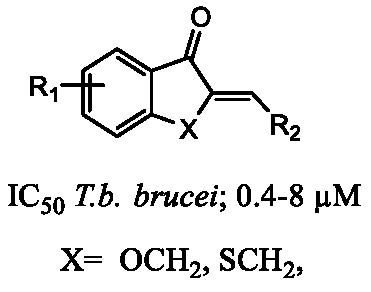{kind=link}
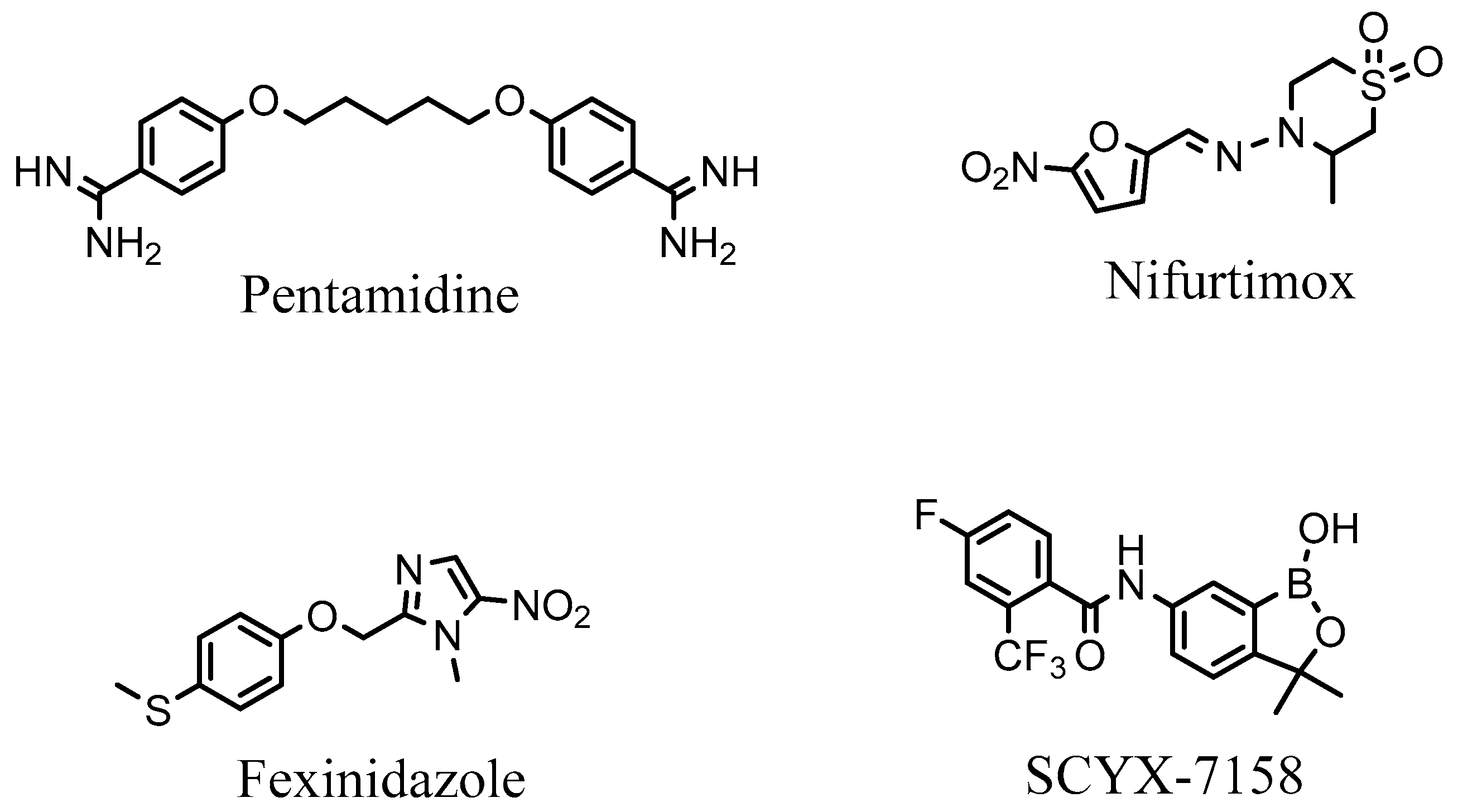{kind=link}
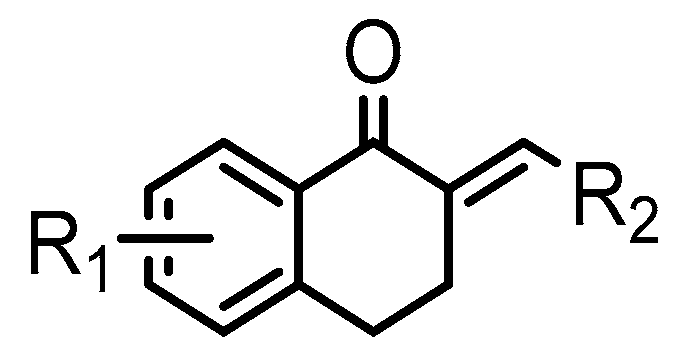{kind=link}
{kind=link}
| Compound | R1 | R2 | % Viability at 20 µM (SD) | ||
|---|---|---|---|---|---|
| T.b. brucei | 3D7 | HeLa Cell | |||
| 1 | - | 3-CN-phenyl | 0.06(0.3) | 113 (3) | 87 (3) |
| 2 | 7-NH2 | 3-CN-phenyl | 95 (3) | 110 (1) | 77 (2) |
| 3 | - | phenyl | 43 (3) | 103 (2) | 116 (5) |
| 4 | - | 4-OCH3-phenyl | 0.5 (0.5) | 100 (9) | 76 (9) |
| 5 | - | 4-F-phenyl | 101 (1) | 107 (5) | 100 (2) |
| 6 | - | 3-F-phenyl | 0.5 (0.2) | 131 (9) | 121 (16) |
| 7 | 6-OH | phenyl | 0.6 (0.2) | 89 (4) | 54 (1) |
| 8 | 5-OH | phenyl | 0.5 (0.1) | 21 (12) | 35 (1) |
| 9 | 7-OH | phenyl | −4 (0.3) | 101 (4) | 9 (0.1) |
| 10 | 7-OCH3 | phenyl | 0.1 (0.1) | 113 (1) | 65 (3) |
| 11 | 6-NH2 | phenyl | 97 (0.3) | 111 (1) | 86 (9) |
| 12 | 7-OH | 4-F-phenyl | −0.6 (0.1) | 11 (6) | 29 (0.1) |
| 13 | 7-OH | 3-F-phenyl | −0.9 (0.1) | 13 (1) | 21 (1) |
| 14 | 7-OH | 4-Cl-phenyl | 0.1 (0.1) | 13 (14) | 27 (2) |
| 15 | 7-OH | 3-Cl-phenyl | 0.6 (0.01) | −3 (6) | 18 (0.4) |
| 16 | 7-OH | 4-Br-phenyl | 0.1 (0.1) | 43 (8) | 62 (2) |
| 17 | - | 3-Cl-phenyl | 0.2 (0.3) | 100 (9) | 41 (13) |
| 18 | 7-OH | 3-Br-phenyl | 1.2 (0.31) | 99 (8) | 26 (3) |
| 19 | 7-OH | 4-OH-phenyl | 1 (0.2) | 104 (12) | 44 (4) |
| 20 | 7-OH | 4-CH3-phenyl | −0.4 (0.1) | 116 (16) | 69 (10) |
| 21 | 7-OH | 4-N(CH3)2-phenyl | 25 (2) | 142 (9) | 51 (4) |
| 22 | 7-OH | 3,4-diCl-phenyl | 0.9 (0.2) | 81 (1) | 5 (0.9) |
| 23 | 7-OH | 4-OCH3-phenyl | 0.2 (0.1) | 88 (10) | 25 (2) |
| 24 | - | 4-Cl-phenyl | 26 (8) | 100 (8) | 100 (3) |
| 25 | - | 3-pyridyl | −0.7 (0.4) | 29 (12) | 24 (0.4) |
| 26 | 7-OCH3 | cyclohexyl | 0.8 (0.5) | 119 (13) | 92 (16) |
| 27 | 7-OCH3 | cyclopentyl | 0.6 (0.02) | 81 (3) | 85 (1.6) |
| 28 | 7-OCH3 | furanyl | 31 (5) | 100 (3) | 97 (9) |
| 29 | 7-OCH3 | pyrrolyl | 103 (1) | 115 (5) | 97 (5) |
| 30 | 7-OCH3 | 2-thiophenyl | 32 (9) | 107 (6) | 106 (2) |
| 31 | 7-OCH3 | 3-pyridyl | 0.2 (0.03) | 85 (12) | 17 (4) |
| 32 | 7-OCH3 | 4-pyridyl | −0.1 (0.7) | 36 (12) | 23 (2) |
| 33 | 7-OCH3 | 2-pyridyl | −0.4 (0.2) | 7 (8) | 20 (0.6) |
| 34 | 7-OCH3 | 2-Cl-3-pyridyl | 98 (2) | 113 (6) | 47 (1.7) |
| 35 | 7-OCH3 | 3-thiophenyl | 22 (2) | 86 (10) | 122 (1) |
| Compound | X | R2 | % Viability at 20 µM (SD) | ||
|---|---|---|---|---|---|
| T. b. brucei | 3D7 | HeLa Cell | |||
| 24 | (CH2)2 | 4-Cl-phenyl | 26 (8) | 100 (8) | 100 (3) |
| 36 | CH2 | 4-Cl-phenyl | 99 (0.1) | 93 (3) | 74 (10) |
| 37 | (CH2)3 | 4-Cl-phenyl | 0.3 (0.3) | 113 (0.6) | 53 (10) |
| 38 | OCH2 | 4-Cl-phenyl | 0.4 (0.2) | 98 (1) | 75 (5) |
| 39 | SCH2 | 4-Cl-phenyl | 0.3 (0.3) | 11 (0.9) | 8 (2) |
| 40 | O | 4-Cl-phenyl | 94 (7) | 90 (6) | 103 (3) |
| 41 | S | 4-Cl-phenyl | 103 (0.3) | 107 (5) | 109 (7) |
| Compound | Structure | IC50 (µM) | MW a | ClogP b | tPSAc |
|---|---|---|---|---|---|
| T. b. brucei | |||||
| 1 |  | 2.3 | 259 | 3.5 | 40 |
| 4 |  | 6.7 | 264 | 4 | 26 |
| 6 |  | 3.1 | 252 | 4 | 17 |
| 7 |  | 0.46 | 250 | 4 | 37 |
| 10 |  | 1.04 | 264 | 4 | 26 |
| 12 |  | 0.68 | 268 | 4 | 37 |
| 16 |  | 1.24 | 329 | 4.8 | 37 |
| 20 |  | 5.3 | 264 | 4.5 | 37 |
| 26 |  | nd | 270 | 5.6 | 26 |
| 27 |  | 5.4 | 256 | 5 | 26 |
| 35 |  | 2.6 | 270 | 4 | 26 |
| 38 |  | 0.4 | 270 | 4.4 | 26 |
| PE | - | 0.0042 | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beteck, R.M.; Legoabe, L.J.; Isaacs, M.; Khanye, S.D.; Laming, D.; Hoppe, H.C. Anti-Trypanosomal and Antimalarial Properties of Tetralone Derivatives and Structurally Related Benzocycloalkanones. Medicina 2019, 55, 206. https://doi.org/10.3390/medicina55050206
Beteck RM, Legoabe LJ, Isaacs M, Khanye SD, Laming D, Hoppe HC. Anti-Trypanosomal and Antimalarial Properties of Tetralone Derivatives and Structurally Related Benzocycloalkanones. Medicina. 2019; 55(5):206. https://doi.org/10.3390/medicina55050206
Chicago/Turabian StyleBeteck, Richard M., Lesetja J. Legoabe, Michelle Isaacs, Setshaba D. Khanye, Dustin Laming, and Heinrich C. Hoppe. 2019. "Anti-Trypanosomal and Antimalarial Properties of Tetralone Derivatives and Structurally Related Benzocycloalkanones" Medicina 55, no. 5: 206. https://doi.org/10.3390/medicina55050206