An Atypical Case of Idiopathic Pulmonary Fibrosis in a Patient from Africa


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
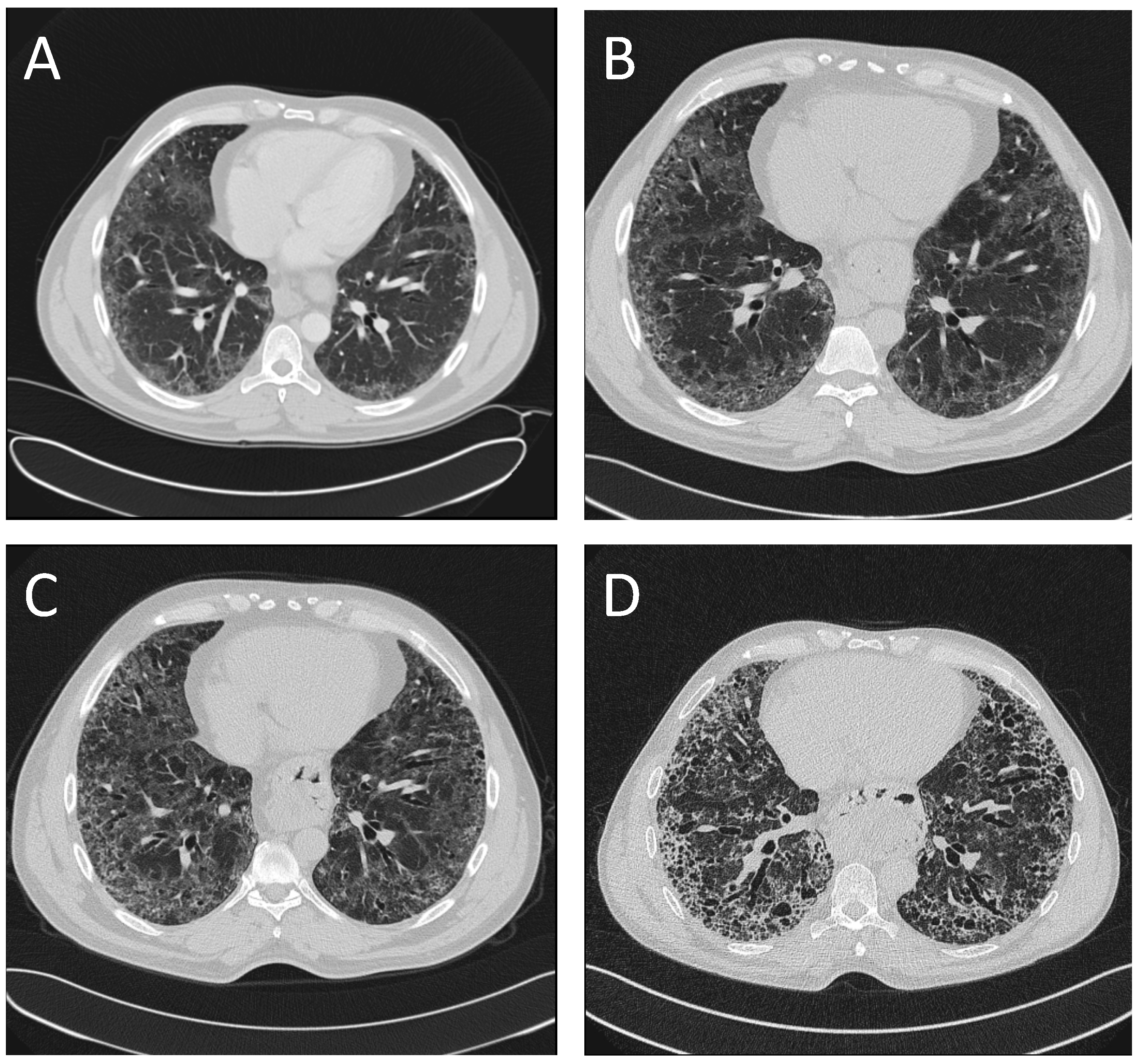
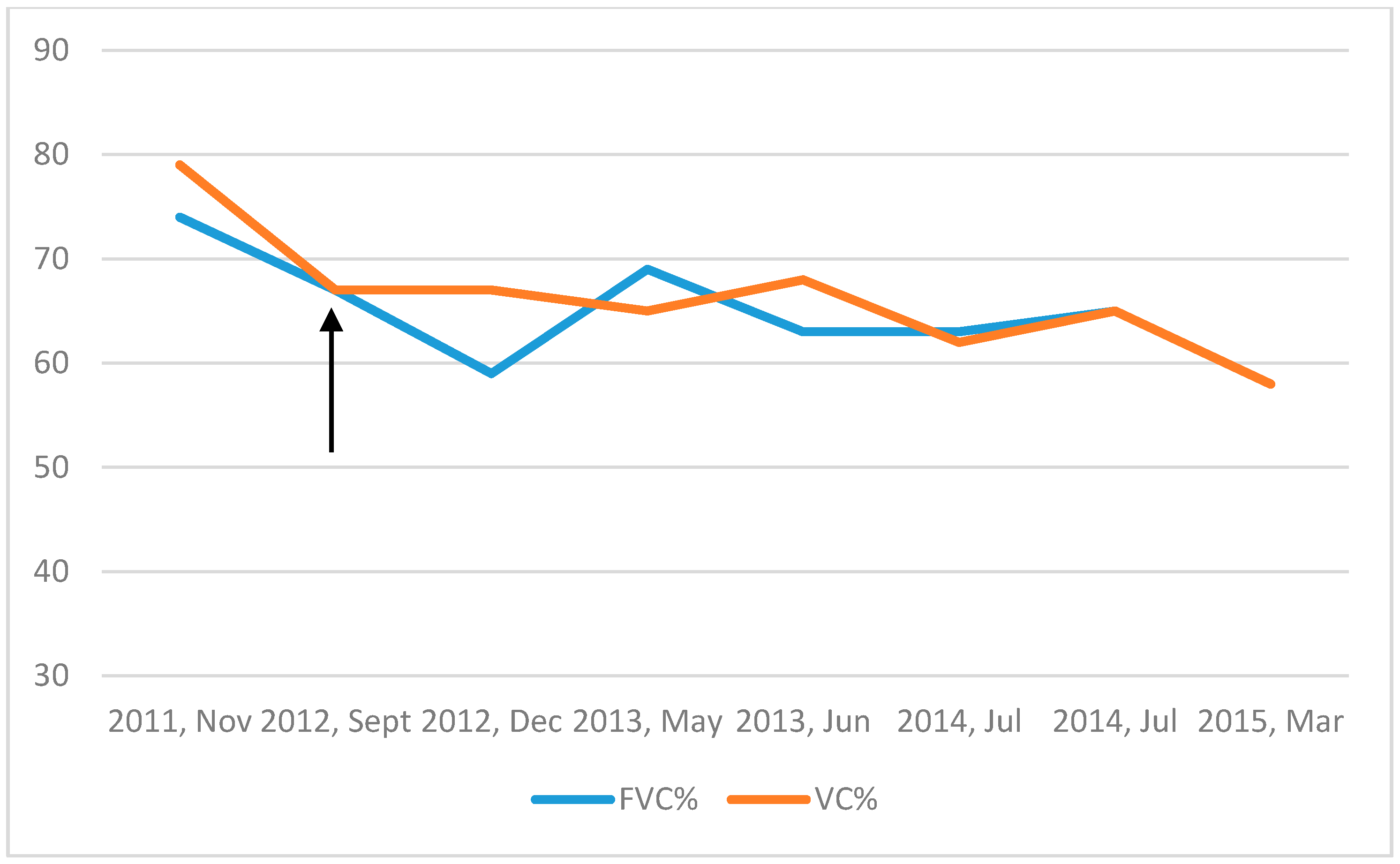
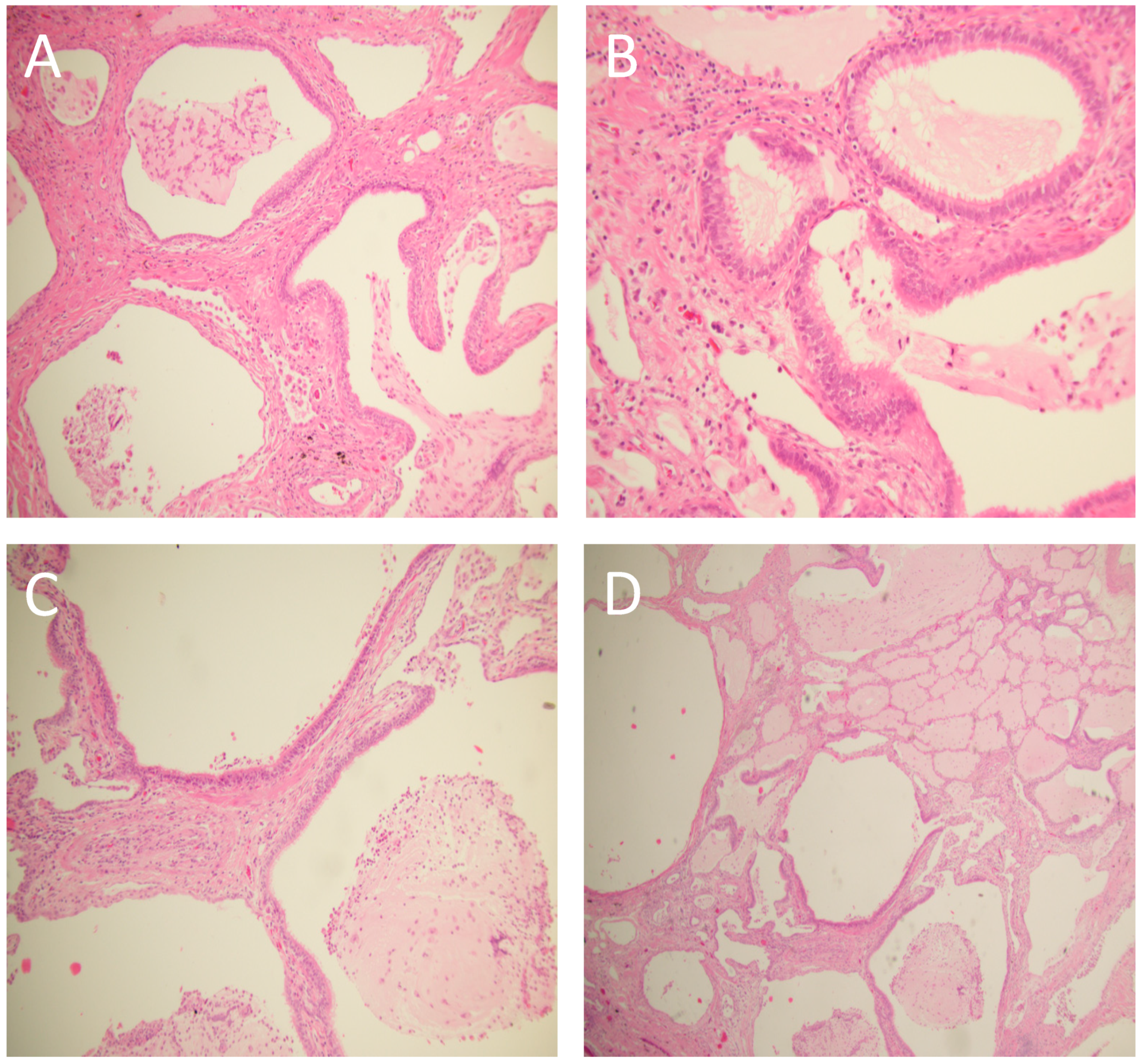
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Lasky, J.A.; Hagiwara, K.; Kondoh, Y. Ethnic differences in idiopathic pulmonary fibrosis: The Japanese perspective. Respir. Investig. 2018, 56, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Duchemann, B.; Annesi-Maesano, I.; Jacobe de Naurois, C.; Sanyal, S.; Brillet, P.Y.; Brauner, M.; Kambouchner, M.; Huynh, S.; Naccache, J.M.; Borie, R.; et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur. Respir. J. 2017, 50, 1602419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swigris, J.J.; Olson, A.L.; Huie, T.J.; Fernandez-Perez, E.R.; Solomon, J.; Sprunger, D.; Brown, K.K. Ethnic and racial differences in the presence of idiopathic pulmonary fibrosis at death. Respir. Med. 2012, 106, 588–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koegelenberg, C.F.; Ainslie, G.M.; Dheda, K.; Allwood, B.W.; Wong, M.L.; Lalloo, U.G.; Abdool-Gaffar, M.S.; Khalfey, H.; Irusen, E.M. Recommendations for the management of idiopathic pulmonary fibrosis in South Africa: A position statement of the South African Thoracic Society. J. Thorac. Dis. 2016, 8, 3711–3719. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, J.; Franks, T.J.; Colby, T.V.; Flaherty, K.R.; Galvin, J.R.; Hayden, D.; Gochuico, B.R.; Kazerooni, E.A.; Martinez, F.; Travis, W.D. Peribronchiolar metaplasia: A common histologic lesion in diffuse lung disease and a rare cause of interstitial lung disease: Clinicopathologic features of 15 cases. Am. J. Surg. Pathol. 2005, 29, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Fawibe, A.E.; Odeigah, L.O.; Saka, M.J. Reference equations for spirometric indices from a sample of the general adult population in Nigeria. BMC Pulm. Med. 2017, 17, 48. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pesonen, I.; Ortiz, C.; Ferrara, G. An Atypical Case of Idiopathic Pulmonary Fibrosis in a Patient from Africa. Medicina 2019, 55, 67. https://doi.org/10.3390/medicina55030067
Pesonen I, Ortiz C, Ferrara G. An Atypical Case of Idiopathic Pulmonary Fibrosis in a Patient from Africa. Medicina. 2019; 55(3):67. https://doi.org/10.3390/medicina55030067
Chicago/Turabian StylePesonen, Ida, Cristian Ortiz, and Giovanni Ferrara. 2019. "An Atypical Case of Idiopathic Pulmonary Fibrosis in a Patient from Africa" Medicina 55, no. 3: 67. https://doi.org/10.3390/medicina55030067