
Association between KRAS and PIK3CA Mutations and Progesterone Resistance in Endometriotic Epithelial Cell Line
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Progesterone Therapy Model of Endometriotic Cell Line
2.2. RNA Extraction and Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
2.3. Western Blot Analysis
2.4. Dienogest (DNG) and Medroxyprogesterone Acetate (MPA) Therapy
2.5. Migration Assay
2.6. Matrigel Invasion Assay
2.7. Cell Proliferation Assay
2.8. Real-Time Quantitative PCR
2.9. Statistical Analysis
3. Results
3.1. Western Blot Analysis and RT-PCR
3.2. Migration, Invasion, and Proliferation Assay
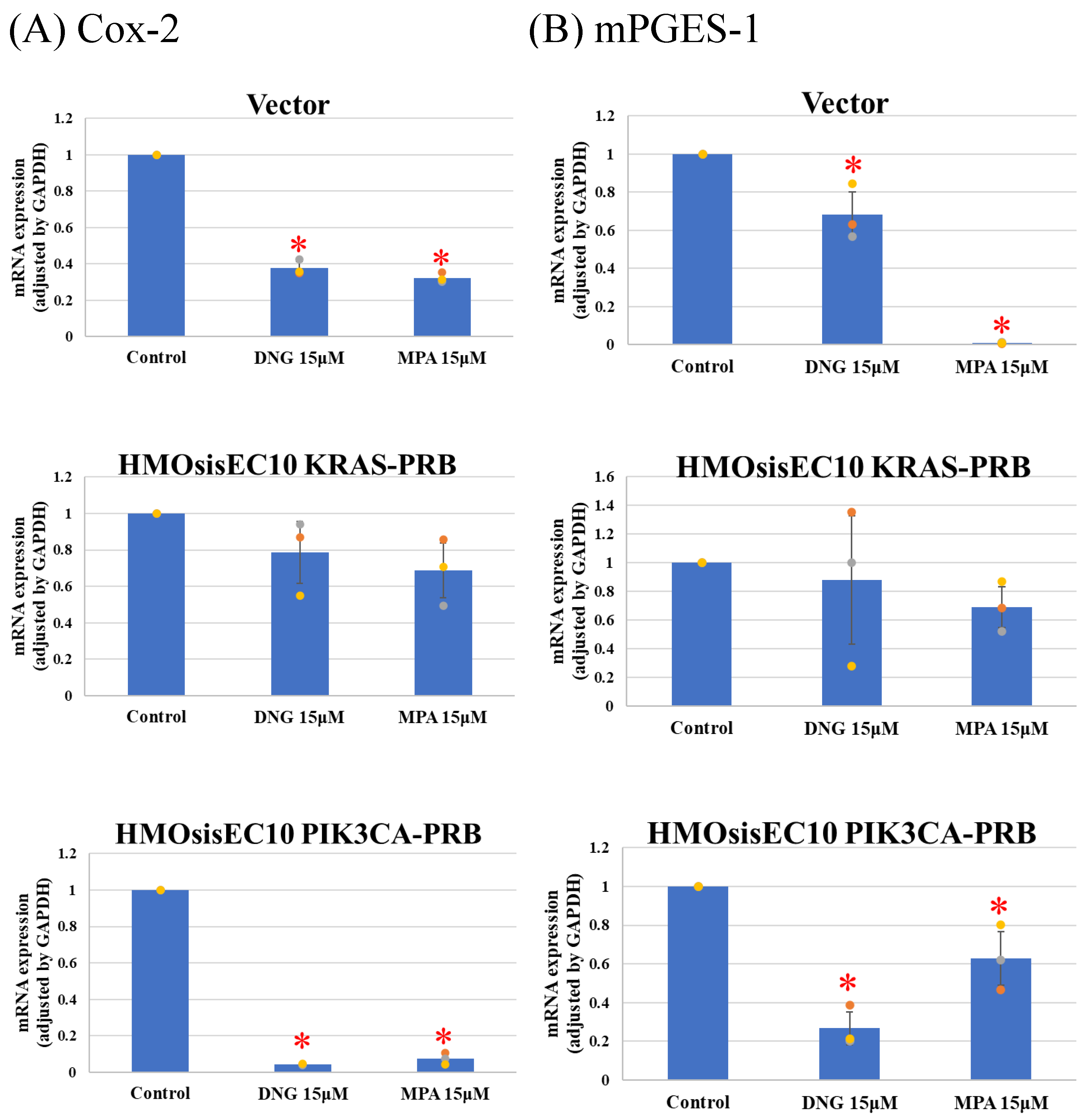
3.3. Real-Time Quantitative PCR (Real-Time qPCR)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Falcone, T.; Flyckt, R. Clinical Management of Endometriosis. Obstet. Gynecol. 2018, 131, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Viganò, P.; Parazzini, F.; Somigliana, E.; Vercellini, P. Endometriosis: Epidemiology and Aetiological Factors. Best Pract. Res. Clin. Obstet. Gynaecol. 2004, 18, 177–200. [Google Scholar] [CrossRef] [PubMed]
- Olive, D.L.; Pritts, E.A. Treatment of Endometriosis. N. Engl. J. Med. 2001, 345, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Giudice, L.C.; Kao, L.C. Endometriosis. Lancet 2004, 364, 1789–1799. [Google Scholar] [CrossRef]
- Liakopoulou, M.-K.; Tsarna, E.; Eleftheriades, A.; Arapaki, A.; Toutoudaki, K.; Christopoulos, P. Medical and Behavioral Aspects of Adolescent Endometriosis: A Review of the Literature. Children 2022, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Nnoaham, K.E.; Hummelshoj, L.; Webster, P.; d’Hooghe, T.; de Cicco Nardone, F.; de CiccoNardone, C.; Jenkinson, C.; Kennedy, S.H.; Zondervan, K.T. Impact of Endometriosis on Quality of Life and Work Productivity: A Multicenter Study across Ten Countries. Fertil. Steril. 2011, 96, 366–373.e8. [Google Scholar] [CrossRef] [PubMed]
- Sanfilippo, J.; Erb, T. Evaluation and Management of Dysmenorrhea in Adolescents. Clin. Obstet. Gynecol. 2008, 51, 257. [Google Scholar] [CrossRef] [PubMed]
- Song, S.Y.; Jung, Y.W.; Shin, W.; Park, M.; Lee, G.W.; Jeong, S.; An, S.; Kim, K.; Ko, Y.B.; Lee, K.H.; et al. Endometriosis-Related Chronic Pelvic Pain. Biomedicines 2023, 11, 2868. [Google Scholar] [CrossRef] [PubMed]
- Gruber, T.M.; Mechsner, S. Pathogenesis of Endometriosis: The Origin of Pain and Subfertility. Cells 2021, 10, 1381. [Google Scholar] [CrossRef] [PubMed]
- Nanda, A.; Thangapandi, T.; Banerjee, P.; Dutta, M.; Wangdi, T.; Sharma, P.; Chaudhury, K.; Jana, S.K. Cytokines, Angiogenesis, and Extracellular Matrix Degradation Are Augmented by Oxidative Stress in Endometriosis. Ann. Lab. Med. 2020, 40, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Attar, E.; Tokunaga, H.; Imir, G.; Yilmaz, M.B.; Redwine, D.; Putman, M.; Gurates, B.; Attar, R.; Yaegashi, N.; Hales, D.B.; et al. Prostaglandin E2 Via Steroidogenic Factor-1 Coordinately Regulates Transcription of Steroidogenic Genes Necessary for Estrogen Synthesis in Endometriosis. J. Clin. Endocrinol. Metab. 2009, 94, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Saunders, P.T.K.; Horne, A.W. Endometriosis: Etiology, Pathobiology, and Therapeutic Prospects. Cell 2021, 184, 2807–2824. [Google Scholar] [CrossRef] [PubMed]
- Vannuccini, S.; Clemenza, S.; Rossi, M.; Petraglia, F. Hormonal Treatments for Endometriosis: The Endocrine Background. Rev. Endocr. Metab. Disord. 2022, 23, 333–355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, G. Progesterone Resistance in Endometriosis: Current Evidence and Putative Mechanisms. Int. J. Mol. Sci. 2023, 24, 6992. [Google Scholar] [CrossRef] [PubMed]
- Reis, F.M.; Coutinho, L.M.; Vannuccini, S.; Batteux, F.; Chapron, C.; Petraglia, F. Progesterone Receptor Ligands for the Treatment of Endometriosis: The Mechanisms behind Therapeutic Success and Failure. Hum. Reprod. Update 2020, 26, 565–585. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; Papadopoulos, N.; Ayhan, A.; Nazeran, T.M.; Noë, M.; Horlings, H.M.; Lum, A.; Jones, S.; Senz, J.; Seckin, T.; et al. Cancer-Associated Mutations in Endometriosis without Cancer. N. Engl. J. Med. 2017, 376, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Yachida, N.; Yoshihara, K.; Suda, K.; Nakaoka, H.; Ueda, H.; Sugino, K.; Yamaguchi, M.; Mori, Y.; Yamawaki, K.; Tamura, R.; et al. Biological Significance of KRAS Mutant Allele Expression in Ovarian Endometriosis. Cancer Sci. 2021, 112, 2020–2032. [Google Scholar] [CrossRef]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Tamura, R.; Mori, Y.; Yamawaki, K.; Adachi, S.; Takahashi, T.; Kase, H.; et al. Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep. 2018, 24, 1777–1789. [Google Scholar] [CrossRef]
- Koppolu, A.; Maksym, R.B.; Paskal, W.; Machnicki, M.; Rak, B.; Pępek, M.; Garbicz, F.; Pełka, K.; Kuśmierczyk, Z.; Jacko, J.; et al. Epithelial Cells of Deep Infiltrating Endometriosis Harbor Mutations in Cancer Driver Genes. Cells 2021, 10, 749. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.-M.; Kurman, R.J. Ovarian Tumorigenesis. Am. J. Pathol 2004, 164, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines 2018, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Nakaoka, H.; Suda, K.; Yoshihara, K.; Ishiguro, T.; Yachida, N.; Saito, K.; Ueda, H.; Sugino, K.; Mori, Y.; et al. Spatiotemporal Dynamics of Clonal Selection and Diversification in Normal Endometrial Epithelium. Nat. Commun. 2022, 13, 943. [Google Scholar] [CrossRef] [PubMed]
- Yachida, N.; Yoshihara, K.; Yamaguchi, M.; Suda, K.; Tamura, R.; Enomoto, T. How Does Endometriosis Lead to Ovarian Cancer? The Molecular Mechanism of Endometriosis-Associated Ovarian Cancer Development. Cancers 2021, 13, 1439. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.M.; Nakayama, K.; Shanta, K.; Razia, S.; Ishikawa, M.; Ishibashi, T.; Yamashita, H.; Sato, S.; Iida, K.; Kanno, K.; et al. Establishment of a Novel In Vitro Model of Endometriosis with Oncogenic KRAS and PIK3CA Mutations for Understanding the Underlying Biology and Molecular Pathogenesis. Cancers 2021, 13, 3174. [Google Scholar] [CrossRef]
- Bono, Y.; Kyo, S.; Takakura, M.; Maida, Y.; Mizumoto, Y.; Nakamura, M.; Nomura, K.; Kiyono, T.; Inoue, M. Creation of Immortalised Epithelial Cells from Ovarian Endometrioma. Br. J. Cancer 2012, 106, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- A p16INK4a-Insensitive CDK4 Mutant Targeted by Cytolytic T Lymphocytes in a Human Melanoma|Science. Available online: https://www.science.org/doi/10.1126/science.7652577 (accessed on 18 March 2024).
- Nakamura, K.; Aimono, E.; Tanishima, S.; Imai, M.; Nagatsuma, A.K.; Hayashi, H.; Yoshimura, Y.; Nakayama, K.; Kyo, S.; Nishihara, H. Intratumoral Genomic Heterogeneity May Hinder Precision Medicine Strategies in Patients with Serous Ovarian Carcinoma. Diagnostics 2020, 10, 200. [Google Scholar] [CrossRef] [PubMed]
- Koninckx, P.R.; Ussia, A.; Adamyan, L.; Wattiez, A.; Gomel, V.; Martin, D.C. Pathogenesis of Endometriosis: The Genetic/Epigenetic Theory. Fertil. Steril. 2019, 111, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Hirata, T.; Koga, K.; Osuga, Y. Extra-pelvic Endometriosis: A Review. Reprod. Med. Biol. 2020, 19, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Keckstein, J.; Hudelist, G. Classification of Deep Endometriosis (DE) Including Bowel Endometriosis: From r-ASRM to #Enzian-Classification. Best Pract. Res. Clin. Obstet. Gynaecol. 2021, 71, 27–37. [Google Scholar] [CrossRef]
- Sampson, J.A. Metastatic or Embolic Endometriosis, Due to the Menstrual Dissemination of Endometrial Tissue into the Venous Circulation. Am. J. Pathol. 1927, 3, 93–110.43. [Google Scholar] [PubMed]
- Koninckx, P.R.; Barlow, D.; Kennedy, S. Implantation versus Infiltration: The Sampson versus the Endometriotic Disease Theory. Gynecol. Obs. Investig. 1999, 47 (Suppl. S1), 3–9; discussion 9–10. [Google Scholar] [CrossRef] [PubMed]
- Fung, J.N.; Rogers, P.A.W.; Montgomery, G.W. Identifying the Biological Basis of GWAS Hits for Endometriosis1. Biol. Reprod. 2015, 92, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Moayedi, M.; Davis, K.D. Theories of Pain: From Specificity to Gate Control. J. Neurophysiol. 2013, 109, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, A. Prostaglandin E2 and Pain-An Update. Biol. Pharm. Bull. 2011, 34, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.; Barcena De Arellano, M.L.; Rüster, C.; Vercellino, G.F.; Chiantera, V.; Schneider, A.; Mechsner, S. Imbalance between Sympathetic and Sensory Innervation in Peritoneal Endometriosis. Brain Behav. Immun. 2012, 26, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Velho, R.V.; Taube, E.; Sehouli, J.; Mechsner, S. Neurogenic Inflammation in the Context of Endometriosis—What Do We Know? Int. J. Mol. Sci. 2021, 22, 13102. [Google Scholar] [CrossRef]
- Kalaitzopoulos, D.R.; Samartzis, N.; Kolovos, G.N.; Mareti, E.; Samartzis, E.P.; Eberhard, M.; Dinas, K.; Daniilidis, A. Treatment of Endometriosis: A Review with Comparison of 8 Guidelines. BMC Womens Health 2021, 21, 397. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-W. Recurrence of Endometriosis and Its Control. Hum. Reprod. Update 2009, 15, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.-S.; Yuk, J.-S.; Cho, Y.-K.; Shin, J.-Y. Dienogest and the Risk of Reoperation in Endometriosis. J. Pers. Med. 2021, 11, 924. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Takahashi, K.; Nakamura, K.; Otake, A.; Sasamoto, N.; Miyoshi, Y.; Shioji, M.; Yamamoto, Y.; Fujitani, M.; Wakimoto, A.; et al. Postoperative Administration of Dienogest for Suppressing Recurrence of Disease and Relieving Pain in Subjects with Ovarian Endometriomas. Gynecol. Endocrinol. 2016, 32, 646–649. [Google Scholar] [CrossRef]
- Vercellini, P.; Cortesi, I.; Crosignani, P.G. Progestins for Symptomatic Endometriosis: A Critical Analysis of the Evidence. Fertil. Steril. 1997, 68, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, R.M.; Kim, T.H.; Shin, J.-H.; Jeong, J.-W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? Int. J. Mol. Sci. 2019, 20, 3822. [Google Scholar] [CrossRef] [PubMed]
- Flores, V.A.; Vanhie, A.; Dang, T.; Taylor, H.S. Progesterone Receptor Status Predicts Response to Progestin Therapy in Endometriosis. J. Clin. Endocrinol. Metab. 2018, 103, 4561–4568. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.L.; Unno, K.; Caraveo, M.; Lu, Z.; Kim, J.J. Increased AKT or MEK1/2 Activity Influences Progesterone Receptor Levels and Localization in Endometriosis. J. Clin. Endocrinol. Metab. 2013, 98, E1871–E1879. [Google Scholar] [CrossRef] [PubMed]
- Barragan, F.; Irwin, J.C.; Balayan, S.; Erikson, D.W.; Chen, J.C.; Houshdaran, S.; Piltonen, T.T.; Spitzer, T.L.B.; George, A.; Rabban, J.T.; et al. Human Endometrial Fibroblasts Derived from Mesenchymal Progenitors Inherit Progesterone Resistance and Acquire an Inflammatory Phenotype in the Endometrial Niche in Endometriosis1. Biol. Reprod. 2016, 94, 118. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.-Y.; Kim, T.H.; Fazleabas, A.T.; Palomino, W.A.; Ahn, S.H.; Tayade, C.; Schammel, D.P.; Young, S.L.; Jeong, J.-W.; Lessey, B.A. KRAS Activation and Over-Expression of SIRT1/BCL6 Contributes to the Pathogenesis of Endometriosis and Progesterone Resistance. Sci. Rep. 2017, 7, 6765. [Google Scholar] [CrossRef] [PubMed]
- Petz, L.N.; Ziegler, Y.S.; Schultz, J.R.; Kim, H.; Kemper, J.K.; Nardulli, A.M. Differential Regulation of the Human Progesterone Receptor Gene through an Estrogen Response Element Half Site and Sp1 Sites. J. Steroid Biochem. Mol. Biol. 2004, 88, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Méar, L.; Herr, M.; Fauconnier, A.; Pineau, C.; Vialard, F. Polymorphisms and Endometriosis: A Systematic Review and Meta-Analyses. Hum. Reprod. Update 2020, 26, 73–103. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Strawn, E.; Basir, Z.; Halverson, G.; Guo, S.-W. Promoter Hypermethylation of Progesterone Receptor Isoform B (PR-B) in Endometriosis. Epigenetics 2006, 1, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Lee, H.-C.; Liu, S.; Quick, C.M.; Fernandes, L.M.; Simmen, F.A.; Tsai, S.-J.; Simmen, R.C.M. Notch-1 Signaling Activation and Progesterone Receptor Expression in Ectopic Lesions of Women With Endometriosis. J. Endocr. Soc. 2018, 2, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Su, R.-W.; Strug, M.R.; Jeong, J.-W.; Miele, L.; Fazleabas, A.T. Aberrant Activation of Canonical Notch1 Signaling in the Mouse Uterus Decreases Progesterone Receptor by Hypermethylation and Leads to Infertility. Proc. Natl. Acad. Sci. USA 2016, 113, 2300–2305. [Google Scholar] [CrossRef]
- Inoue, S.; Hirota, Y.; Ueno, T.; Fukui, Y.; Yoshida, E.; Hayashi, T.; Kojima, S.; Takeyama, R.; Hashimoto, T.; Kiyono, T.; et al. Uterine Adenomyosis Is an Oligoclonal Disorder Associated with KRAS Mutations. Nat. Commun. 2019, 10, 5785. [Google Scholar] [CrossRef] [PubMed]
- Kastner, P.; Krust, A.; Turcotte, B.; Stropp, U.; Tora, L.; Gronemeyer, H.; Chambon, P. Two Distinct Estrogen-Regulated Promoters Generate Transcripts Encoding the Two Functionally Different Human Progesterone Receptor Forms A and B. EMBO J. 1990, 9, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Ichioka, M.; Mita, S.; Shimizu, Y.; Imada, K.; Kiyono, T.; Bono, Y.; Kyo, S. Dienogest, a Synthetic Progestin, down-Regulates Expression of CYP19A1 and Inflammatory and Neuroangiogenesis Factors through Progesterone Receptor Isoforms A and B in Endometriotic Cells. J. Steroid Biochem. Mol. Biol. 2015, 147, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Tung, L.; Mohamed, M.K.; Hoeffler, J.P.; Takimoto, G.S.; Horwitz, K.B. Antagonist-Occupied Human Progesterone B-Receptors Activate Transcription without Binding to Progesterone Response Elements and Are Dominantly Inhibited by A-Receptors. Mol. Endocrinol. 1993, 7, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Vegeto, E.; Shahbaz, M.M.; Wen, D.X.; Goldman, M.E.; O’Malley, B.W.; McDonnell, D.P. Human Progesterone Receptor A Form Is a Cell- and Promoter-Specific Repressor of Human Progesterone Receptor B Function. Mol. Endocrinol. 1993, 7, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Xu, B.; Suganuma, I.; Kusuki, I.; Mita, S.; Shimizu, Y.; Mizuguchi, K.; Kitawaki, J. Dienogest Inhibits Aromatase and Cyclooxygenase-2 Expression and Prostaglandin E2 Production in Human Endometriotic Stromal Cells in Spheroid Culture. Fertil. Steril. 2012, 97, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Mita, S.; Takeuchi, T.; Notsu, T.; Mizuguchi, K.; Kyo, S. Dienogest, a Synthetic Progestin, Inhibits Prostaglandin E2 Production and Aromatase Expression by Human Endometrial Epithelial Cells in a Spheroid Culture System. Steroids 2011, 76, 60–67. [Google Scholar] [CrossRef]
- Mita, S.; Shimizu, Y.; Notsu, T.; Imada, K.; Kyo, S. Dienogest Inhibits Toll-like Receptor 4 Expression Induced by Costimulation of Lipopolysaccharide and High-Mobility Group Box 1 in Endometrial Epithelial Cells. Fertil. Steril. 2011, 96, 1485–1489.e4. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Okamoto, R.; Tsuzuki, T.; Tsuji, S.; Yasuda, K.; Kanzaki, H. Progestins Inhibit Estradiol-Induced Vascular Endothelial Growth Factor and Stromal Cell–Derived Factor 1 in Human Endometrial Stromal Cells. Fertil. Steril. 2011, 96, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Igarashi, S.; Sasaki, M.; Tanaka, T. Distribution of Cyclooxygenase-2 in Eutopic and Ectopic Endometrium in Endometriosis and Adenomyosis. Hum. Reprod. 2001, 16, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Rakhila, H.; Carli, C.; Daris, M.; Lemyre, M.; Leboeuf, M.; Akoum, A. Identification of Multiple and Distinct Defects in Prostaglandin Biosynthetic Pathways in Eutopic and Ectopic Endometrium of Women with Endometriosis. Fertil. Steril. 2013, 100, 1650–1659.e2. [Google Scholar] [CrossRef]
- Moriyama, T.; Higashi, T.; Togashi, K.; Iida, T.; Segi, E.; Sugimoto, Y.; Tominaga, T.; Narumiya, S.; Tominaga, M. Sensitization of TRPV1 by EP 1 and IP Reveals Peripheral Nociceptive Mechanism of Prostaglandins. Mol. Pain 2005, 1, 1744–8069-1–3. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.M.; Grosser, T.; Wang, M.; Yu, Y.; FitzGerald, G.A. Prostanoids in Health and Disease. J. Lipid Res. 2009, 50, S423–S428. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Sakurai, T.; Kogo, H. Relationship between Prostaglandin E2 and Vascular Endothelial Growth Factor (VEGF) in Angiogenesis in Human Vascular Endothelial Cells. Vasc. Pharmacol. 2006, 44, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Banu, S.K.; Rodriguez, R.; Starzinski-Powitz, A.; Arosh, J.A. Selective Blockade of Prostaglandin E2 Receptors EP2 and EP4 Signaling Inhibits Proliferation of Human Endometriotic Epithelial Cells and Stromal Cells through Distinct Cell Cycle Arrest. Fertil. Steril. 2010, 93, 2498–2506. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Banu, S.K.; Burghardt, R.C.; Starzinski-Powitz, A.; Arosh, J.A. Selective Inhibition of Prostaglandin E2 Receptors EP2 and EP4 Inhibits Adhesion of Human Endometriotic Epithelial and Stromal Cells through Suppression of Integrin-Mediated Mechanisms1. Biol. Reprod. 2013, 88, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Banu, S.K.; Subbarao, T.; Starzinski-Powitz, A.; Arosh, J.A. Selective Inhibition of Prostaglandin E2 Receptors EP2 and EP4 Inhibits Invasion of Human Immortalized Endometriotic Epithelial and Stromal Cells through Suppression of Metalloproteinases. Mol. Cell. Endocrinol. 2011, 332, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Orr, N.L.; Albert, A.; Liu, Y.D.; Lum, A.; Hong, J.; Ionescu, C.L.; Senz, J.; Nazeran, T.M.; Lee, A.F.; Noga, H.; et al. KRAS Mutations and Endometriosis Burden of Disease. J. Pathol. Clin. Res. 2023, 9, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Osuga, Y.; Morimoto, C.; Hirata, T.; Hirota, Y.; Yano, T.; Taketani, Y. Dienogest Inhibits BrdU Uptake with G0/G1 Arrest in Cultured Endometriotic Stromal Cells. Fertil. Steril. 2008, 89, 1344–1347. [Google Scholar] [CrossRef] [PubMed]
- Okada, H. The Inhibitory Effect of Dienogest, a Synthetic Steroid, on the Growth of Human Endometrial Stromal Cells in Vitro. Mol. Hum. Reprod. 2001, 7, 341–347. [Google Scholar] [CrossRef] [PubMed]
- MacLean, J.A.; Hayashi, K. Progesterone Actions and Resistance in Gynecological Disorders. Cells 2022, 11, 647. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, F.; Mansouri, N.; Shahhoseini, M.; Heidari Khoei, H.; Mikaeeli, G.; Vankelecom, H.; Baharvand, H. Endometriosis Organoids: Prospects and Challenges. Reprod. BioMedicine Online 2022, 45, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Gnecco, J.S.; Brown, A.; Buttrey, K.; Ives, C.; Goods, B.A.; Baugh, L.; Hernandez-Gordillo, V.; Loring, M.; Isaacson, K.B.; Griffith, L.G. Organoid Co-Culture Model of the Human Endometrium in a Fully Synthetic Extracellular Matrix Enables the Study of Epithelial-Stromal Crosstalk. Med 2023, 4, 554–579.e9. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.H.; Wilde, M.I. Dienogest. Drugs 1998, 56, 825–833. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, K.; Nakayama, K.; Razia, S.; Islam, S.H.; Farzana, Z.U.; Sonia, S.B.; Yamashita, H.; Ishikawa, M.; Ishibashi, T.; Imamura, K.; et al. Association between KRAS and PIK3CA Mutations and Progesterone Resistance in Endometriotic Epithelial Cell Line. Curr. Issues Mol. Biol. 2024, 46, 3579-3594. https://doi.org/10.3390/cimb46040224
Kanno K, Nakayama K, Razia S, Islam SH, Farzana ZU, Sonia SB, Yamashita H, Ishikawa M, Ishibashi T, Imamura K, et al. Association between KRAS and PIK3CA Mutations and Progesterone Resistance in Endometriotic Epithelial Cell Line. Current Issues in Molecular Biology. 2024; 46(4):3579-3594. https://doi.org/10.3390/cimb46040224
Chicago/Turabian StyleKanno, Kosuke, Kentaro Nakayama, Sultana Razia, Sohel Hasibul Islam, Zahan Umme Farzana, Shahataj Begum Sonia, Hitomi Yamashita, Masako Ishikawa, Tomoka Ishibashi, Kayo Imamura, and et al. 2024. "Association between KRAS and PIK3CA Mutations and Progesterone Resistance in Endometriotic Epithelial Cell Line" Current Issues in Molecular Biology 46, no. 4: 3579-3594. https://doi.org/10.3390/cimb46040224