
Identifying Network Biomarkers in Early Diagnosis of Hepatocellular Carcinoma via miRNA–Gene Interaction Network Analysis
Abstract
:1. Introduction
2. Materials and Methods
2.1. Screening Differentially Expressed Genes and miRNAs
2.2. Functional Enrichment Analysis
2.3. miRNA–Gene Integrated Analysis
2.4. Construct miRNA–Gene Integrated Network
2.5. Survival Analysis of Hub Nodes
3. Results
3.1. Screening Differentially Expressed Genes
3.2. Function Enrichment
3.3. Identification of cDEM
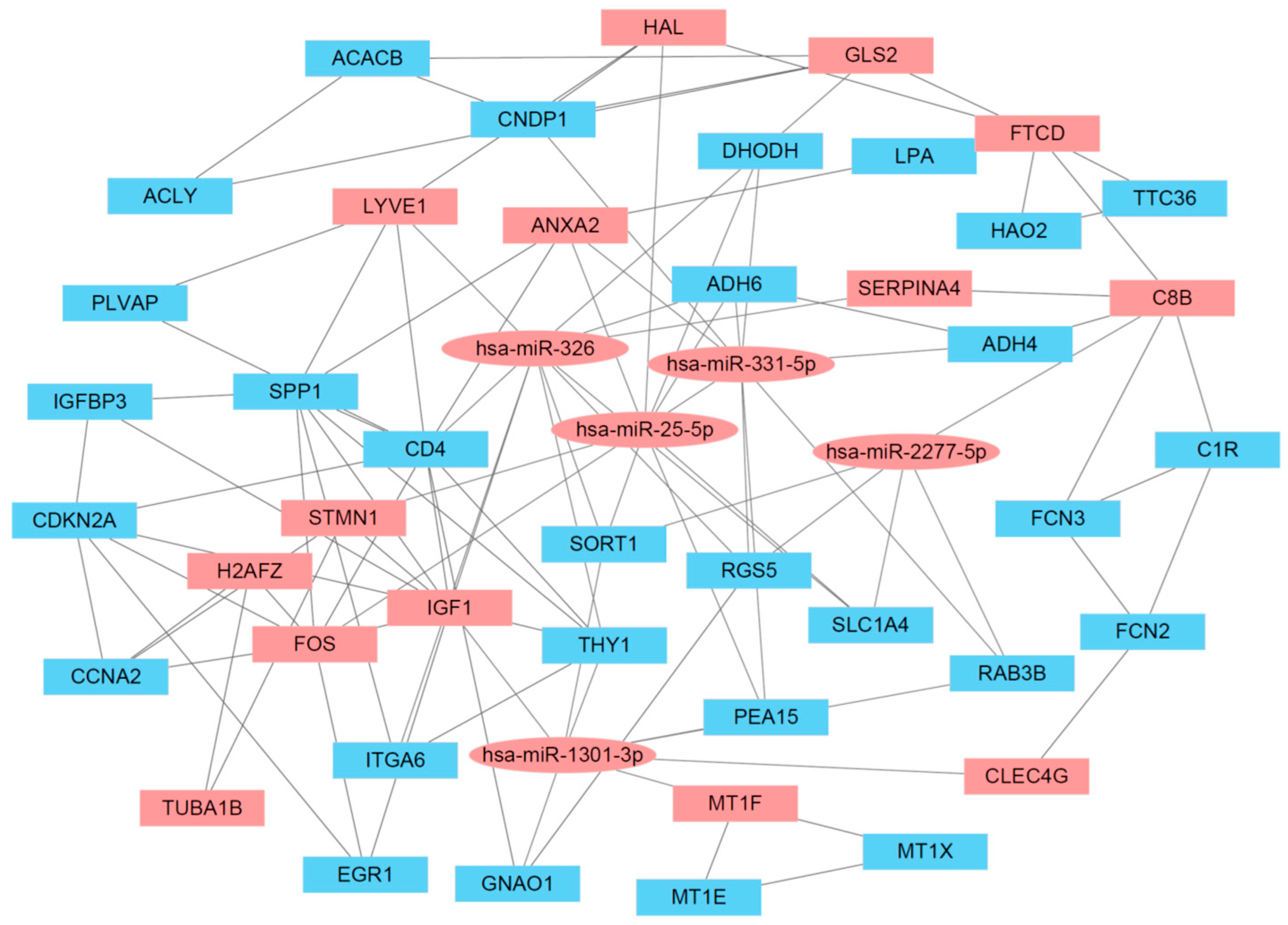
3.4. cDEG–cDEM Interaction Network
3.5. Target Enrichment Analysis of cDEM
3.6. Network Analysis of the cDEG–cDEM Network
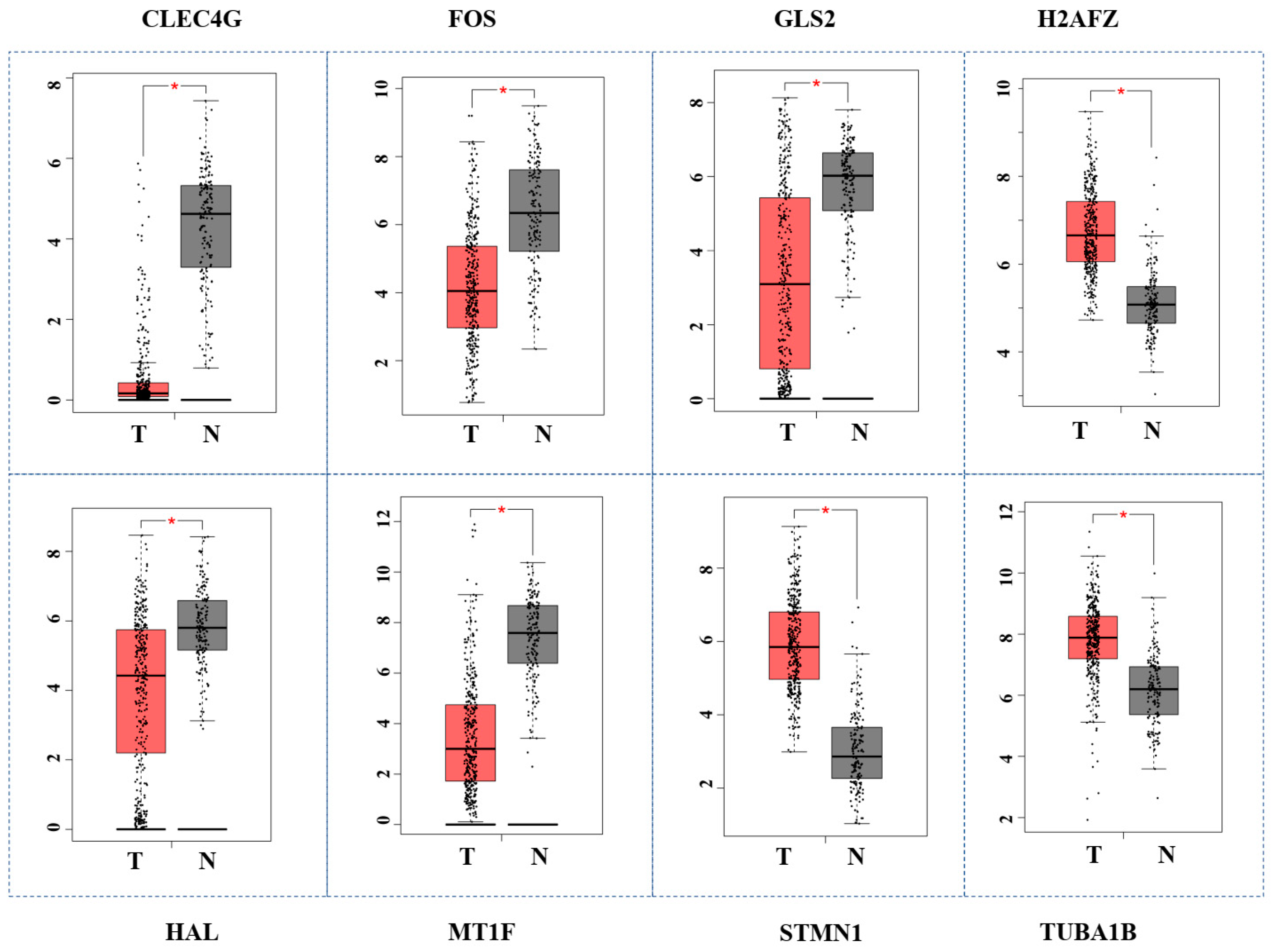
3.7. Validation of Hub Genes by GEPIA
3.8. Survival Analysis of Hub Genes Using KMplot
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Wangensteen, K.J.; Chang, K.M. Multiple Roles for Hepatitis B and C Viruses and the Host in the Development of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. 1), 27–37. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, J.; Ziol, M.; Paradis, V.; Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 2019, 71, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Galle, P.R.; Foerster, F.; Kudo, M.; Chan, S.L.; Llovet, J.M.; Qin, S.; Schelman, W.R.; Chintharlapalli, S.; Abada, P.B.; Sherman, M.; et al. Biology and significance of alpha-fetoprotein in hepatocellular carcinoma. Liver Int. 2019, 39, 2214–2229. [Google Scholar] [CrossRef]
- Pattan, V.; Kashyap, R.; Bansal, V.; Candula, N.; Koritala, T.; Surani, S. Genomics in medicine: A new era in medicine. World J. Methodol. 2021, 11, 231–242. [Google Scholar] [CrossRef]
- Vargas, A.J.; Harris, C.C. Biomarker development in the precision medicine era: Lung cancer as a case study. Nat. Rev. Cancer 2016, 16, 525–537. [Google Scholar] [CrossRef]
- Macias, R.I.R.; Kornek, M.; Rodrigues, P.M.; Paiva, N.A.; Castro, R.E.; Urban, S.; Pereira, S.P.; Cadamuro, M.; Rupp, C.; Loosen, S.H.; et al. Diagnostic and prognostic biomarkers in cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. 1), 108–122. [Google Scholar] [CrossRef] [PubMed]
- Panizza, E.; Regalado, B.D.; Wang, F.; Nakano, I.; Vacanti, N.M.; Cerione, R.A.; Antonyak, M.A. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Sci. Alliance 2023, 6, e202201680. [Google Scholar] [CrossRef]
- Forterre, A.; Komuro, H.; Aminova, S.; Harada, M. A Comprehensive Review of Cancer MicroRNA Therapeutic Delivery Strategies. Cancers 2020, 12, 1852. [Google Scholar] [CrossRef]
- Afshari, A.; Yaghobi, R.; Karimi, M.H.; Mowla, J. Alterations in MicroRNA gene expression profile in liver transplant patients with hepatocellular carcinoma. BMC Gastroenterol. 2021, 21, 262. [Google Scholar] [CrossRef]
- Yang, C.; Ma, X.; Guan, G.; Liu, H.; Yang, Y.; Niu, Q.; Wu, Z.; Jiang, Y.; Bian, C.; Zang, Y.; et al. MicroRNA-766 promotes cancer progression by targeting NR3C2 in hepatocellular carcinoma. FASEB J. 2019, 33, 1456–1467. [Google Scholar] [CrossRef]
- Yalçınkaya, B.; Güzel, E.; Taştekin, D.; Pençe, S. Role of mir-33a, mir-203b, mir361-3p, and mir-424 in hepatocellular carcinoma. Turk. J. Med. Sci. 2021, 51, 638–643. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.L.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W.Z. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Lin, Y.C.D.; Cui, S.D.; Huang, Y.X.; Tang, Y.; Xu, J.T.; Bao, J.Y.; Li, Y.L.; Wen, J.; Zuo, H.L.; et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef] [PubMed]
- Kehl, T.; Kern, F.; Backes, C.; Fehlmann, T.; Stockel, D.; Meese, E.; Lenhof, H.P.; Keller, A. miRPathDB 2.0: A novel release of the miRNA Pathway Dictionary Database. Nucleic Acids Res. 2020, 48, D142–D147. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Lu, L.; Zhang, M. Edge Betweenness Centrality. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; pp. 647–648. [Google Scholar]
- Valera-Calero, J.A.; Arendt-Nielsen, L.; Cigaran-Mendez, M.; Fernandez-De-Las-Penas, C.; Varol, U. Network Analysis for Better Understanding the Complex Psycho-Biological Mechanisms behind Fibromyalgia Syndrome. Diagnostics 2022, 12, 1845. [Google Scholar] [CrossRef]
- Li, C.W.; Tang, Z.F.; Zhang, W.J.; Ye, Z.C.; Liu, F. GEPIA2021: Integrating multiple deconvolution-based analysis into GEPIA. Nucleic Acids Res. 2021, 49, W242–W246. [Google Scholar] [CrossRef] [PubMed]
- Lanczky, A.; Gyorffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, J.; Yuan, R.; Wang, Y.; Zhang, J.; Lin, Y.; Wang, L.; Zhu, X.; Zhu, W.; Bai, J.; et al. Dynamic 3D genome reorganization during development and metabolic stress of the porcine liver. Cell Discov. 2022, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar]
- Kotsiliti, E. Understanding HCC sex disparities. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 147. [Google Scholar] [CrossRef]
- Wei, G.Y.; Hu, M.; Zhao, L.; Guo, W.S. MiR-451a suppresses cell proliferation, metastasis and EMT via targeting YWHAZ in hepatocellular carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5158–5167. [Google Scholar]
- Gan, Y.; Ye, F.; He, X.X. The role of YWHAZ in cancer: A maze of opportunities and challenges. J. Cancer 2020, 11, 2252–2264. [Google Scholar] [CrossRef]
- Duron, C.; Pan, Y.; Gutmann, D.H.; Hardin, J.; Radunskaya, A. Variability of Betweenness Centrality and Its Effect on Identifying Essential Genes. Bull. Math. Biol. 2019, 81, 3655–3673. [Google Scholar] [CrossRef]
- Yerukala, S.; Ho, S.Y. Novel miRNA signature for predicting the stage of hepatocellular carcinoma. Sci. Rep. 2020, 10, 14452. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Honarmand Tamizkar, K.; Hussen, B.M.; Taheri, M. MicroRNA signature in liver cancer. Pathol. Res. Pract. 2021, 219, 153369. [Google Scholar] [CrossRef] [PubMed]
- Bongolo, C.C.; Thokerunga, E.; Yan, Q.; Yacouba, M.B.M.; Wang, C. Exosomes Derived from microRNA-27a-3p Overexpressing Mesenchymal Stem Cells Inhibit the Progression of Liver Cancer through Suppression of Golgi Membrane Protein 1. Stem Cells Int. 2022, 12, 9748714. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Ha, D.; Lee, J.; Kim, I.; Park, M.; Im, S.H.; Shin, K.; Kim, S. Network-based machine learning approach to predict immunotherapy response in cancer patients. Nat. Commun. 2022, 13, 3703. [Google Scholar] [CrossRef] [PubMed]
- Shee, S.; Koh, R.; Voon, K.; Chye, S.; Othman, I.; Ng, K. The roles of microRNA-331 Family in Cancers. J. Cancer Res. Pract. 2019, 6, 1–6. [Google Scholar]
- Aizarani, N.; Saviano, A.; Sagar, N.; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef]
- Suzuki, S.; Venkatesh, D.; Kanda, H.; Nakayama, A.; Hosokawa, H.; Lee, E.Y.; Miki, T.; Stockwell, B.R.; Yokote, K.; Tanaka, T.; et al. GLS2 Is a Tumor Suppressor and a Regulator of Ferroptosis in Hepatocellular Carcinoma. Cancer Res. 2022, 82, 3209–3222. [Google Scholar] [CrossRef]
- Bongiovanni, L.; Andriessen, A.; Silvestri, S.; Porcellato, I.; Brachelente, C.; De Bruin, A. H2AFZ: A Novel Prognostic Marker in Canine Melanoma and a Predictive Marker for Resistance to CDK4/6 Inhibitor Treatment. Front. Vet. Sci. 2021, 8, 705359. [Google Scholar] [CrossRef] [PubMed]
- Žujović Janko, T.; Stojanović Marko, M.; Brzački Vesna, M.; Kujović Aleksandar, D.; Đorđević Miodrag, N.; Jančić Snežana, A.; Milenković Sanja, M.; Milošević Velimir, S. Influence of stathmin 1 (STMN1) expression on neoangiogenesis in colorectal adenocarcinoma. Pathol. Res. Pract. 2022, 238, 154057. [Google Scholar] [CrossRef]
- Leca, I.; Phillips, A.W.; Ushakova, L.; Cushion, T.D.; Keays, D.A. Codon modification of Tuba1a alters mRNA levels and causes a severe neurodevelopmental phenotype in mice. Sci. Rep. 2023, 13, 1215. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Term | Count | Proportion | p-Value |
|---|---|---|---|---|
| UP_TISSUE | Liver | 54 | 57.45% | 1.71 × 10−26 |
| UP_TISSUE | Kidney | 15 | 15.96% | 1.77 × 10−02 |
| UP_TISSUE | Plasma | 13 | 13.83% | 1.88 × 10−08 |
| UP_TISSUE | PCR rescued clones | 9 | 9.57% | 3.28 × 10−02 |
| UP_TISSUE | Hippocampus | 7 | 7.45% | 2.52 × 10−02 |
| UP_TISSUE | Fetal liver | 6 | 6.38% | 3.46 × 10−03 |
| UP_TISSUE | Myometrium | 2 | 2.13% | 4.25 × 10−02 |
| GO Biological Processes | Monocarboxylic acid metabolic process | 21 | 22.34% | 2.94 × 10−18 |
| GO Biological Processes | Alcohol metabolic process | 13 | 13.83% | 1.99 × 10−11 |
| GO Biological Processes | Fatty-acid metabolic process | 13 | 13.83% | 2.82 × 10−11 |
| Reactome Gene Sets | Biological oxidation | 12 | 12.77% | 5.20 × 10−12 |
| GO Biological Processes | Olefinic compound metabolic process | 11 | 11.70% | 9.69 × 10−13 |
| GO Biological Processes | Terpenoid metabolic process | 10 | 10.64% | 5.26 × 10−13 |
| GO Biological Processes | Isoprenoid metabolic process | 10 | 10.64% | 2.98 × 10−12 |
| GO Biological Processes | Cellular hormone metabolic process | 10 | 10.64% | 7.48 × 10−12 |
| GO Biological Processes | Epoxygenase P450 pathway | 6 | 6.38% | 2.03 × 10−11 |
| Reactome Gene Sets | Synthesis of hydroxyeicosatetraenoic acids | 5 | 5.32% | 3.27 × 10−11 |
| No. | miRNA | Transcribed mRNA | Gene Symbol | Binding Energy |
|---|---|---|---|---|
| 1 | hsa-miR-106a-5p | NM_001361 | DHODH | −22.8 |
| 2 | hsa-miR-106b-5p | NM_001102470 | ADH6 | −21.4 |
| 3 | hsa-miR-1180-3p | NM_001145454 | STMN1 | −27.5 |
| 4 | hsa-miR-1301-3p | NM_033304 | ADRA1A | −26.9 |
| 5 | hsa-miR-136-5p | NM_001237 | CCNA2 | −20.8 |
| 6 | hsa-miR-15b-5p | NM_001280797 | GLS2 | −19.0 |
| 7 | hsa-miR-18a-5p | NM_152545 | RASGEF1B | −20.9 |
| 8 | hsa-miR-2277-5p | NM_016593 | CYP39A1 | −29.3 |
| 9 | hsa-miR-25-5p | NM_018281 | ECHDC2 | −24.2 |
| 10 | hsa-miR-324-5p | NM_001257 | CDH13 | −23.9 |
| 11 | hsa-miR-326 | NM_001289033 | SERPINA4 | −29.0 |
| 12 | hsa-miR-331-5p | NM_001306171 | ADH4 | −27.4 |
| 13 | hsa-miR-374b-3p | NM_001144911 | CLEC4M | −17.1 |
| 14 | hsa-miR-379-5p | NM_001363587 | CYP4A11 | −22.4 |
| 15 | hsa-miR-17-5p | NM_001297576 | PEA15 | −18.7 |
| 16 | hsa-miR-301a-3p | NM_001297576 | PEA15 | −16.4 |
| 17 | hsa-miR-369-5p | NM_001205228 | SORT1 | −16.8 |
| 18 | hsa-miR-376c-3p | NM_152545 | RASGEF1B | −18.0 |
| 19 | hsa-miR-381-3p | NM_001311160 | THY1 | −23.3 |
| 20 | hsa-miR-451a | NM_001311160 | THY1 | −17.0 |
| miRNA | Database | Pathway | Hits | p-Value |
|---|---|---|---|---|
| hsa-miR-451a | WikiPathways | Hepatitis C and Hepatocellular Carcinoma | 5 | 2.3 × 10−6 |
| hsa-miR-106a-5p | WikiPathways | Hepatitis C and Hepatocellular Carcinoma | 13 | 9.1 × 10−5 |
| hsa-miR-106b-5p | WikiPathways | Hepatitis C and Hepatocellular Carcinoma | 7 | 2.1 × 10−4 |
| hsa-miR-17-5p | WikiPathways | Hepatitis C and Hepatocellular Carcinoma | 8 | 5.6 × 10−4 |
| hsa-miR-15b-5p | WikiPathways | Hepatitis C and Hepatocellular Carcinoma | 10 | 2.0 × 10−3 |
| hsa-miR-18a-5p | WikiPathways | Hepatitis C and Hepatocellular Carcinoma | 5 | 0.012 |
| hsa-miR-376c-3p | WikiPathways | Hepatitis C and Hepatocellular Carcinoma | 2 | 0.025 |
| hsa-miR-326 | WikiPathway | Hepatitis C and Hepatocellular Carcinoma | 3 | 0.037 |
| hsa-miR-381-3p | WikiPathways | Hepatitis C and Hepatocellular Carcinoma | 30 | 0.049 |
| No. | Edge | Edge Betweenness | Type |
|---|---|---|---|
| 1 | FOS interacts with H2AFZ | 512.84 | 2 |
| 2 | C8B interacts with FTCD | 423.06 | 2 |
| 3 | STMN1 interacts with TUBA1B | 408.92 | 2 |
| 4 | hsa-miR-1301-3p interacts with MT1F | 387.96 | 1 |
| 5 | hsa-miR-25-5p interacts with HAL | 380.97 | 1 |
| 6 | hsa-miR-331-5p interacts with FOS | 377.00 | 1 |
| 7 | hsa-miR-326 interacts with GLS2 | 370.50 | 1 |
| 8 | FTCD interacts with HAL | 369.99 | 2 |
| 9 | hsa-miR-1301-3p interacts with CLEC4G | 348.00 | 1 |
| 10 | hsa-miR-25-5p interacts with STMN1 | 343.13 | 1 |
| 11 | hsa-miR-326 interacts with SERPINA4 | 313.91 | 1 |
| 12 | hsa-miR-2277-5p interacts with C8B | 307.48 | 1 |
| 13 | hsa-miR-1301-3p interacts with IGF1 | 279.78 | 1 |
| 14 | hsa-miR-25-5p interacts with ANXA2 | 275.02 | 1 |
| 15 | HAL interacts with LYVE1 | 270.90 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Qi, Y.; Wang, Y.; Chen, X.; Wang, Y.; Zhang, X. Identifying Network Biomarkers in Early Diagnosis of Hepatocellular Carcinoma via miRNA–Gene Interaction Network Analysis. Curr. Issues Mol. Biol. 2023, 45, 7374-7387. https://doi.org/10.3390/cimb45090466
Yang Z, Qi Y, Wang Y, Chen X, Wang Y, Zhang X. Identifying Network Biomarkers in Early Diagnosis of Hepatocellular Carcinoma via miRNA–Gene Interaction Network Analysis. Current Issues in Molecular Biology. 2023; 45(9):7374-7387. https://doi.org/10.3390/cimb45090466
Chicago/Turabian StyleYang, Zhiyuan, Yuanyuan Qi, Yijing Wang, Xiangyun Chen, Yuerong Wang, and Xiaoli Zhang. 2023. "Identifying Network Biomarkers in Early Diagnosis of Hepatocellular Carcinoma via miRNA–Gene Interaction Network Analysis" Current Issues in Molecular Biology 45, no. 9: 7374-7387. https://doi.org/10.3390/cimb45090466