
QTL Mapping and Genome-Wide Association Study Reveal Genetic Loci and Candidate Genes Related to Soluble Solids Content in Melon

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Phenotypic Data Analysis
2.2. DNA Extraction and Whole-Genome Resequencing
2.3. Linkage Analysis
2.4. Association Analysis
2.5. Candidate Genes Identification and qRT-PCR Analysis
3. Results
3.1. Phenotypic Variation of Soluble Solids Content in Two Populations
3.2. Population Sequencing and Linkage Map Construction
3.3. Identification of QTLs for Soluble Solids Content
3.4. GWAS for Soluble Solids Content
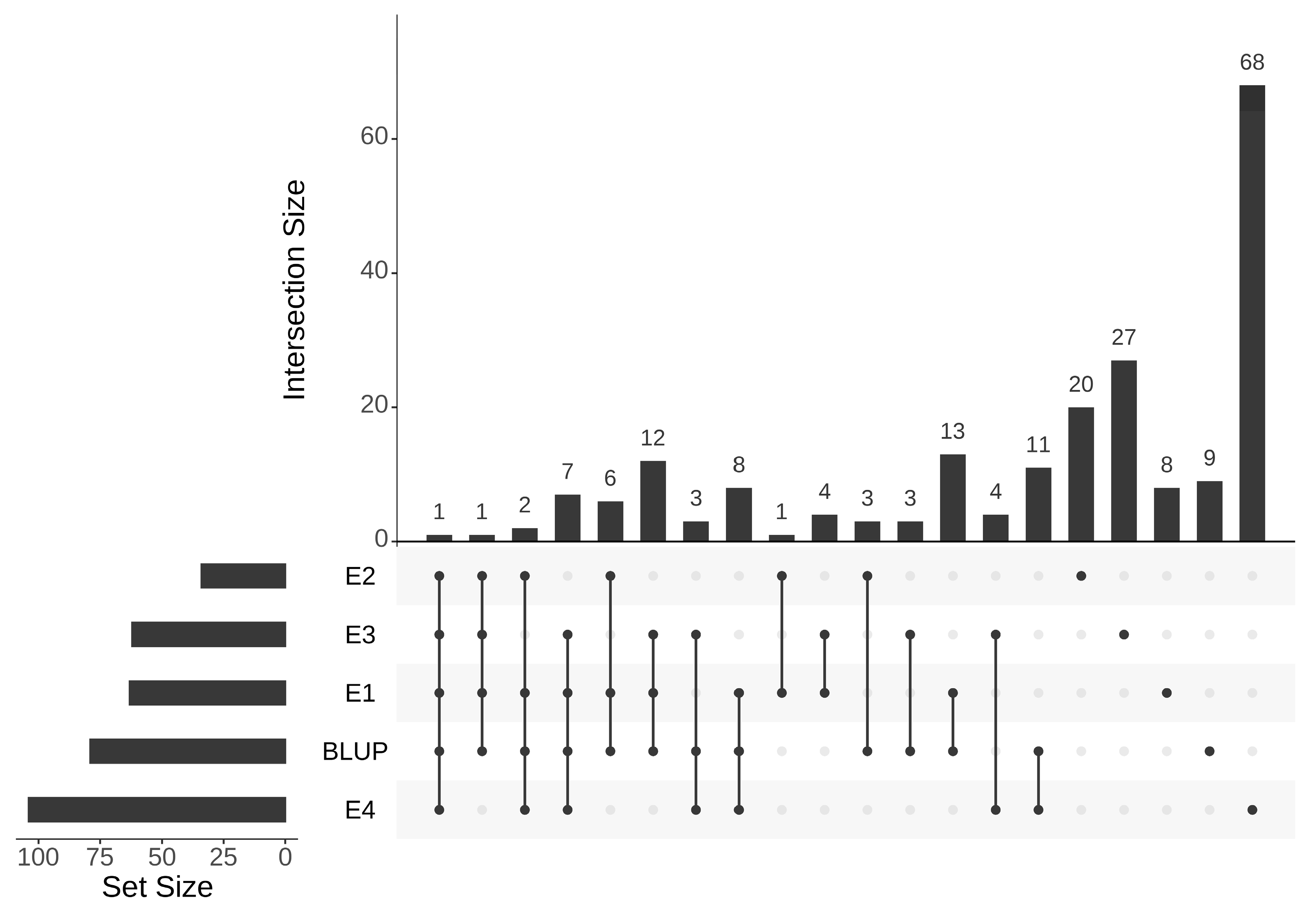
3.5. Co-Detected Regions by QTL Mapping and GWAS
3.6. Identification of Candidate Genes Related to SSC
4. Discussion
4.1. Phenotypic Data over Multiple Years and High-Density Markers Improved the Accuracy of QTL Mapping and Gwas
4.2. QTL Intervals and SNP Loci for SSC Identified via Linkage Mapping and GWAS
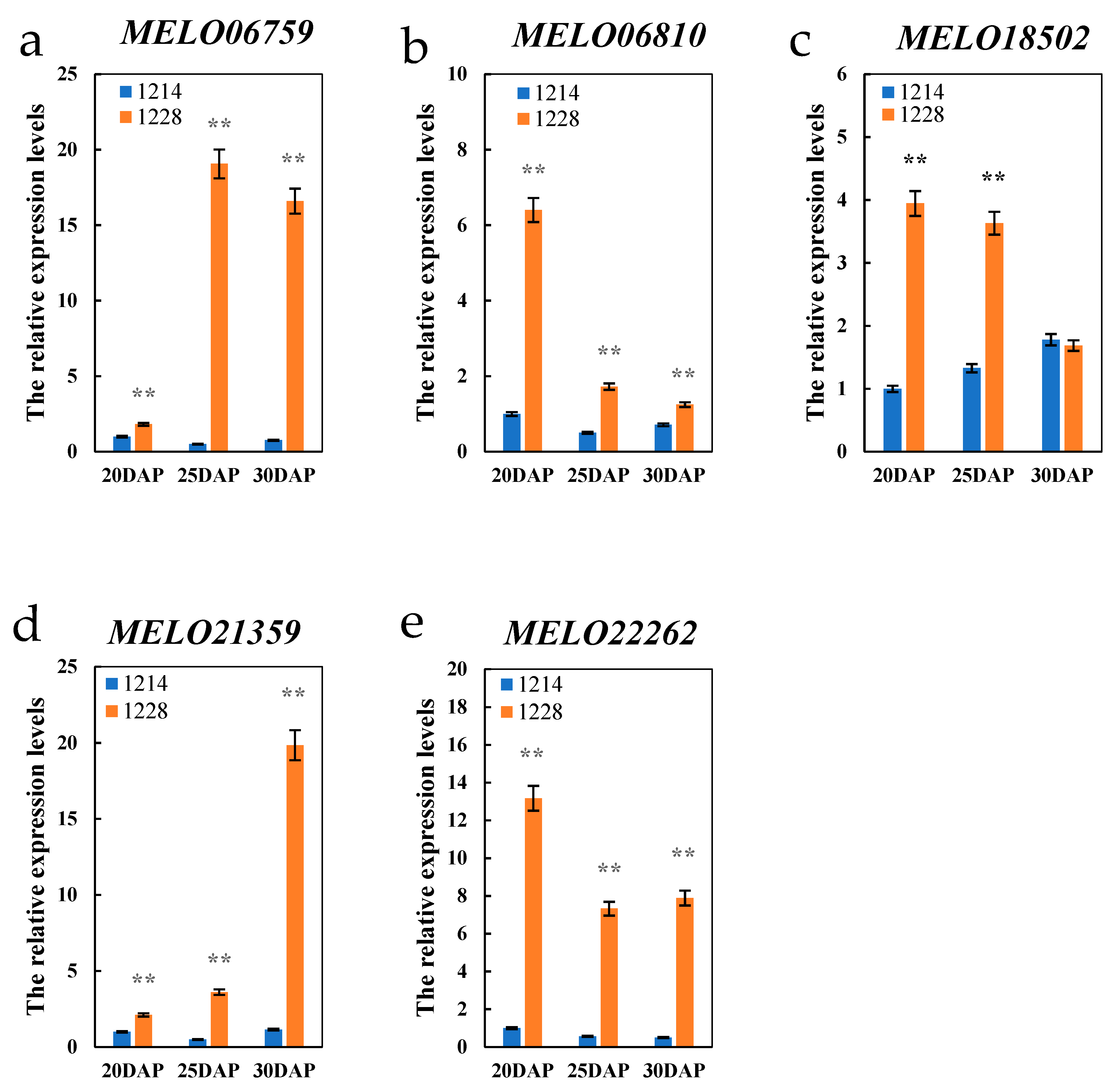
4.3. Further Analysis of Candidate Genes Differentially Expressed between Parents at Different Fruit Growth Stages
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jeffrey, C. A review of the Cucurbitaceae. Bot. J. Linn. Soc. 1980, 81, 233–247. [Google Scholar] [CrossRef]
- Pitrat, M.; Hanelt, P.; Hammer, K. Some Comments on Infraspecific Classification of Cultivars of Melon. Acta Hortic. 2000, 510, 29–36. [Google Scholar] [CrossRef]
- Monforte, A.J.; Oliver, M.; Gonzalo, M.J.; Alvarez, J.M.; Dolcet-Sanjuan, R.; Arús, P. Identification of quantitative trait loci involved in fruit quality traits in melon (Cucumis melo L.). Theor. Appl. Genet. 2003, 108, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Eduardo, I.; Arús, P.; Monforte, A.J.; Obando, J.; Fernández-Trujillo, J.P.; Martínez, J.A.; Alarcón, A.L.; Álvarez, J.M.; van der Knaap, E. Estimating the Genetic Architecture of Fruit Quality Traits in Melon Using a Genomic Library of Near Isogenic Lines. J. Am. Soc. Hortic. Sci. 2007, 132, 80–89. [Google Scholar] [CrossRef]
- Paris, M.K.; Zalapa, J.E.; McCreight, J.D.; Staub, J.E. Genetic dissection of fruit quality components in melon (Cucumis melo L.) using a RIL population derived from exotic × elite US Western Shipping germplasm. Mol. Breed. 2008, 22, 405–419. [Google Scholar] [CrossRef]
- Harel-Beja, R.; Tzuri, G.; Portnoy, V.; Lotan-Pompan, M.; Lev, S.; Cohen, S.; Dai, N.; Yeselson, L.; Meir, A.; Libhaber, S.E.; et al. A genetic map of melon highly enriched with fruit quality QTLs and EST markers, including sugar and carotenoid metabolism genes. Theor. Appl. Genet. 2010, 121, 511–533. [Google Scholar] [CrossRef]
- Diaz, A.; Fergany, M.; Formisano, G.; Ziarsolo, P.; Blanca, J.; Fei, Z.; Staub, J.E.; Zalapa, J.E.; Cuevas, H.E.; Dace, G.; et al. A consensus linkage map for molecular markers and Quantitative Trait Loci associated with economically important traits in melon (Cucumis melo L.). BMC Plant Biol. 2011, 11, 111. [Google Scholar] [CrossRef]
- Ramamurthy, R.K.; Waters, B.M. Identification of fruit quality and morphology QTLs in melon (Cucumis melo) using a population derived from flexuosus and cantalupensis botanical groups. Euphytica 2015, 204, 163–177. [Google Scholar] [CrossRef]
- Baloch, A.; Baloch, A.W.; Liu, S.; Gao, P.; Baloch, M.J.; Wang, X.; Davis, A.; Ali, M.; Luan, F. Linkage map construction and qtl analysis of fruit traits in melon (Cucumis melo L.) based on caps markers. Pak. J. Bot. 2016, 48, 1579–1584. [Google Scholar]
- Argyris, J.M.; Díaz, A.; Ruggieri, V.; Fernández, M.; Jahrmann, T.; Gibon, Y.; Picó, B.; Martín-Hernández, A.M.; Monforte, A.J.; Garcia-Mas, J. QTL Analyses in Multiple Populations Employed for the Fine Mapping and Identification of Candidate Genes at a Locus Affecting Sugar Accumulation in Melon (Cucumis melo L.). Front. Plant Sci. 2017, 8, 1679. [Google Scholar] [CrossRef]
- Amanullah, S.; Liu, S.; Gao, P.; Zhu, Z.; Zhu, Q.; Fan, C.; Luan, F. QTL mapping for melon (Cucumis melo L.) fruit traits by assembling and utilization of novel SNPs based CAPS markers. Sci. Hortic. 2018, 236, 18–29. [Google Scholar] [CrossRef]
- Galpaz, N.; Gonda, I.; Shem-Tov, D.; Barad, O.; Tzuri, G.; Lev, S.; Fei, Z.; Xu, Y.; Mao, L.; Jiao, C.; et al. Deciphering genetic factors that determine melon fruit-quality traits using RNA -Seq-based high-resolution QTL and eQTL mapping. Plant J. 2018, 94, 169–191. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Ruggieri, V.; Pérez, S.; Alexiou, K.G.; Fernández, M.; Jahrmann, T.; Pujol, M.; Garcia-Mas, J. QTL mapping of melon fruit quality traits using a high-density GBS-based genetic map. BMC Plant Biol. 2018, 18, 324. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Domingo, M.S.; Argyris, J.; Mayobre, C.; Valverde, L.; Martín-Hernández, A.M.; Pujol, M.; Garcia-Mas, J. A novel introgression line collection to unravel the genetics of climacteric ripening and fruit quality in melon. Sci. Rep. 2021, 11, 11364. [Google Scholar] [CrossRef] [PubMed]
- Gur, A.; Tzuri, G.; Meir, A.; Sa’ar, U.; Portnoy, V.; Katzir, N.; Schaffer, A.A.; Li, L.; Burger, J.; Tadmor, Y. Genome-Wide Linkage-Disequilibrium Mapping to the Candidate Gene Level in Melon (Cucumis melo). Sci. Rep. 2017, 7, 9770. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Lian, Q.; Zhang, Z.; Fu, Q.; He, Y.; Ma, S.; Ruggieri, V.; Monforte, A.J.; Wang, P.; Julca, I.; et al. A comprehensive genome variation map of melon identifies multiple domestication events and loci influencing agronomic traits. Nat. Genet. 2019, 51, 1607–1615. [Google Scholar] [CrossRef]
- Liu, S.; Gao, P.; Zhu, Q.; Zhu, Z.; Liu, H.; Wang, X.; Weng, Y.; Gao, M.; Luan, F. Resequencing of 297 melon accessions reveals the genomic history of improvement and loci related to fruit traits in melon. Plant Biotechnol. J. 2020, 18, 2545–2558. [Google Scholar] [CrossRef]
- Pavan, S.; Marcotrigiano, A.; Ciani, E.; Mazzeo, R.; Zonno, V.; Ruggieri, V.; Lotti, C.; Ricciardi, L. Genotyping-by-sequencing of a melon (Cucumis melo L.) germplasm collection from a secondary center of diversity highlights patterns of genetic variation and genomic features of different gene pools. BMC Genom. 2017, 18, 59. [Google Scholar] [CrossRef]
- Oren, E.; Dafna, A.; Tzuri, G.; Halperin, I.; Isaacson, T.; Elkabetz, M.; Meir, A.; Saar, U.; Ohali, S.; La, T.; et al. Pan-genome and multi-parental framework for high-resolution trait dissection in melon (Cucumis melo). Plant J. 2022, 112, 1525–1542. [Google Scholar] [CrossRef]
- Tripodi, P.; Soler, S.; Campanelli, G.; Díez, M.J.; Esposito, S.; Sestili, S.; Figàs, M.R.; Leteo, F.; Casanova, C.; Platani, C.; et al. Genome wide association mapping for agronomic, fruit quality, and root architectural traits in tomato under organic farming conditions. BMC Plant Biol. 2021, 21, 481. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, C.; Ge, P.; Li, F.; Zhu, L.; Wang, Y.; Tao, J.; Zhang, X.; Dong, H.; Gai, W.; et al. A 21-bp InDel in the promoter of STP1 selected during tomato improvement accounts for soluble solid content in fruits. Hortic. Res. 2023, 10, uhad009. [Google Scholar] [CrossRef] [PubMed]
- Ferik, F.; Ates, D.; Ercisli, S.; Erdogan, A.; Orhan, E.; Tanyolac, M.B. Genome-wide association links candidate genes to fruit firmness, fruit flesh color, flowering time, and soluble solid content in apricot (Prunus armeniaca L.). Mol. Biol. Rep. 2021, 49, 5283–5291. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Zhou, Z.; Wang, Q.; Guo, J.; Zhao, P.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wang, X.; et al. Genome-wide association study of 12 agronomic traits in peach. Nat. Commun. 2016, 7, 13246. [Google Scholar] [CrossRef]
- Guo, S.; Zhao, S.; Sun, H.; Wang, X.; Wu, S.; Lin, T.; Ren, Y.; Gao, L.; Deng, Y.; Zhang, J.; et al. Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat. Genet. 2019, 51, 1616–1623. [Google Scholar] [CrossRef]
- Leida, C.; Moser, C.; Esteras, C.; Sulpice, R.; Lunn, J.E.; De Langen, F.; Monforte, A.J.; Picó, B. Variability of candidate genes, genetic structure and association with sugar accumulation and climacteric behavior in a broad germplasm collection of melon (Cucumis melo L.). BMC Genet. 2015, 16, 28. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ando, K.; Wu, S.; Reddy, U.K.; Tamang, P.; Bao, K.; Hammar, S.A.; Grumet, R.; McCreight, J.D.; Fei, Z. Genetic characterization of melon accessions in the U.S. National Plant Germplasm System and construction of a melon core collection. Mol. Hortic. 2021, 1, 11. [Google Scholar] [CrossRef]
- Yang, X.; Yan, J.; Zheng, Y.; Yu, J.; Li, J. Reviews of association analysis for quantitative traits in plants. Acta Agron. Sin. 2007, 33, 523. [Google Scholar]
- Yu, J.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef]
- Wei, Z.; Yuan, Q.; Lin, H.; Li, X.; Zhang, C.; Gao, H.; Zhang, B.; He, H.; Liu, T.; Jie, Z.; et al. Linkage analysis, GWAS, transcriptome analysis to identify candidate genes for rice seedlings in response to high temperature stress. BMC Plant Biol. 2021, 21, 85. [Google Scholar] [CrossRef]
- Xu, J.; Ni, Z.; Chen, F.; Fu, X.; Yu, F. Integrated linkage mapping and genome-wide association study to dissect the genetic basis of zinc deficiency tolerance in maize at seedling stage. Crop. J. 2022, 10, 1807–1818. [Google Scholar] [CrossRef]
- Hu, D.; Li, X.; Yang, Z.; Liu, S.; Hao, D.; Chao, M.; Zhang, J.; Yang, H.; Su, X.; Jiang, M.; et al. Downregulation of a gibberellin 3β-hydroxylase enhances photosynthesis and increases seed yield in soybean. New Phytol. 2022, 235, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Nimmakayala, P.; Tomason, Y.R.; Abburi, V.L.; Alvarado, A.; Saminathan, T.; Vajja, V.G.; Salazar, G.; Panicker, G.K.; Levi, A.; Wechter, W.P.; et al. Genome-Wide Differentiation of Various Melon Horticultural Groups for Use in GWAS for Fruit Firmness and Construction of a High Resolution Genetic Map. Front. Plant Sci. 2016, 7, 1437. [Google Scholar] [CrossRef] [PubMed]
- Perpiñá, G.; Esteras, C.; Gibon, Y.; Monforte, A.J.; Picó, B. A new genomic library of melon introgression lines in a cantaloupe genetic background for dissecting desirable agronomical traits. BMC Plant Biol. 2016, 16, 154. [Google Scholar] [CrossRef]
- Oren, E.; Tzuri, G.; Dafna, A.; Meir, A.; Kumar, R.; Katzir, N.; Elkind, Y.; Freilich, S.; Schaffer, A.A.; Tadmor, Y.; et al. High-density NGS-based map construction and genetic dissection of fruit shape and rind netting in Cucumis melo. Theor. Appl. Genet. 2020, 133, 1927–1945. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Liu, H.; Zhu, Z.; Liu, S.; Song, Z.; Xia, L.; Zhao, J.; Luan, F.; Liu, S. Identification of Candidate Chromosome Region Related to Melon (Cucumis melo L.) Fruit Surface Groove Trait Through Biparental Genetic Mapping and Genome-Wide Association Study. Front. Plant Sci. 2022, 13, 828287. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, X.; Ayre, B.G.; Turgeon, R. The Origin and Composition of Cucurbit “Phloem” Exudate. Plant Physiol. 2012, 158, 1873–1882. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.-Y.; Ho, L.-H.; Neuhaus, H.E.; Guo, W.-J. Transporter SlSWEET15 unloads sucrose from phloem and seed coat for fruit and seed development in tomato. Plant Physiol. 2021, 187, 2230–2245. [Google Scholar] [CrossRef]
- Li, Y.; Liu, H.; Yao, X.; Wang, J.; Feng, S.; Sun, L.; Ma, S.; Xu, K.; Chen, L.-Q.; Sui, X. Hexose transporter CsSWEET7a in cucumber mediates phloem unloading in companion cells for fruit development. Plant Physiol. 2021, 186, 640–654. [Google Scholar] [CrossRef]
- Wen, S.; Neuhaus, H.E.; Cheng, J.; Bie, Z. Contributions of sugar transporters to crop yield and fruit quality. J. Exp. Bot. 2022, 73, 2275–2289. [Google Scholar] [CrossRef]
- Ren, Y.; Sun, H.; Zong, M.; Guo, S.; Ren, Z.; Zhao, J.; Li, M.; Zhang, J.; Tian, S.; Wang, J.; et al. Localization shift of a sugar transporter contributes to phloem unloading in sweet watermelons. New Phytol. 2020, 227, 1858–1871. [Google Scholar] [CrossRef]
- Peng, Q.; Cai, Y.; Lai, E.; Nakamura, M.; Liao, L.; Zheng, B.; Ogutu, C.; Cherono, S.; Han, Y. The sucrose transporter MdSUT4.1 participates in the regulation of fruit sugar accumulation in apple. BMC Plant Biol. 2020, 20, 191. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Guo, S.; Zhang, J.; He, H.; Sun, H.; Tian, S.; Gong, G.; Zhang, H.; Levi, A.; Tadmor, Y.; et al. A Tonoplast Sugar Transporter Underlies a Sugar Accumulation QTL in Watermelon. Plant Physiol. 2017, 176, 836–850. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wen, S.; Xiao, S.; Lu, B.; Ma, M.; Bie, Z. Overexpression of the tonoplast sugar transporter CmTST2 in melon fruit increases sugar accumulation. J. Exp. Bot. 2017, 69, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Hu, B.; Liu, J.; Zhou, Y.; Liu, S. Identification and Characterization of Tonoplast Sugar Transporter (TST) Gene Family in Cucumber. Hortic. Plant J. 2020, 6, 145–157. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, X.; Yang, J.; Li, H.; Ma, B.; Zhang, K.; Zhang, Y.; Cheng, L.; Ma, F.; Li, M. Heterologous expression of the apple hexose transporter Md HT 2.2 altered sugar concentration with increasing cell wall invertase activity in tomato fruit. Plant Biotechnol. J. 2019, 18, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, B.; Wu, L.; Li, H.; Wang, Z.; Wei, X.; Ma, B.; Zhang, Y.; Ma, F.; Ruan, Y.-L.; et al. MdERDL6-mediated glucose efflux to the cytosol promotes sugar accumulation in the vacuole through up-regulating TSTs in apple and tomato. Proc. Natl. Acad. Sci. USA 2020, 118, e2022788118. [Google Scholar] [CrossRef]
- Lunn, J.E.; Delorge, I.; Figueroa, C.M.; Van Dijck, P.; Stitt, M. Trehalose metabolism in plants. Plant J. 2014, 79, 544–567. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, Y.; Song, X.; Miao, M. Trehalose-6-phosphate and SNF1-related protein kinase 1 are involved in the first-fruit inhibition of cucumber. J. Plant Physiol. 2014, 177, 110–120. [Google Scholar] [CrossRef]
- Dai, N.; Cohen, S.; Portnoy, V.; Tzuri, G.; Harel-Beja, R.; Pompan-Lotan, M.; Carmi, N.; Zhang, G.; Diber, A.; Pollock, S.; et al. Metabolism of soluble sugars in developing melon fruit: A global transcriptional view of the metabolic transition to sucrose accumulation. Plant Mol. Biol. 2011, 76, 1–18. [Google Scholar] [CrossRef]
- Yang, J.; Deng, G.; Lian, J.; Garraway, J.; Niu, Y.; Hu, Z.; Yu, J.; Zhang, M. The Chromosome-Scale Genome of Melon Dissects Genetic Architecture of Important Agronomic Traits. iScience 2020, 23, 101422. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Genome Project Data Processing S: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhang, X.; Miao, C.; Zhang, J.; Ming, R.; Schnable, J.C.; Schnable, P.S.; Lyons, E.; Lu, J. ALLMAPS: Robust scaffold ordering based on multiple maps. Genome Biol. 2015, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 2014, 11, 407–409. [Google Scholar] [CrossRef]
- Lex, A.; Gehlenborg, N.; Strobelt, H.; Vuillemot, R.; Pfister, H. UpSet: Visualization of Intersecting Sets. IEEE Trans. Vis. Comput. Graph. 2014, 20, 1983–1992. [Google Scholar] [CrossRef]
- Cheng, H.; Kong, W.; Tang, T.; Ren, K.; Zhang, K.; Wei, H.; Lin, T. Identification of Key Gene Networks Controlling Soluble Sugar and Organic Acid Metabolism During Oriental Melon Fruit Development by Integrated Analysis of Metabolic and Transcriptomic Analyses. Front. Plant Sci. 2022, 13, 830517. [Google Scholar] [CrossRef]
- Ohkawa, W.; Kanayama, Y.; Daibo, N.; Sato, T.; Nishiyama, M.; Kanahama, K. Metabolic process of the 14C-sugars on the translocation pathways of cucumber plants. Sci. Hortic. 2010, 124, 46–50. [Google Scholar] [CrossRef]
- Saladié, M.; Cañizares, J.; Phillips, M.A.; Rodriguez-Concepcion, M.; Larrigaudière, C.; Gibon, Y.; Stitt, M.; Lunn, J.E.; Garcia-Mas, J. Comparative transcriptional profiling analysis of developing melon (Cucumis melo L.) fruit from climacteric and non-climacteric varieties. BMC Genom. 2015, 16, 440. [Google Scholar] [CrossRef]
- Shin, A.-Y.; Kim, Y.-M.; Koo, N.; Lee, S.M.; Nahm, S.; Kwon, S.-Y. Transcriptome analysis of the oriental melon (Cucumis melo L. var. makuwa) during fruit development. PeerJ 2017, 5, e2834. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Ni, D.-A.; Ruan, Y.-L. Posttranslational Elevation of Cell Wall Invertase Activity by Silencing Its Inhibitor in Tomato Delays Leaf Senescence and Increases Seed Weight and Fruit Hexose Level. Plant Cell 2009, 21, 2072–2089. [Google Scholar] [CrossRef]
- Zanor, M.I.; Osorio, S.; Nunes-Nesi, A.; Carrari, F.; Lohse, M.; Usadel, B.; Kühn, C.; Bleiss, W.; Giavalisco, P.; Willmitzer, L.; et al. RNA Interference of LIN5 in Tomato Confirms Its Role in Controlling Brix Content, Uncovers the Influence of Sugars on the Levels of Fruit Hormones, and Demonstrates the Importance of Sucrose Cleavage for Normal Fruit Development and Fertility. Plant Physiol. 2009, 150, 1204–1218. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Li, M.; Guo, S.; Sun, H.; Zhao, J.; Zhang, J.; Liu, G.; He, H.; Tian, S.; Yu, Y.; et al. Evolutionary gain of oligosaccharide hydrolysis and sugar transport enhanced carbohydrate partitioning in sweet watermelon fruits. Plant Cell 2021, 33, 1554–1573. [Google Scholar] [CrossRef]
- Liu, H.; Liu, X.; Zhao, Y.; Nie, J.; Yao, X.; Lv, L.; Yang, J.; Ma, N.; Guo, Y.; Li, Y.; et al. Alkaline α-galactosidase 2 (CsAGA2) plays a pivotal role in mediating source–sink communication in cucumber. Plant Physiol. 2022, 189, 1501–1518. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, T.; Zhang, C.; Zhang, W.; Deng, N.; Dirk, L.M.A.; Downie, A.B.; Zhao, T. Raffinose positively regulates maize drought tolerance by reducing leaf transpiration. Plant J. 2023, 114, 55–67. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, M.; Cao, L.; Dang, Z.; Ruan, N.; Wang, Y.; Huang, Y.; Wu, J.; Zhang, M.; Xu, Z.; et al. OsUGE3-mediated cell wall polysaccharides accumulation improves biomass production, mechanical strength, and salt tolerance. Plant Cell Environ. 2022, 45, 2492–2507. [Google Scholar] [CrossRef]
- Bavnhøj, L.; Paulsen, P.A.; Flores-Canales, J.C.; Schiøtt, B.; Pedersen, B.P. Molecular mechanism of sugar transport in plants unveiled by structures of glucose/H+ symporter STP10. Nat. Plants 2021, 7, 1409–1419. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Environment a | Parents | Mean ± SE (%) | SD b | Range (%) | CV c (%) | Skewness | Kurtosis | G d | E d | G × E d | H2 e | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1214 | 1228 | ||||||||||||
| RILs | 2020 SPR | 7.43 | 12.40 | 10.43 ± 0.13 | 1.56 | 5.67–14.50 | 14.96 | −0.15 | 0.13 | ** | ** | ** | 0.65 |
| 2021 SPR | 9.26 | 13.48 | 11.01 ± 0.13 | 1.55 | 5.83–14.07 | 14.07 | −0.35 | 0.46 | |||||
| 2022 SPR | 9.62 | 14.35 | 11.29 ± 0.13 | 1.46 | 7.15–14.80 | 12.93 | −0.34 | −0.01 | |||||
| Accessions | 2020 SPR | - | - | 10.01 ± 0.20 | 2.62 | 3.00–17.30 | 26.17 | −0.39 | −0.17 | ** | ** | ** | 0.72 |
| 2021 SPR | - | - | 9.86 ± 0.20 | 2.87 | 3.00–17.43 | 29.01 | −0.17 | −0.57 | |||||
| 2021 AUT | - | - | 8.97 ± 0.21 | 2.71 | 3.55–15.50 | 30.21 | 0.16 | −0.57 | |||||
| 2022 SPR | - | - | 10.68 ± 0.23 | 3.12 | 3.80–18.98 | 29.21 | −0.03 | −0.75 | |||||
| LGs | Number of Bins | Map Length (cM) | Average Distance (cM) | Max Gap (cM) | <5 cM | >5 cM |
|---|---|---|---|---|---|---|
| LG01 | 148 | 133.75 | 0.90 | 4.27 | 148 | 0 |
| LG02 | 109 | 84.73 | 0.78 | 3.00 | 109 | 0 |
| LG03 | 119 | 105.39 | 0.89 | 3.84 | 119 | 0 |
| LG04 | 96 | 90.16 | 0.94 | 3.84 | 96 | 0 |
| LG05 | 152 | 126.17 | 0.83 | 3.42 | 152 | 0 |
| LG06 | 158 | 128.21 | 0.81 | 4.27 | 158 | 0 |
| LG07 | 114 | 105.84 | 0.93 | 3.84 | 114 | 0 |
| LG08 | 109 | 101.16 | 0.93 | 4.27 | 109 | 0 |
| LG09 | 118 | 105.97 | 0.90 | 3.00 | 118 | 0 |
| LG10 | 43 | 58.46 | 1.36 | 14.26 | 42 | 1 |
| LG11 | 149 | 133.92 | 0.90 | 3.84 | 149 | 0 |
| LG12 | 112 | 80.59 | 0.72 | 3.84 | 112 | 0 |
| total | 1427 | 1254.34 | 0.88 | 14.26 | 1426 | 1 |
| Chr. | Environment | LOD | Position Interval (cM) | Physical Position (bp) | SNP Loci | −log10(p) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2020SPR | 2021SPR | 2021AUT | 2022SPR | BLUP | ||||||
| 3 | 2020SPR | 4.5 | 95.2–104.3 | 28,509,689–28,965,705 | 28,672,844 | 6.01 | ||||
| 3 | 2022SPR | 5.5 | 93.7–99.5 | 28,446,470–28,845,998 | 28,694,392 | 6.27 | 6.05 | 6.27 | ||
| 3 | BLUP | 5.7 | 95.6–102.9 | 28,509,689–28,947,998 | 28,711,496 | 6.47 | 6.51 | |||
| 28,714,701 | 6.19 | |||||||||
| 28,724,776 | 6.08 | 6.67 | ||||||||
| 3 | BLUP | 5.2 | 102.9–105.1 | 28,947,998–29,341,849 | 29,267,782 | 6.68 | 6.04 | 8.49 | 7.65 | |
| 29,283,426 | 6.14 | |||||||||
| 29,289,157 | 6.22 | |||||||||
| 29,330,788 | 7.77 | 8.33 | 7.88 | 7.88 | ||||||
| 4 | 2021SPR | 3.5 | 20.9–28.6 | 2,334,788–3,020,226 | 2,491,434 | 6.74 | 6.80 | |||
| 4 | BLUP | 3.4 | 51.7–54.2 | 8,961,531–10,360,279 | 10,141,036 | 7.13 | 6.63 | |||
| 10,275,303 | 7.06 | |||||||||
| 6 | 2021SPR | 3.5 | 95.5–101 | 32,217,543–32,935,488 | 32,656,543 | 6.56 | ||||
| Gene | Chromosome | Start | End | Functional Annotation |
|---|---|---|---|---|
| MELO03698 | chr2 | 14,397,615 | 14,406,002 | Sucrose-phosphate synthase |
| MELO06759 | chr3 | 28,826,329 | 28,829,358 | Beta-fructofuranosidase, insoluble isoenzyme CWINV3 |
| MELO06810 | chr3 | 29,119,436 | 29,126,262 | Alpha-galactosidase |
| MELO12562 | chr6 | 6,109,512 | 6,114,363 | Trehalose 6-phosphate phosphatase |
| MELO18502 | chr8 | 16,852,497 | 16,859,087 | Raffinose synthase |
| MELO20547 | chr9 | 14,794,455 | 14,800,212 | Sucrose synthase |
| MELO21359 | chr9 | 23,772,893 | 23,777,340 | UDP-glucose 4′-epimerase |
| MELO22262 | chr10 | 5,145,518 | 5,147,793 | Sugar transport protein 10-like |
| MELO27354 | chr12 | 20,598,706 | 20,606,584 | Trehalose-6-phosphate synthase |
| MELO27634 | chr12 | 23,004,024 | 23,006,015 | Sugar transporter SWEET12-like |
| MELO27635 | chr12 | 23,013,800 | 23,015,623 | Sugar transporter SWEET12-like |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, H.; Wang, K.; Wang, M.; Feng, L.; Zhang, H.; Wei, X. QTL Mapping and Genome-Wide Association Study Reveal Genetic Loci and Candidate Genes Related to Soluble Solids Content in Melon. Curr. Issues Mol. Biol. 2023, 45, 7110-7129. https://doi.org/10.3390/cimb45090450
Yan H, Wang K, Wang M, Feng L, Zhang H, Wei X. QTL Mapping and Genome-Wide Association Study Reveal Genetic Loci and Candidate Genes Related to Soluble Solids Content in Melon. Current Issues in Molecular Biology. 2023; 45(9):7110-7129. https://doi.org/10.3390/cimb45090450
Chicago/Turabian StyleYan, Honglang, Kang Wang, Manman Wang, Lulu Feng, Huimin Zhang, and Xiaoyun Wei. 2023. "QTL Mapping and Genome-Wide Association Study Reveal Genetic Loci and Candidate Genes Related to Soluble Solids Content in Melon" Current Issues in Molecular Biology 45, no. 9: 7110-7129. https://doi.org/10.3390/cimb45090450