
Genome-Wide SNP and Indel Discovery in Abaca (Musa textilis Née) and among Other Musa spp. for Abaca Genetic Resources Management
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction and Sequencing
2.3. Quality Control and Trimming of Sequence Reads
2.4. Mapping of Reads to the Reference Genome
2.5. Calling of Genome-Wide SNPs and InDels
2.6. Quality Filtering of SNPs and InDels
2.7. Analysis of Genome-Wide Variation and Phylogenetic Relationships
3. Results
3.1. DNA Extracts and Quality of Generated Sequence Reads
3.2. Mapping Quality Statistics
3.3. Variant Calling and Filtering Statistics
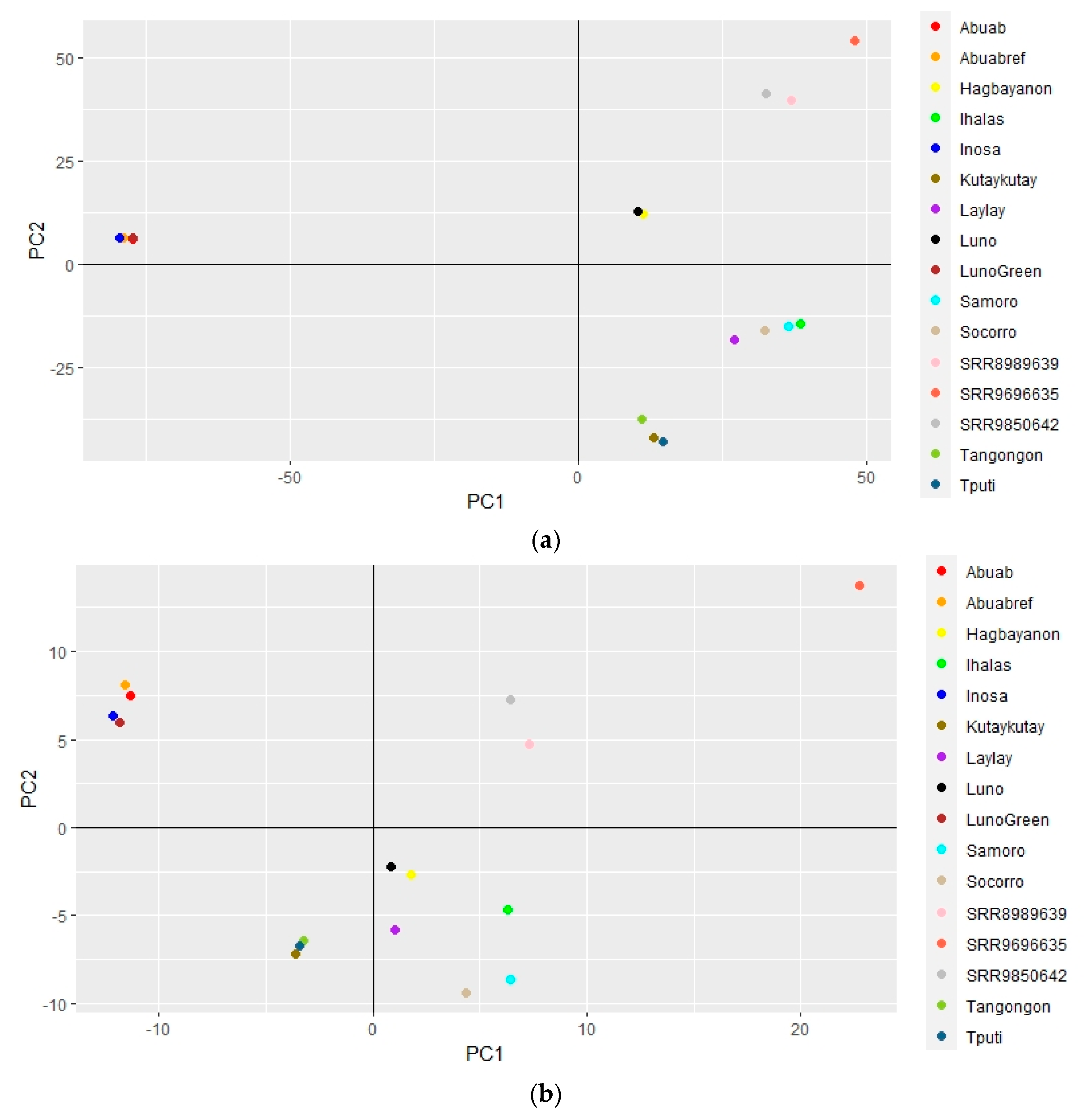
3.4. Genome-Wide Variation and Phylogenetic Relationships within Musa textilis
3.5. Genome-Wide Variation and Phylogenetic Relationships among Musa Species
3.6. Genetic Characterization of the Musa Accessions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lasalita-Zapico, F.; Aguilar, C.H. Elucidating plant genetic diversity and evolution through bioinformatics. Int. Proc. Chem. Biol. Environ. Eng. 2014, 63, 28–34. [Google Scholar]
- Philippine Fiber Industry Development Authority (PhilFIDA) Annual Report; Philippine Fiber Industry Development Authority: Quezon City, Philippines, 2020.
- Spencer, J.E. The abacá plant and its fiber, Manila Hemp. Econ. Bot. 1953, 7, 195–213. [Google Scholar] [CrossRef]
- Bledzki, A.K.; Mamun, A.A.; Faruk, O. Abaca fibre reinforced PP composites and comparison with jute and flax fibre PP composites. Express Polym. Lett. 2007, 1, 755–762. [Google Scholar] [CrossRef]
- Barba, B.J.; Madrid, J.; Penaloza, D.J.P. A review of abaca fiber-reinforced polymer composites: Different modes of preparation and their applications. J. Chil. Chem. Soc. 2020, 65, 4919–4924. [Google Scholar] [CrossRef]
- Liu, K.; Takagi, H.; Yang, Z. Dependence of tensile properties of abaca fiber fragments and its unidirectional composites on the fragment height in the fiber stem. Compos. Part A Appl. Sci. 2013, 45, 14–22. [Google Scholar] [CrossRef]
- Philippine Fiber Industry Development Authority (PhilFIDA) Abaca Sustainability Manual; PhilFIDA: Quezon City, Philippines; Deutsche Gesellschaft für Internationale Zusammenarbeit (GIZ) GmbH: Makati City, Philippines, 2016.
- Karthik, T.; Prabha Karan, C.; Rathinamoorthy, R. Nonwovens: Process, Structure, Properties and Applications; Woodhead Publishing India PVT: Delhi, India, 2017. [Google Scholar]
- Halos, S.C. The Abaca; Department of Agriculture-Biotechnology Program Office: Quezon City, Philippines, 2008.
- Altoveros, N.C.; Borromeo, T.H. The State of Plant Genetic Resources for Food and Agriculture in the Philippines, A Country Report; Bureau of Plant Industry, Department of Agriculture: Malate, Philipines, 2007; Available online: https://www.fao.org/pgrfa-gpa-archive/phl/docs/countryreport2.pdf (accessed on 28 September 2022).
- Philippine Fiber Industry Development Authority (PhilFIDA) (2021) Abaca Technoguide. 2021. Available online: https://philfida.da.gov.ph/images/Publications/Technoguides/abaca-technoguide.pdf (accessed on 29 December 2022).
- Galvez, L.C.; Catalla, J.L.; Borromeo, T.H.; Altoveros, N.C. Abaca Germplasm Collection; Philippine Fiber Industry Development Authority: Quezon City, Philippines, 2018.
- Bajet, N.B.; Magnaye, L. Virus Diseases of Banana and Abaca in the Philippines; PARRFI: Laguna, Philippines, 2000. [Google Scholar]
- Galvez, L.C.; Barbosa, C.F.C.; Koh, R.B.L.; Aquino, V.M. Loop-mediated isothermal amplification (LAMP) assays for the detection of abaca bunchy top virus and banana bunchy top virus in abaca. Crop Prot. 2020, 131, 105101. [Google Scholar] [CrossRef]
- Arcalas, J.Y. El Niño, Storms Slashed Abaca Output in 2019–PhilFida. El Niño Ready Nations. 2020. Available online: https://elninoreadynations.com/el-nino-storms-slashed-abaca-output-in-2019-philfida/ (accessed on 22 January 2022).
- Vishwakarma, M.K.; Kale, S.M.; Sriswathi, M.; Naresh, T.; Shasidhar, Y.; Garg, V.; Pandey, M.K.; Varshney, R.K. Genome-wide discovery and deployment of insertions and deletions markers provided greater insights on species, genomes, and sections relationships in the genus Arachis. Front. Plant Sci. 2017, 8, 2064. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Luo, T.; Zhang, W.; Mason, A.S.; Huang, D.; Huang, X.; Tang, W.; Dou, Y.; Zhang, C.; Xiao, Y. Development of high-density SNP markers and their application in evaluating genetic diversity and population structure in Elaeis guineensis. Front. Plant Sci. 2019, 10, 130. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Li, Z.; Zhang, J.; Bai, S.; Zhao, W.; Tong, C. Investigation of genome-wide InDel distribution and segregation in Populus with restriction-site associated DNA sequencing data. Trop. Plant Biol. 2022, 15, 171–180. [Google Scholar] [CrossRef]
- Korir, N.K.; Han, J.; Shangguan, L.; Wang, C.; Kayesh, E.; Zhang, Y.; Fang, J. Plant variety and cultivar identification: Advances and prospects. Crit. Rev. Biotechnol. 2013, 33, 111–125. [Google Scholar] [CrossRef]
- Garrido-Cardenas, J.A.; Mesa-Valle, C.; Manzano-Agugliaro, F. Trends in plant research using molecular markers. Planta 2018, 247, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jung, J.-K.; Shim, E.-J.; Chung, S.-M.; Park, Y.; Lee, G.P.; Sim, S.-C. Genome-wide SNP discovery and core marker sets for DNA barcoding and variety identification in commercial tomato cultivars. Sci. Hortic. 2021, 276, 109734. [Google Scholar] [CrossRef]
- Fang, W.-P.; Meinhardt, L.W.; Tan, H.-W.; Zhou, L.; Mischke, S.; Zhang, D. Varietal identification of tea (Camellia sinensis) using nanofluidic array of single nucleotide polymorphism (SNP) markers. Hortic. Res. 2014, 1, 14035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yllano, O.B.; Diaz, M.G.Q.; Lalusin, A.G.; Laurena, A.C.; Tecson, E.M. Genetic analyses of abaca (Musa textilis Née) germplasm from its primary center of origin, the Philippines, using Simple Sequence Repeat (SSR) markers. Philipp. Agric. Sci. 2020, 103, 12. [Google Scholar]
- Galvez, L.C.; Meinhardt, L.W.; Goenaga, R.; Zhang, D. Accurate identification of abaca (Musa textilis Née) cultivars using single nucleotide polymorphisms (SNP) markers developed for banana (Musa acuminata Colla). Int. J. Plant Biol. Res. 2021, 9, 1125. [Google Scholar]
- Ratnadewi, D. Identification of Musa textilis by using RAPD markers. In Resource Management: Public-Private Partnership and Knowledge Sharing; Adhi, A.K., Pambudy, R., Burhanuddin, Eds.; SEAG IPB: Bogor, Indonesia, 2001; pp. 75–80. Available online: https://repository.ipb.ac.id/jspui/bitstream/123456789/64194/1/karil1.pdf (accessed on 13 September 2022).
- Boguero, A.P.B.; Parducho, M.A.L.; Mendoza, M.R.; Abustan, M.A.M.; Lalusin, A.G. Molecular screening of abaca (Musa textilis Née) accessions using microsatellite markers associated with resistance to bunchy top virus. Philipp. J. Crop Sci. 2016, 41, 13–19. [Google Scholar]
- Kumar, S.; Banks, T.W.; Cloutier, S. SNP Discovery through Next-Generation Sequencing and Its Applications. Int. J. Plant Genom. 2012, 2012, e831460. [Google Scholar] [CrossRef]
- Hahn, M.W. Molecular Population Genetics; Oxford University Press: Cary, NC, USA, 2018. [Google Scholar]
- Li, J.; Jew, B.; Zhan, L.; Hwang, S.; Coppola, G.; Freimer, N.B.; Sul, J.H. ForestQC: Quality control on genetic variants from next-generation sequencing data using random forest. PLoS Comput. Biol. 2019, 15, e1007556. [Google Scholar] [CrossRef] [Green Version]
- Galvez, L.C.; Koh, R.B.L.; Barbosa, C.F.C.; Asunto, J.C.; Catalla, J.L.; Atienza, R.G.; Costales, K.T.; Aquino, V.M.; Zhang, D. Sequencing and de novo assembly of abaca (Musa textilis Née) var. Abuab genome. Genes 2021, 12, 1202. [Google Scholar] [CrossRef]
- Philippine Fiber Industry Development Authority (PhilFIDA). 3 Abaca Varieties Now Registered with the National Seed Industry Council. 2017. Available online: http://www.philfida.da.gov.ph/index.php/news-articles/116-3-abaca-varities-now-regsitered-with-nsic (accessed on 29 December 2022).
- Koh, R.B.L.; Barbosa, C.F.C.; Aquino, V.M.; Galvez, L.C. Extraction of high molecular weight DNA suitable for next-generation sequencing from the fiber crop abaca. Ind. Crops Prod. 2021, 161, 113194. [Google Scholar] [CrossRef]
- Sambles, C.; Venkatesan, L.; Shittu, O.M.; Harrison, J.; Moore, K.; Tripathi, L.; Grant, M.; Warmington, R.; Studholme, D.J. Genome sequencing data for wild and cultivated bananas, plantains and abacá. Data Brief 2020, 33, 106341. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 18 August 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfield, J.K.; Marshall, J.; Danecek, P.; Li, H.; Ohan, V.; Whitwham, A.; Keane, T.; Davies, R.M. HTSlib: C library for reading/writing high-throughput sequencing data. GigaScience 2021, 10, giab007. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef] [Green Version]
- Lefouili, M.; Nam, K. The evaluation of Bcftools mpileup and GATK HaplotypeCaller for variant calling in non-human species. Sci. Rep. 2022, 12, 1. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.T.L.; Rosenberg, N.A. Mathematical properties of linkage disequilibrium statistics defined by normalization of the coefficient D=pAB−pApB. Hum. Hered. 2019, 84, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G.; Robertson, A. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 1968, 38, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Kachman, S.D.; Garrick, D.; Fernando, R.L.; Cheng, H. Exact Distribution of Linkage Disequilibrium in the Presence of Mutation, Selection, or Minor Allele Frequency Filtering. Front. Genet. 2020, 11, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar] [CrossRef]
- Serrote, C.M.L.; Reiniger, L.R.S.; Silva, K.B.; Rabaiolli, S.M.D.S.; Stefanel, C.M. Determining the Polymorphism Information Content of a molecular marker. Gene 2020, 726, 144175. [Google Scholar] [CrossRef]
- Harris, A.M.; DeGiorgio, M. An unbiased estimator of gene diversity with improved variance for samples containing related and inbred individuals of any ploidy. Genes Genomes Genet. 2017, 7, 671–691. [Google Scholar] [CrossRef] [Green Version]
- Chanock, S.J.; Ostrander, E.A. 22—Discovery and Characterization of Cancer Genetic Susceptibility Alleles. In Abeloff’s Clinical Oncology, 5th ed.; Niederhuber, J.E., Kastan, M.B., Tepper, J.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 309–321. [Google Scholar]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Knaus, B.J.; Grünwald, N.J. VCFR: A package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 2017, 17, 44–53. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis. 2016. Available online: https://ggplot2.tidyverse.org (accessed on 18 August 2022).
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Tabima, J.F.; Knaus, B.J.; Grünwald, N.J. (n.d) GBS Analysis. Available online: https://grunwaldlab.github.io/Population_Genetics_in_R/gbs_analysis.html#:~:text=GBS%20is%20one%20of%20several,recommend%20reading%20Baird%20et%20al (accessed on 28 December 2022).
- Simmonds, N.W. Notes on Banana Varieties in Hawaii. Pac. Sci. 1954, 8, 226–229. [Google Scholar]
- Li, Z.; Wang, J.; Fu, Y.; Jing, Y.; Huang, B.; Chen, Y.; Wang, Q.; Wang, X.B.; Meng, C.; Yang, Q.; et al. The Musa troglodytarum L. genome provides insights into the mechanism of non-climacteric behaviour and enrichment of carotenoids. BMC Biol. 2022, 20, 186. [Google Scholar] [CrossRef] [PubMed]
- Nyine, M.; Uwimana, B.; Akech, V.; Brown, A.; Ortiz, R.; Doležel, J.; Lorenzen, J.; Swennen, R. Association genetics of bunch weight and its component traits in East African highland banana (Musa spp. AAA group). Theor. Appl. Genet. 2019, 132, 3295–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anselmo, C.A.F.; Pinheiro, A. Phylogenetic trees via Hamming distance decomposition tests. J. Stat. Comput. Simul. 2012, 82, 1287–1297. [Google Scholar] [CrossRef]
- Volk, G.M.; Byrne, P.F.; Coyne, C.J.; Flint-Garcia, S.; Reeves, P.A.; Richards, C. Integrating Genomic and Phenomic Approaches to Support Plant Genetic Resources Conservation and Use. Plants 2021, 10, 2260. [Google Scholar] [CrossRef]
- Wu, W.; Ng, W.-L.; Yang, J.-X.; Li, W.-M.; Ge, X.-J. High cryptic species diversity is revealed by genome-wide polymorphisms in a wild relative of banana, Musa itinerans, and implications for its conservation in subtropical China. BMC Plant Biol. 2018, 18, 194. [Google Scholar] [CrossRef]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Foster, J.T.; Bull, R.L.; Keim, P. Ricin forensics: Comparisons to microbial forensics. In Microbial Forensics, 3rd ed.; Budowle, B., Schutzer, S., Morse, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 241–250. [Google Scholar]
- Phillips, C.; Amigo, J.; Tillmar, A.O.; Peck, M.A.; de la Puente, M.; Ruiz-Ramírez, J.; Bittner, F.; Idrizbegović, Š.; Wang, Y.; Parsons, T.J.; et al. A compilation of tri-allelic SNPs from 1000 Genomes and use of the most polymorphic loci for a large-scale human identification panel. Forensic Sci. Int. Genet. 2020, 46, 102232. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; McDowell, I.C.; Nodzenski, M.; Scholtens, D.M.; Allen, A.S.; Lowe, W.L.; Reddy, T.E. Transversions have larger regulatory effects than transitions. BMC Genom. 2017, 18, 394. [Google Scholar] [CrossRef] [Green Version]
- Choudhuri, S. Chapter 9—Phylogenetic Analysis. In Bioinformatics for Beginners; Choudhuri, S., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 209–218. [Google Scholar] [CrossRef]
- Jensen, J.L.; Bohonak, A.J.; Kelley, S.T. Isolation by distance, web service. BMC Genet. 2005, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Roorkiwal, M.; Kale, S.; Garg, V.; Yadala, R.; Varshney, R.K. InDel markers: An extended marker resource for molecular breeding in chickpea. PLoS ONE 2019, 14, e0213999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dela Cueva, F.M.; Perez, N.A.M.; Benjamin, A.F.A.; Yanos, L.A.; Gueco, L.S.; Thomas, J.E. Resistance of Musa balbisiana Accessions of the Philippines to Banana Bunchy Top Virus; The American Phytopathological Society (APS): St. Paul, MN, USA, 2023. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Species | Description | Source of Sequence Reads | Accession Number |
|---|---|---|---|---|
| Abuabref | M. textilis | Reference genome | [30] | N/A |
| Abuab | M. textilis | Commercial abaca variety | This study | SRR22906090 |
| Hagbayanon | M. textilis | Commercial abaca variety | This study | SRR22906084 |
| Ihalas | M. textilis | Wild abaca accession | [32] | N/A |
| Inosa | M. textilis | Commercial abaca variety | This study | SRR22906089 |
| Kutay | M. textilis | Commercial abaca variety | This study | SRR22906087 |
| Laylay | M. textilis | Commercial abaca variety | This study | SRR22906086 |
| Luno | M. textilis | Commercial abaca variety | This study | SRR22906083 |
| LunoGreen | M. textilis | Luno with green inflorescence | This study | SRR22906080 |
| Samoro | M. textilis | Present as a single hill in MFESS | This study | SRR22906088 |
| Socorro | M. textilis | Commercial abaca variety | This study | SRR22906082 |
| Tangongon | M. textilis | Commercial abaca variety | This study | SRR22906085 |
| Tputi | M. textilis | Commercial abaca variety | This study | SRR22906081 |
| Unknown cultivar | M. textilis | Abaca accession | [33] | SRR9696635 |
| Unknown cultivar | M. textilis | Abaca accession | [33] | SRR8989639 |
| Unknown cultivar | M. textilis | Abaca accession | [33] | SRR9850642 |
| Banana | M. acuminata | Wild banana accession | [33] | SRR8989638 |
| Banana | M. acuminata | Wild banana accession | [33] | SRR8989629 |
| Banana | M. acuminata | Wild banana accession | [33] | SRR8989632 |
| Banana | M. balbisiana | Wild banana accession | [33] | SRR8989633 |
| Banana | M. balbisiana | Wild banana accession | [33] | SRR9734079 |
| Banana | M. balbisiana | Wild banana accession | [33] | SRR6147592 |
| Banana | M. balbisiana | Wild banana accession | [33] | SRR9850640 |
| Banana | M. troglodytarum | Wild banana accession | [33] | SRR8989640 |
| Banana | M. troglodytarum | Wild banana accession | [33] | SRR9734080 |
| Banana | M. troglodytarum | Wild banana accession | [33] | SRR9850641 |
| Within M. textilis | Among Musa spp. | |||
|---|---|---|---|---|
| SNPs | InDels | SNPs | InDels | |
| Total | 19,189,434 | 1,400,947 | 42,647,249 | 2,466,646 |
| High quality (MQ > 40), biallelic | 15,410,778 | 1,109,789 | 34,643,663 | 1,933,417 |
| High quality (MQ > 40), multiallelic | 388,338 | 135,230 | 1,814,554 | 2,31,670 |
| Number of transitions (Ts) | 108,067 | N/A | 18,106 | N/A |
| Number of transversions (Tv) | 22,701 | N/A | 13,138 | N/A |
| Ts/Tv ratio | 4.76 | N/A | 1.38 | N/A |
| Retained after LD pruning | 635,945 | 84,711 | 2,130,711 | 192,835 |
| Number of loci with at most 10% missing genotypes | 130,768 | 13,620 | 31,244 | 577 |
| Diversity Metrics | This Study | [24] | |
|---|---|---|---|
| SNPs | InDels | SNPs | |
| PIC | 0.312 ± 0.068 | 0.332 ± 0.076 | - |
| He | 0.396 ± 0.106 | 0.431 ± 0.111 | 0.281 ± 0.135 |
| MAF | 0.310 ± 0.126 | 0.362 ± 0.124 | 0.196 ± 0.132 |
| Accession/Variety | Species | N_SITES | F |
|---|---|---|---|
| SRR8989629 | M. acuminata | 30,123 | 0.93086 |
| SRR6147592 | M. balbisiana | 30,963 | 0.928 |
| SRR9850640 | M. balbisiana | 28,416 | 0.92696 |
| SRR9734079 | M. balbisiana | 30,554 | 0.9252 |
| SRR8989632 | M. acuminata | 30,643 | 0.91983 |
| SRR8989633 | M. balbisiana | 31,192 | 0.91946 |
| SRR8989638 | M. acuminata | 30,648 | 0.91891 |
| SRR9850641 | M. troglodytarum | 25,747 | 0.86544 |
| SRR8989640 | M. troglodytarum | 31,205 | 0.8618 |
| SRR9734080 | M. troglodytarum | 31,204 | 0.85997 |
| Ihalas | M. textilis | 30,374 | 0.57407 |
| Kutaykutay | M. textilis | 31,079 | 0.53542 |
| Tputi | M. textilis | 31,165 | 0.52615 |
| Samoro | M. textilis | 30,835 | 0.46154 |
| Socorro | M. textilis | 30,819 | 0.45137 |
| Tangongon | M. textilis | 31,072 | 0.40478 |
| Laylay | M. textilis | 30,220 | 0.35956 |
| SRR9696635 | M. textilis | 31,032 | 0.04446 |
| SRR8989639 | M. textilis | 31,185 | 0.02363 |
| SRR9850642 | M. textilis | 31,214 | 0.02259 |
| Luno | M. textilis | 30,726 | −0.37979 |
| Hagbayanon | M. textilis | 30,791 | −0.38391 |
| Abuab | M. textilis | 29,625 | −3.893 |
| LunoGreen | M. textilis | 29,761 | −3.90269 |
| Inosa | M. textilis | 31,039 | −3.97664 |
| Abuabref | M. textilis | 31,180 | −3.99309 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa, C.F.C.; Asunto, J.C.; Koh, R.B.L.; Santos, D.M.C.; Zhang, D.; Cao, E.P.; Galvez, L.C. Genome-Wide SNP and Indel Discovery in Abaca (Musa textilis Née) and among Other Musa spp. for Abaca Genetic Resources Management. Curr. Issues Mol. Biol. 2023, 45, 5776-5797. https://doi.org/10.3390/cimb45070365
Barbosa CFC, Asunto JC, Koh RBL, Santos DMC, Zhang D, Cao EP, Galvez LC. Genome-Wide SNP and Indel Discovery in Abaca (Musa textilis Née) and among Other Musa spp. for Abaca Genetic Resources Management. Current Issues in Molecular Biology. 2023; 45(7):5776-5797. https://doi.org/10.3390/cimb45070365
Chicago/Turabian StyleBarbosa, Cris Francis C., Jayson C. Asunto, Rhosener Bhea L. Koh, Daisy May C. Santos, Dapeng Zhang, Ernelea P. Cao, and Leny C. Galvez. 2023. "Genome-Wide SNP and Indel Discovery in Abaca (Musa textilis Née) and among Other Musa spp. for Abaca Genetic Resources Management" Current Issues in Molecular Biology 45, no. 7: 5776-5797. https://doi.org/10.3390/cimb45070365