

Insight into the lncRNA–mRNA Co-Expression Profile and ceRNA Network in Lipopolysaccharide-Induced Acute Lung Injury
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Data Collection and Sample Preparation
2.2. Cell Culture and Treatment
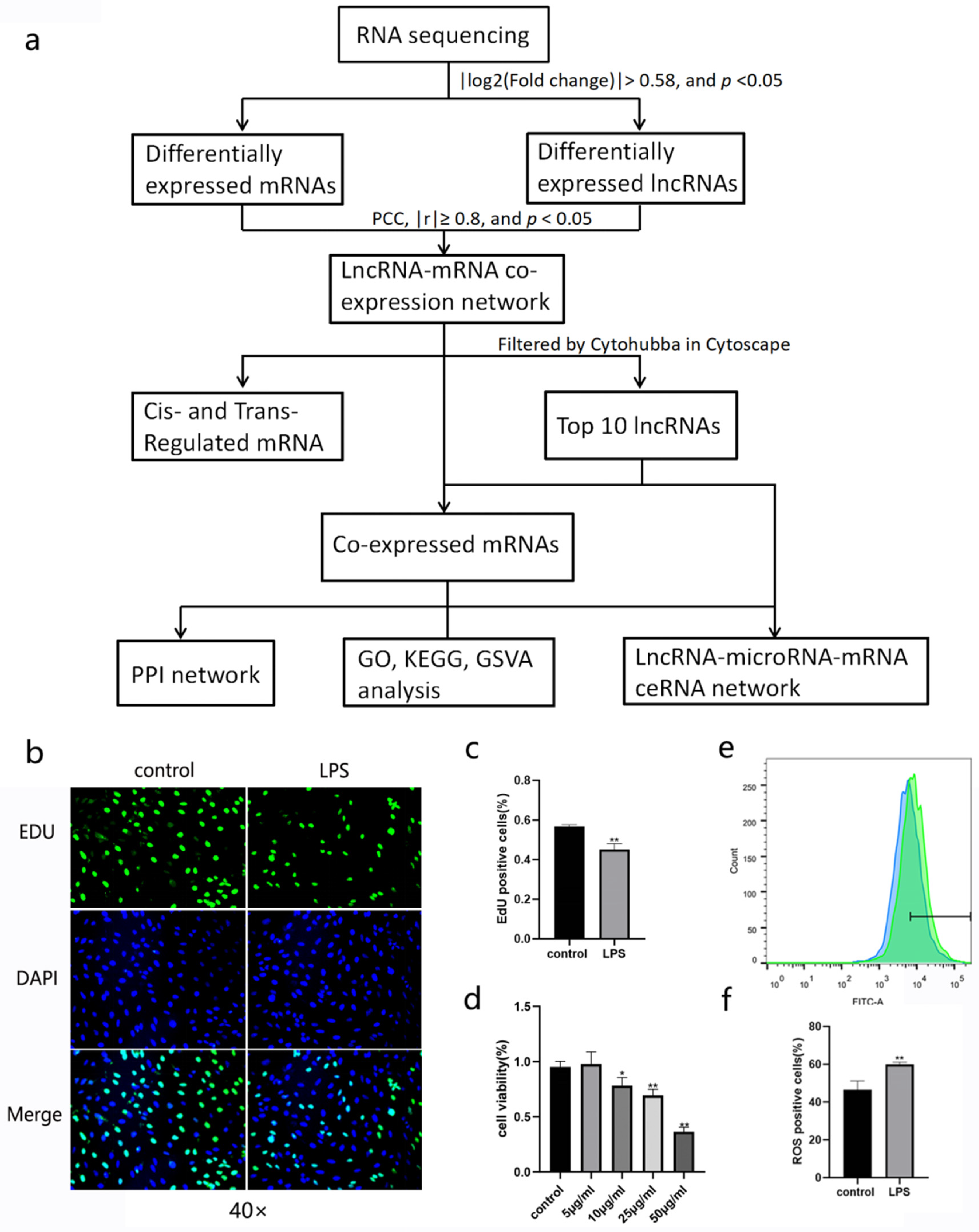
2.3. EDU Detection
2.4. Cell Viability Assay
2.5. Reactive Oxygen Species (ROS) Detection
2.6. RNA Isolation and Library Preparation
2.7. RNA-Seq Analysis
2.8. The lncRNA–mRNA Co-Expression Network
2.9. Cis- and Trans-Regulated Gene Analysis
2.10. Gene Functional Enrichment Analysis
2.11. Gene Set Variation Analysis (GSVA)
2.12. Construction of Protein–Protein Interaction (PPI) Network
2.13. LncRNA–microRNA–mRNA ceRNA Network Construction
2.14. Expression Profile Validation Using RT-qPCR
2.15. Statistical Analysis
3. Results
3.1. Establishment of ALI Model in BEAS-2B Cells
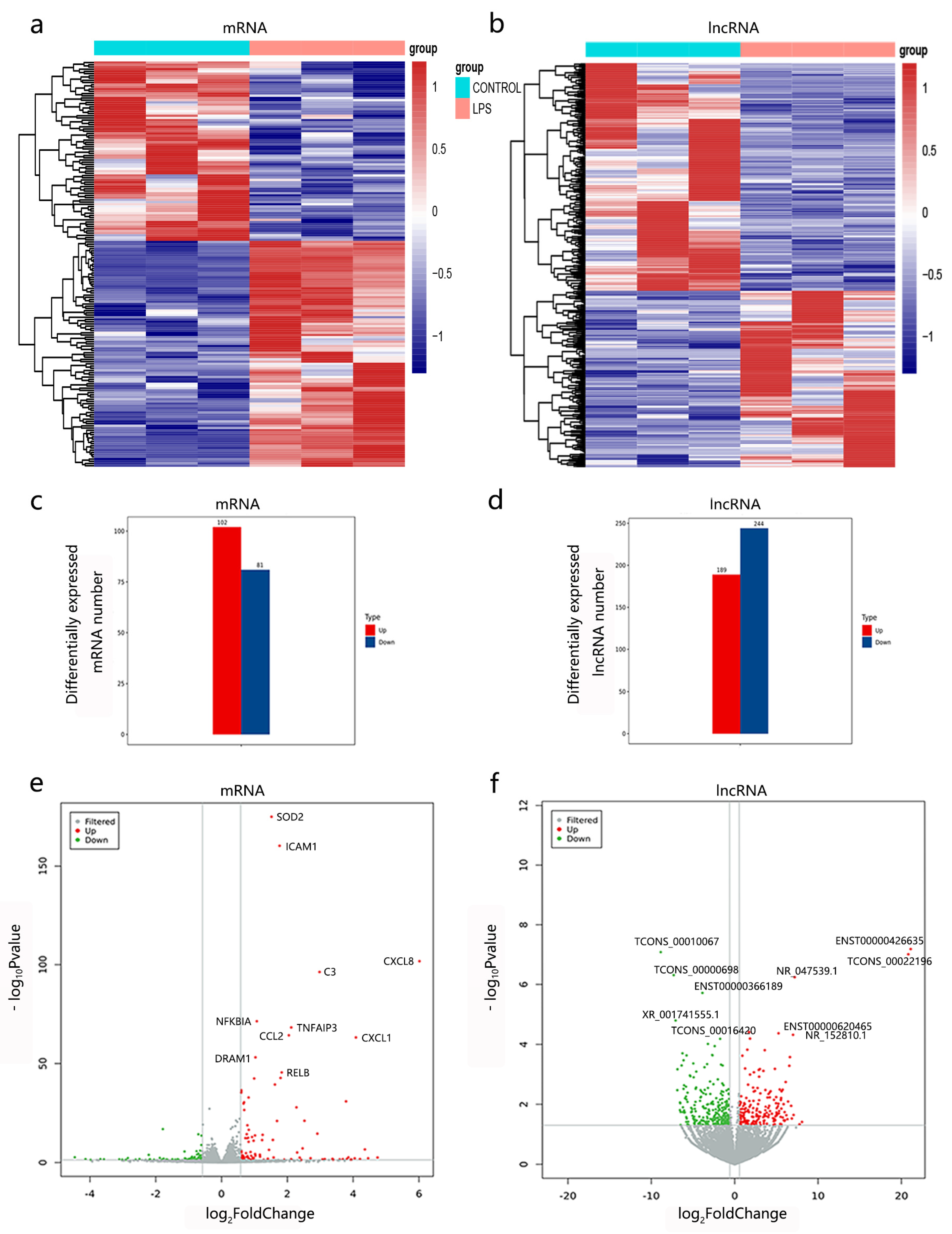
3.2. Characterization of lncRNAs and mRNAs
3.3. Expression Profiles of lncRNAs and mRNAs
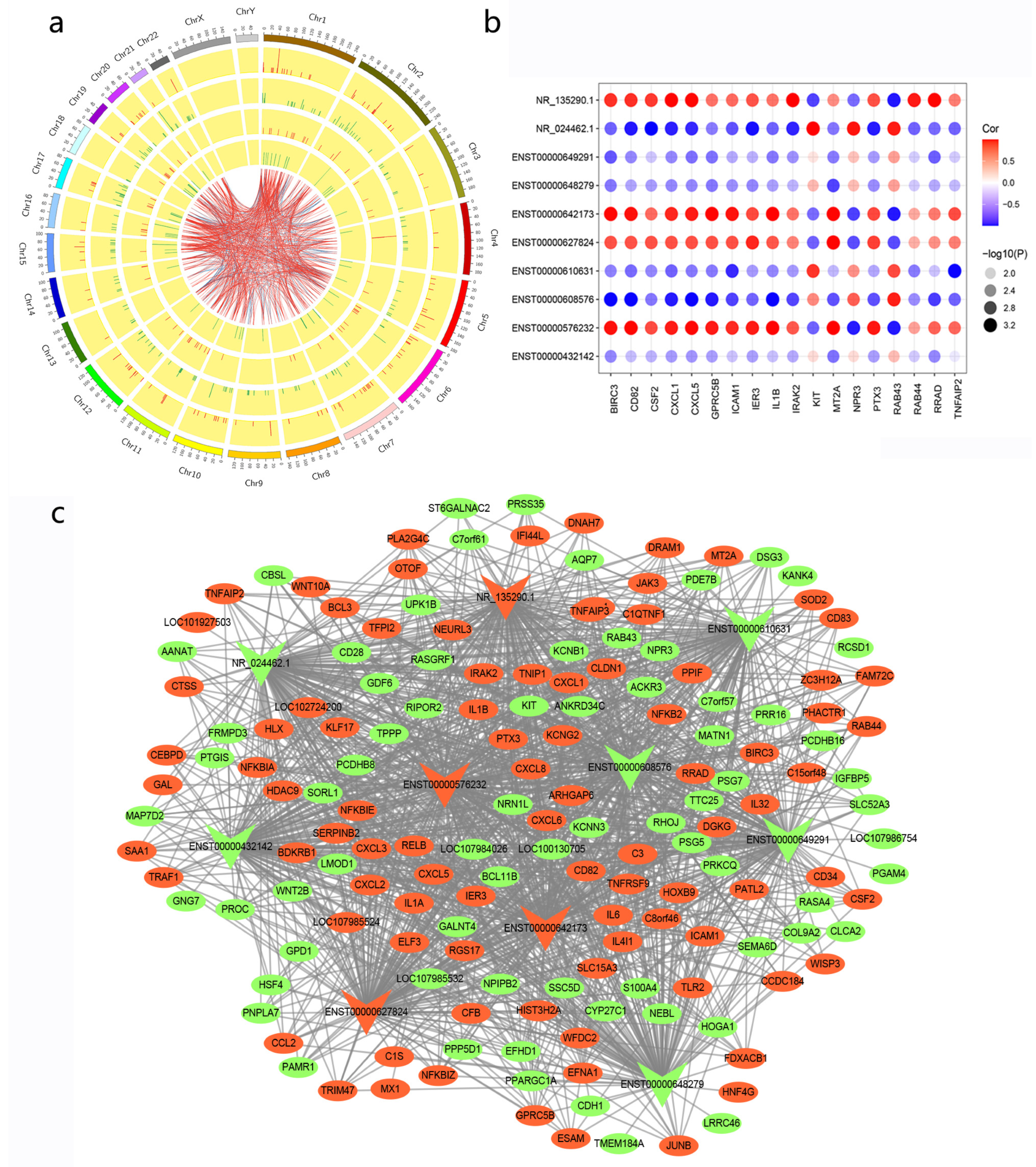
3.4. Co-Expression Network of lncRNA–mRNA
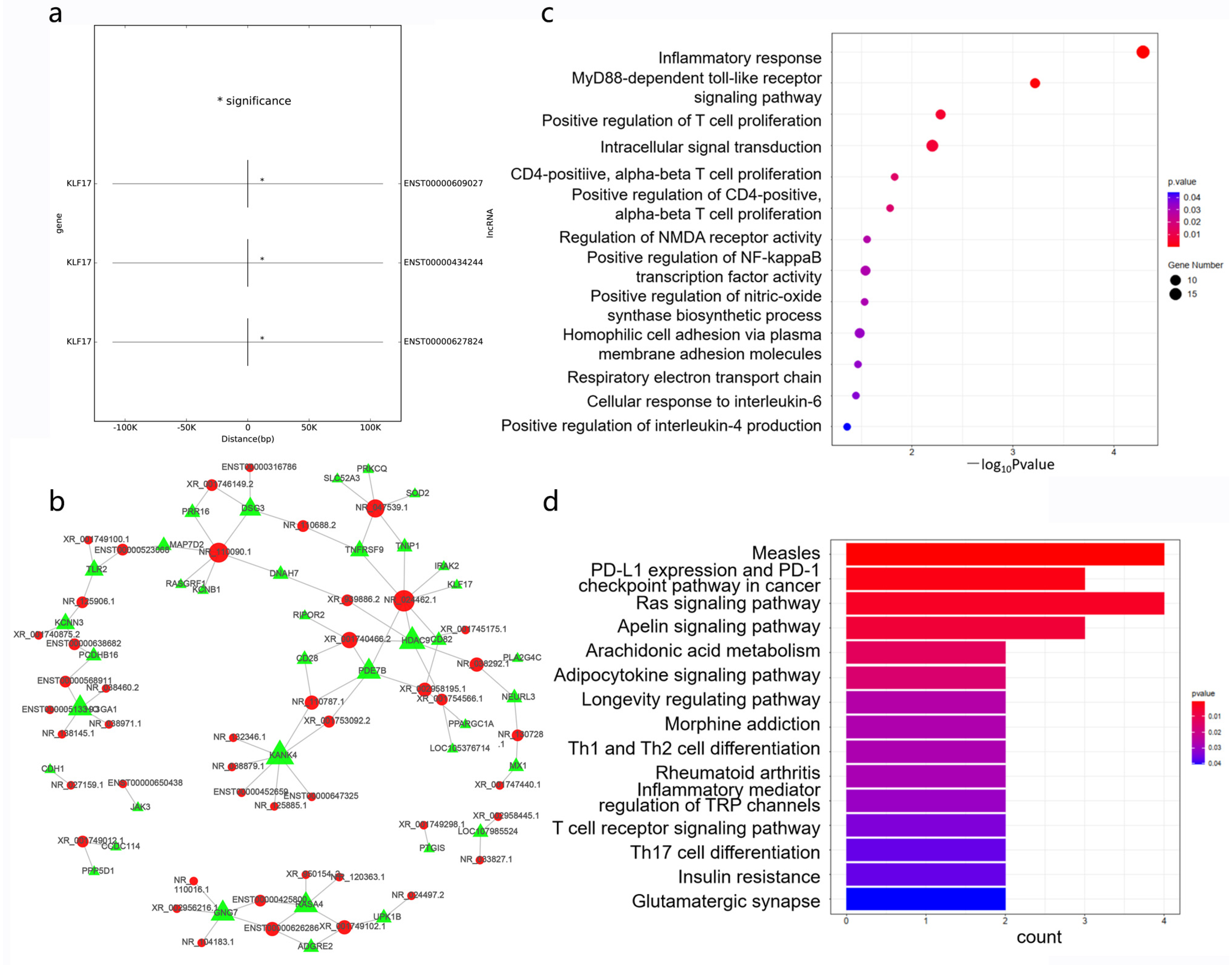
3.5. Cis- and Trans-Regulated Gene Analysis
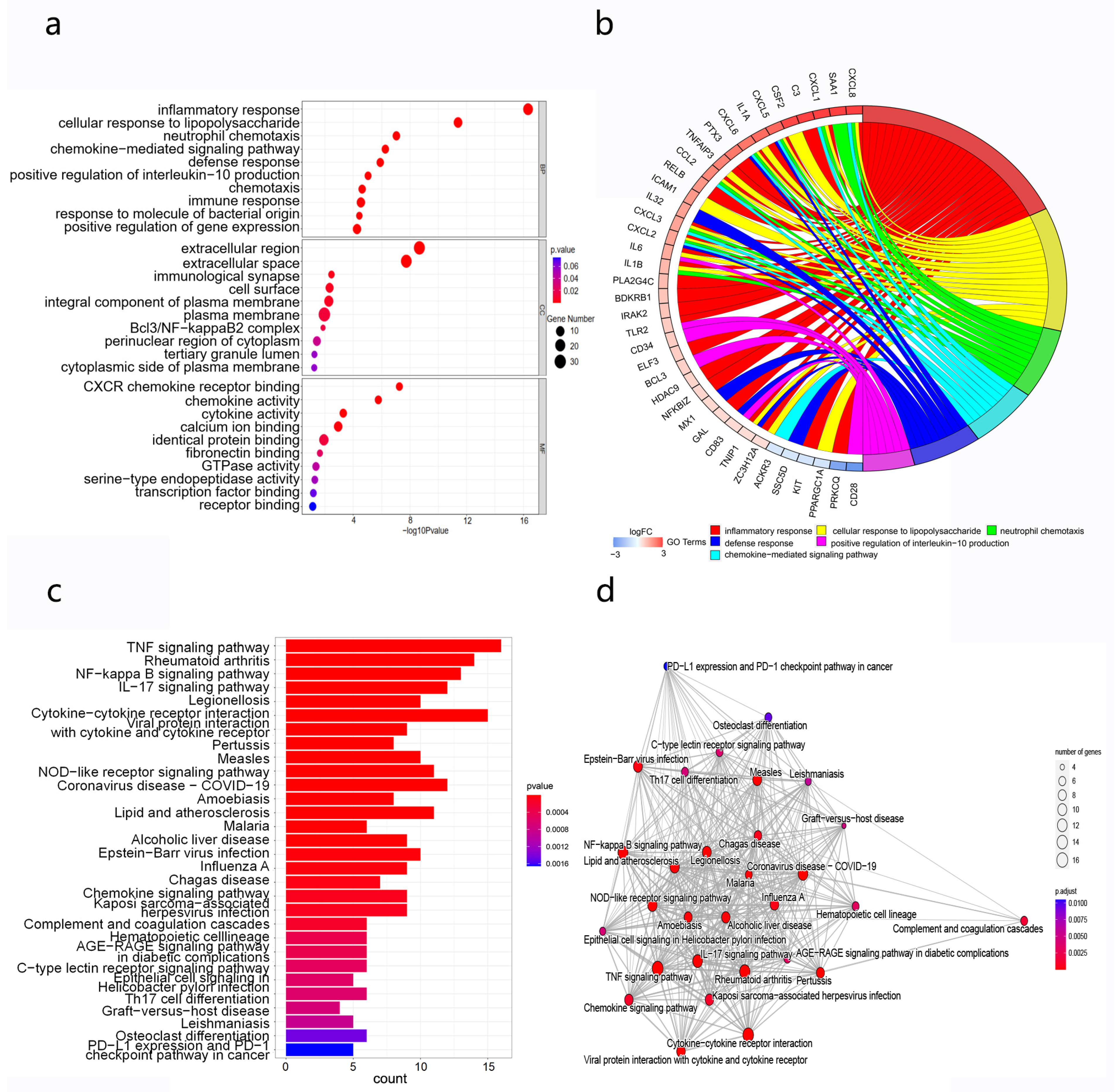
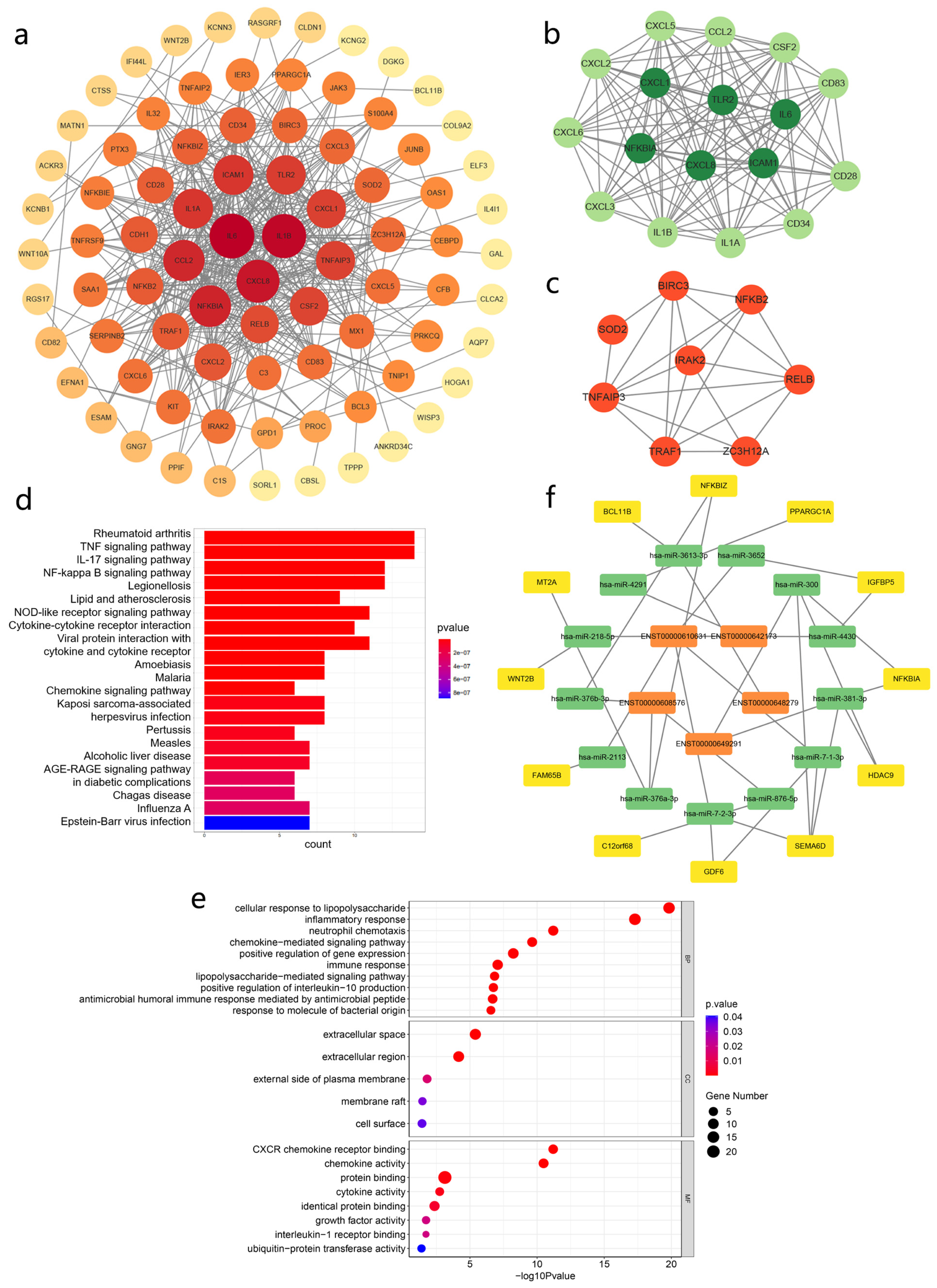
3.6. Enrichment Analysis of Differentially Expressed Genes
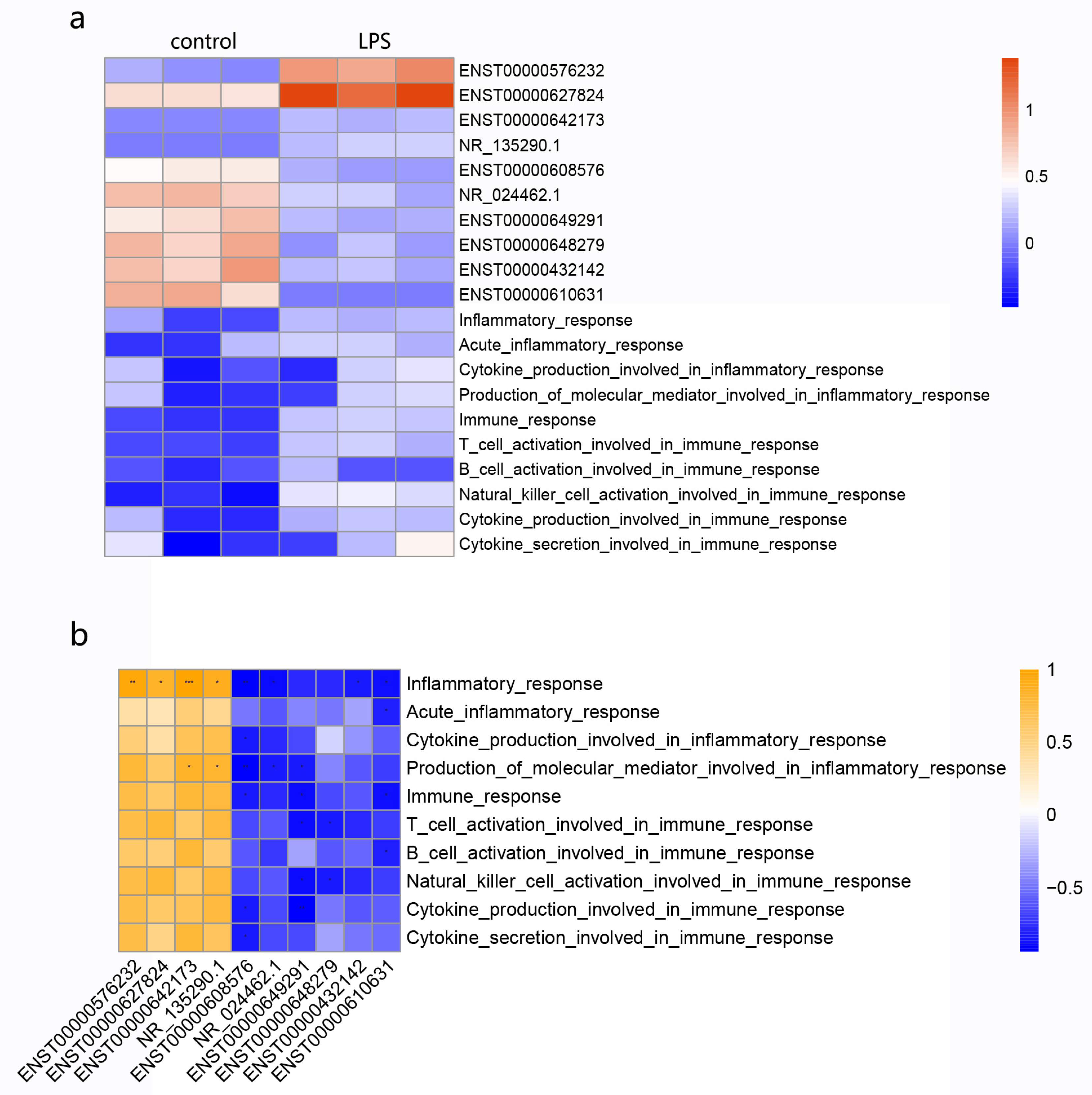
3.7. Relationship of Candidate lncRNAs with Immune and Inflammatory Responses
3.8. PPI Analysis
3.9. Establishment of a lncRNA–microRNA–mRNA ceRNA Regulatory Network
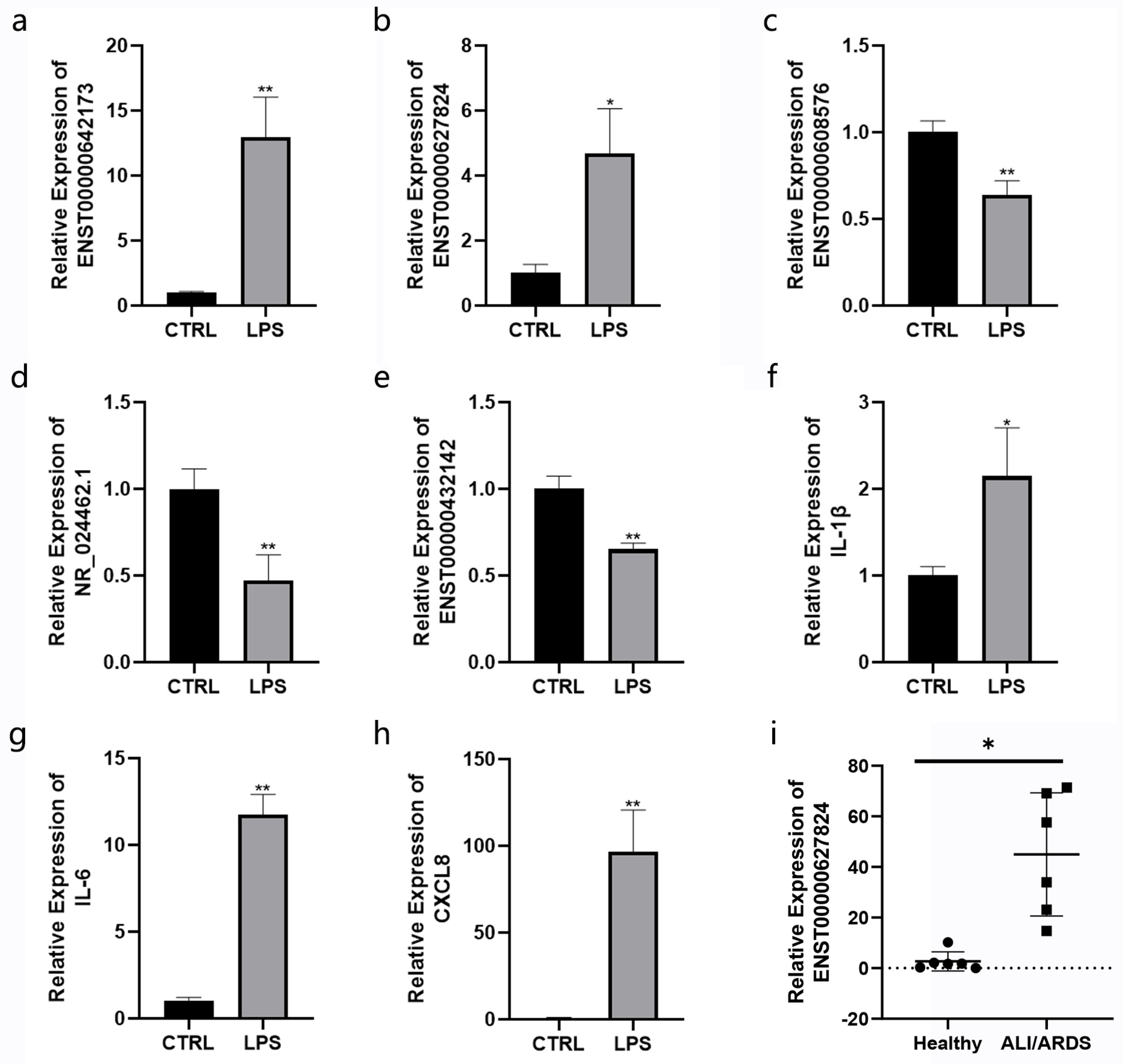
3.10. Validation of Selected lncRNA Using RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, A.S.; Slutsky. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [PubMed]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, N.J.; Gattinoni, L.; Calfee, C.S. Acute respiratory distress syndrome. Lancet 2021, 398, 622–637. [Google Scholar] [CrossRef]
- Calfee, C.S.; Delucchi, K.; Parsons, P.E.; Thompson, B.T.; Ware, L.B.; Matthay, M.A. Subphenotypes in acute respiratory distress syndrome: Latent class analysis of data from two randomised controlled trials. Lancet Respir. Med. 2014, 2, 611–620. [Google Scholar] [CrossRef]
- Famous, K.R.; Delucchi, K.; Ware, L.B.; Kangelaris, K.N.; Liu, K.D.; Thompson, B.T.; Calfee, C.S. Acute Respiratory Distress Syndrome Subphenotypes Respond Differently to Randomized Fluid Management Strategy. Am. J. Respir. Crit. Care Med. 2017, 195, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Rinn, J.L.; Chang, H.Y. Long Noncoding RNAs: Molecular Modalities to Organismal Functions. Annu. Rev. Biochem. 2020, 89, 283–308. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Prabhakar, N.; Kumar, L.; Bhattacharjee, A.; Kar, S.; Malik, S.; Kumar, D.; Ruokolainen, J.; Negi, A.; Jha, N.K.; et al. Crosstalk between long noncoding RNA and microRNA in Cancer. Cell Oncol. 2023, 24, 2211–3428. [Google Scholar] [CrossRef]
- Farokhian, A.; Rajabi, A.; Sheida, A.; Abdoli, A.; Rafiei, M.; Jazi, Z.H.; Asouri, S.A.; Morshedi, M.A.; Hamblin, M.R.; Adib-Hajbagheri, P.; et al. Apoptosis and myocardial infarction: Role of ncRNAs and exosomal ncRNAs. Epigenomics 2023, 15, 307–334. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, N.K. Long Non-Coding RNAs and Proliferative Retinal Diseases. Pharmaceutics 2023, 15, 1454. [Google Scholar] [CrossRef]
- Mosca, N.; Russo, A.; Potenza, N. Making Sense of Antisense lncRNAs in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 8886. [Google Scholar] [CrossRef]
- Zaki, A.; Ali, M.S.; Hadda, V.; Ali, S.M.; Chopra, A.; Fatma, T. Long non-coding RNA (lncRNA): A potential therapeutic target in acute lung injury. Genes Dis. 2022, 9, 1258–1268. [Google Scholar] [CrossRef]
- Hu, C.; Li, J.; Tan, Y.; Liu, Y.; Bai, C.; Gao, J.; Zhao, S.; Yao, M.; Lu, X.; Qiu, L.; et al. Tanreqing Injection Attenuates Macrophage Activation and the Inflammatory Response via the lncRNA-SNHG1/HMGB1 Axis in Lipopolysaccharide-Induced Acute Lung Injury. Front. Immunol. 2022, 13, 820718. [Google Scholar] [CrossRef]
- Ayeldeen, G.; Shaker, O.G.; Amer, E.; Zaafan, M.A.; Herzalla, M.R.; Keshk, M.A.; Abdelhamid, A.M. The impact of lncRNA-GAS5/miRNA-200/ACE2 molecular pathway on the severity of COVID-19. Curr. Med. Chem. 2023. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Anders, W.H.S. Differential expression of RNA-Seq data at the gene level-the DESeq package. EMBL 2012, 24, 10. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Wucher, V.; Legeai, F.; Hédan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef] [Green Version]
- Hunter, D.R.; Handcock, M.S.; Butts, C.T.; Goodreau, S.M.; Morris, M. ergm: A Package to Fit, Simulate and Diagnose Exponential-Family Models for Networks. J. Stat. Softw. 2008, 24, nihpa54860. [Google Scholar] [CrossRef] [Green Version]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W321. [Google Scholar] [CrossRef] [PubMed]
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L.; et al. The multiMiR R package and database: Integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Menden, H.; Xia, S.; Mabry, S.M.; Noel-MacDonnell, J.; Rajasingh, J.; Ye, S.Q.; Sampath, V. Histone deacetylase 6 regulates endothelial MyD88-dependent canonical TLR signaling, lung inflammation, and alveolar remodeling in the developing lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L332–L346. [Google Scholar] [CrossRef]
- Parekh, D.; Dancer, R.C.; Thickett, D.R. Acute lung injury. Clin. Med. 2011, 11, 615–618. [Google Scholar] [CrossRef]
- Serkova, N.J.; Standiford, T.J.; Stringer, K.A. The emerging field of quantitative blood metabolomics for biomarker discovery in critical illnesses. Am. J. Respir. Crit. Care Med. 2011, 184, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Husain, S.; Sage, A.T.; Del Sorbo, L.; Cypel, M.; Martinu, T.; Juvet, S.C.; Mariscal, A.; Wright, J.; Chao, B.T.; Shamandy, A.A.; et al. A biomarker assay to risk-stratify patients with symptoms of respiratory tract infection. Eur. Respir. J. 2022, 60, 6. [Google Scholar] [CrossRef]
- Patel, M.C.; Shirey, K.A.; Boukhvalova, M.S.; Vogel, S.N.; Blanco, J.C.G. Serum High-Mobility-Group Box 1 as a Biomarker and a Therapeutic Target during Respiratory Virus Infections. mBio 2018, 9, e00246-18. [Google Scholar] [CrossRef] [Green Version]
- Bonidia, R.P.; Sampaio, L.D.H.; Domingues, D.S.; Paschoal, A.R.; Lopes, F.M.; de Carvalho, A.; Sanches, D.S. Feature extraction approaches for biological sequences: A comparative study of mathematical features. Brief. Bioinform. 2021, 22, bbab011. [Google Scholar] [CrossRef]
- Lu, Z.; Chang, W.; Meng, S.; Xu, X.; Xie, J.; Guo, F.; Yang, Y.; Qiu, H.; Liu, L. Mesenchymal stem cells induce dendritic cell immune tolerance via paracrine hepatocyte growth factor to alleviate acute lung injury. Stem Cell Res. Ther. 2019, 10, 372. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V. Pulmonary Innate Immune Response Determines the Outcome of Inflammation during Pneumonia and Sepsis-Associated Acute Lung Injury. Front. Immunol. 2020, 11, 1722. [Google Scholar] [CrossRef]
- Liang, W.J.; Zeng, X.Y.; Jiang, S.L.; Tan, H.Y.; Yan, M.Y.; Yang, H.Z. Long non-coding RNA MALAT1 sponges miR-149 to promote inflammatory responses of LPS-induced acute lung injury by targeting MyD88. Cell Biol. Int. 2019, 44, 317–326. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, Q.; Yang, X.; Ding, C.; Wang, Q.; Xiong, Y. LncRNA LINC00649 recruits TAF15 and enhances MAPK6 expression to promote the development of lung squamous cell carcinoma via activating MAPK signaling pathway. Cancer Gene Ther. 2022, 29, 1285–1295. [Google Scholar] [CrossRef]
- Khan, M.R.; Xiang, S.; Song, Z.; Wu, M. The p53-inducible long noncoding RNA TRINGS protects cancer cells from necrosis under glucose starvation. Embo J. 2017, 6, 3483–3500. [Google Scholar] [CrossRef]
- Toubiana, J.; Courtine, E.; Pène, F.; Viallon, V.; Asfar, P.; Daubin, C.; Rousseau, C.; Chenot, C.; Ouaaz, F.; Grimaldi, D.; et al. IRAK1 functional genetic variant affects severity of septic shock. Crit. Care Med. 2010, 38, 2287–2294. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Chen, Y.; Liu, J.; Yang, J.; Yang, C.; Zhang, T.; Jiang, K.; Wu, Z.; Shaukat, A. Deng, miR-497a-5p attenuates lipopolysaccharide-induced inflammatory injury by targeting IRAK2. J. Cell Physiol. 2019, 234, 22874–22883. [Google Scholar] [CrossRef]
- Habacher, C.; Ciosk, R. ZC3H12A/MCPIP1/Regnase-1-related endonucleases: An evolutionary perspective on molecular mechanisms and biological functions. Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaku, A.; Inagaki, T.; Asano, R.; Okazawa, M.; Mori, H.; Sato, A.; Hia, F.; Masaki, T.; Manabe, Y.; Ishibashi, T.; et al. Takeuchi, Regnase-1 Prevents Pulmonary Arterial Hypertension through mRNA Degradation of Interleukin-6 and Platelet-Derived Growth Factor in Alveolar Macrophages. Circulation 2022, 146, 1006–1022. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Huang, Y.; Lin, J.; Sun, S.; Che, K.; Shen, J.; Liao, J.; Chen, Y.; Chen, K.; Lin, Z.; et al. Mxi1 participates in the progression of lung cancer via the microRNA-300/KLF9/GADD34 Axis. Cell Death Dis. 2022, 13, 425. [Google Scholar] [CrossRef] [PubMed]
- Gunyuz, Z.E.; Sahi-Ilhan, E.; Kucukkose, C.; Ipekgil, D.; Tok, G.; Mese, G.; Ozcivici, E. Yalcin-Ozuysal, SEMA6D Differentially Regulates Proliferation, Migration, and Invasion of Breast Cell Lines. ACS Omega 2022, 7, 15769–15778. [Google Scholar] [CrossRef]
- Naito, M.; Nakanishi, Y.; Motomura, Y.; Takamatsu, H.; Koyama, S.; Nishide, M.; Naito, Y.; Izumi, M.; Mizuno, Y.; Yamaguchi, Y.; et al. Semaphorin 6D-expressing mesenchymal cells regulate IL-10 production by ILC2s in the lung. Life Sci. Alliance 2022, 5, e202201486. [Google Scholar] [CrossRef]
- Aisyah, R.; Sadewa, A.H.; Patria, S.Y.; Wahab, A. The PPARGC1A Is the Gene Responsible for Thrifty Metabolism Related Metabolic Diseases: A Scoping Review. Genes 2022, 13, 1894. [Google Scholar] [CrossRef]
- Shah, D.; Torres, C.; Bhandari, V. Adiponectin deficiency induces mitochondrial dysfunction and promotes endothelial activation and pulmonary vascular injury. Faseb J. 2019, 33, 13617–13631. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Zhu, L.; Liu, J.; Li, C.; Xu, Z.; Liu, J.; Zhang, H. MIR3142HG promotes lipopolysaccharide-induced acute lung injury by regulating miR-450b-5p/HMGB1 axis. Mol. Cell. Biochem. 2021, 476, 4205–4215. [Google Scholar] [CrossRef]
- Gao, Y.; Li, S.; Dong, R.; Li, X. Long noncoding RNA MIR3142HG accelerates lipopolysaccharide-induced acute lung injury via miR-95-5p/JAK2 axis. Hum. Cell 2022, 35, 856–870. [Google Scholar] [CrossRef]
- Yu, F.; Liang, M.; Wu, W.; Huang, Y.; Zheng, J.; Zheng, B.; Chen, C. Upregulation of Long Non-Coding RNA GCC2-AS1 Facilitates Malignant Phenotypes and Correlated with Unfavorable Prognosis for Lung Adenocarcinoma. Front. Oncol. 2020, 10, 628608. [Google Scholar] [CrossRef]
- Cheng, C.; Zhang, Z.; Cheng, F.; Shao, Z. Exosomal lncRNA RAMP2-AS1 Derived from Chondrosarcoma Cells Promotes Angiogenesis Through miR-2355-5p/VEGFR2 Axis. OncoTargets Ther. 2020, 13, 3291–3301. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript ID | Gene Symbol | log2Fold Change | Regulation | p-Value | Chrom | Length | Type |
|---|---|---|---|---|---|---|---|
| ENST00000576232 | MRPL20-AS1 | 3.44576434 | Up | 0.0007 | Chr1 | 585 | lincRNA |
| ENST00000627824 | AL139220.2 | 1.26417905 | Up | 0.0006 | Chr1 | 1796 | sense_intronic |
| ENST00000642173 | MIR3142HG | 3.082571424 | Up | 0.0043 | Chr5 | 2204 | lincRNA |
| NR_135290.1 | GCC2-AS1 | 4.688605503 | Up | 0.0369 | Chr2 | 569 | antisense |
| ENST00000608576 | AC073529.1 | −1.970778414 | Down | 0.0124 | ChrX | 1080 | lincRNA |
| NR_024462.1 | RAMP2-AS1 | −1.642451334 | Down | 0.0043 | Chr17 | 1985 | antisense |
| ENST00000649291 | AL138920.1 | −1.731306 | Down | 0.0000 | Chr10 | 3626 | lincRNA |
| ENST00000648279 | AL161756.1 | −2.383783 | Down | 0.0001 | Chr14 | 2132 | sense_exonic |
| ENST00000432142 | LINC01376 | −2.008642 | Down | 0.0272 | Chr2 | 571 | lincRNA |
| ENST00000610631 | AC145423.2 | −4.84318185 | Down | 0.0257 | Chr12 | 239 | sense_intronic |
| Parameters | Healthy (n = 6) | ALI/ARDS (n = 6) | p-Value |
|---|---|---|---|
| Age | 50.83 ± 9.68 | 58.17 ± 12.19 | 0.28 |
| Sex | 0.99 | ||
| Female | 2 (33.3%) | 3 (50%) | |
| Male | 4 (66.7%) | 3 (50%) | |
| Smoke | 0.99 | ||
| Yes | 2 (33.3%) | 2 (33.3%) | |
| No | 4 (66.7%) | 4 (66.7%) | |
| Hypertension | 0.55 | ||
| Yes | 1 (16.7%) | 3 (50%) | |
| No | 5 (83.3%) | 3 (50%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.; Gong, L.; Xu, F.; Wang, S.; Liu, H.; Wang, Y.; Hu, L.; Zhu, L. Insight into the lncRNA–mRNA Co-Expression Profile and ceRNA Network in Lipopolysaccharide-Induced Acute Lung Injury. Curr. Issues Mol. Biol. 2023, 45, 6170-6189. https://doi.org/10.3390/cimb45070389
Shen Y, Gong L, Xu F, Wang S, Liu H, Wang Y, Hu L, Zhu L. Insight into the lncRNA–mRNA Co-Expression Profile and ceRNA Network in Lipopolysaccharide-Induced Acute Lung Injury. Current Issues in Molecular Biology. 2023; 45(7):6170-6189. https://doi.org/10.3390/cimb45070389
Chicago/Turabian StyleShen, Yue, Linjing Gong, Fan Xu, Sijiao Wang, Hanhan Liu, Yali Wang, Lijuan Hu, and Lei Zhu. 2023. "Insight into the lncRNA–mRNA Co-Expression Profile and ceRNA Network in Lipopolysaccharide-Induced Acute Lung Injury" Current Issues in Molecular Biology 45, no. 7: 6170-6189. https://doi.org/10.3390/cimb45070389