
Selective Concurrence of the Long Non-Coding RNA MALAT1 and the Polycomb Repressive Complex 2 to Promoter Regions of Active Genes in MCF7 Breast Cancer Cells
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
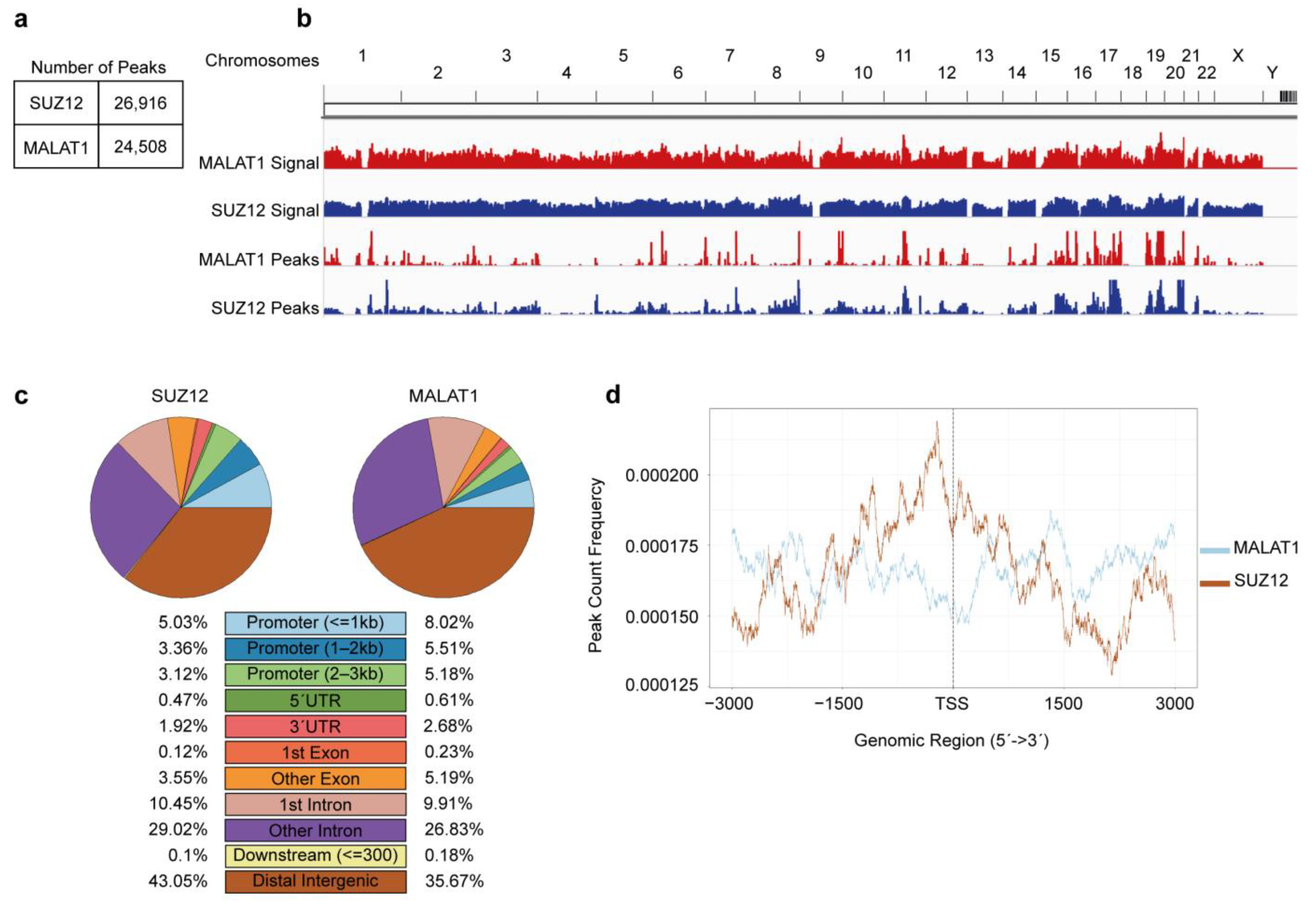
2.1. CHIP- and CHART-Seq Datasets
2.2. Quality Control, Alignment and Peak Calling
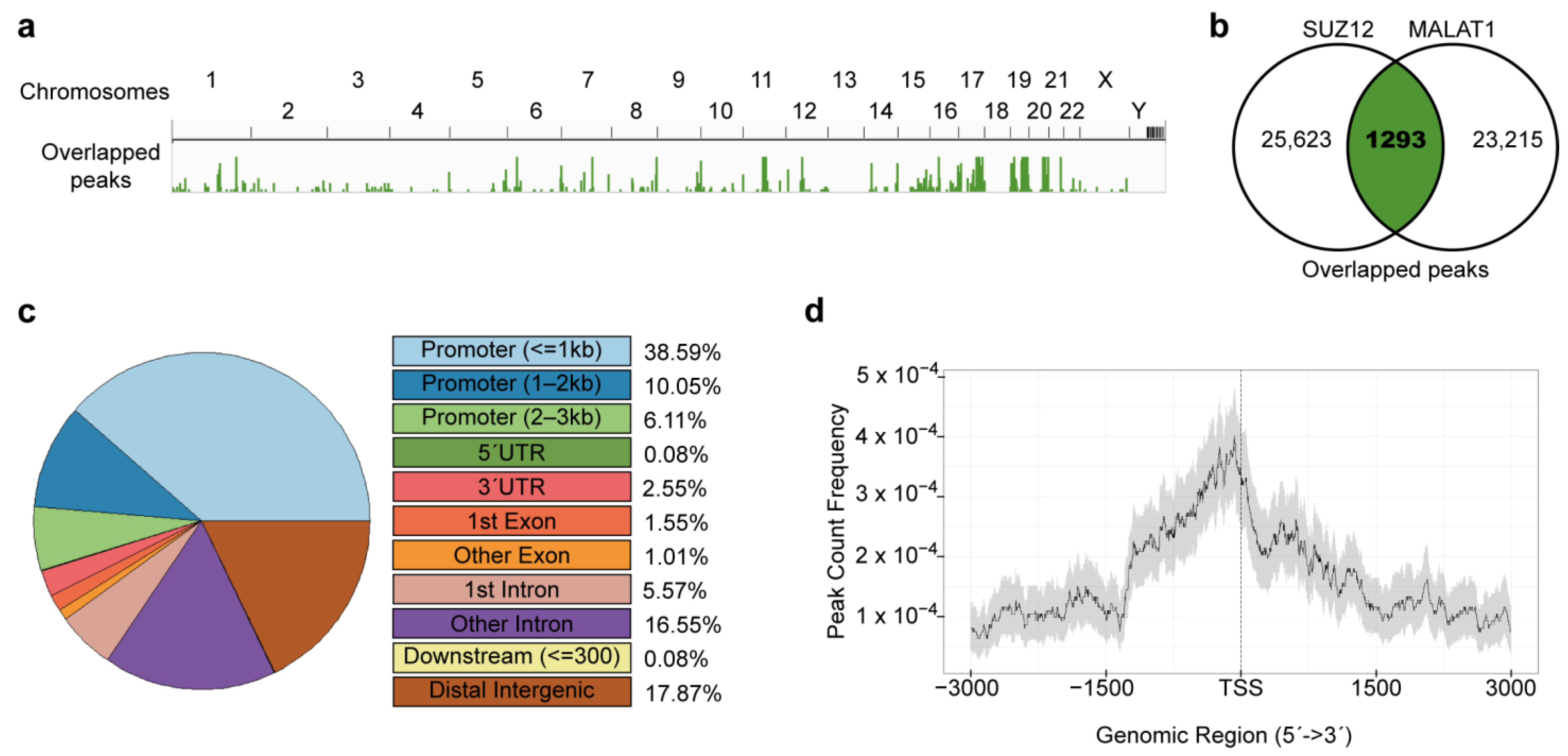
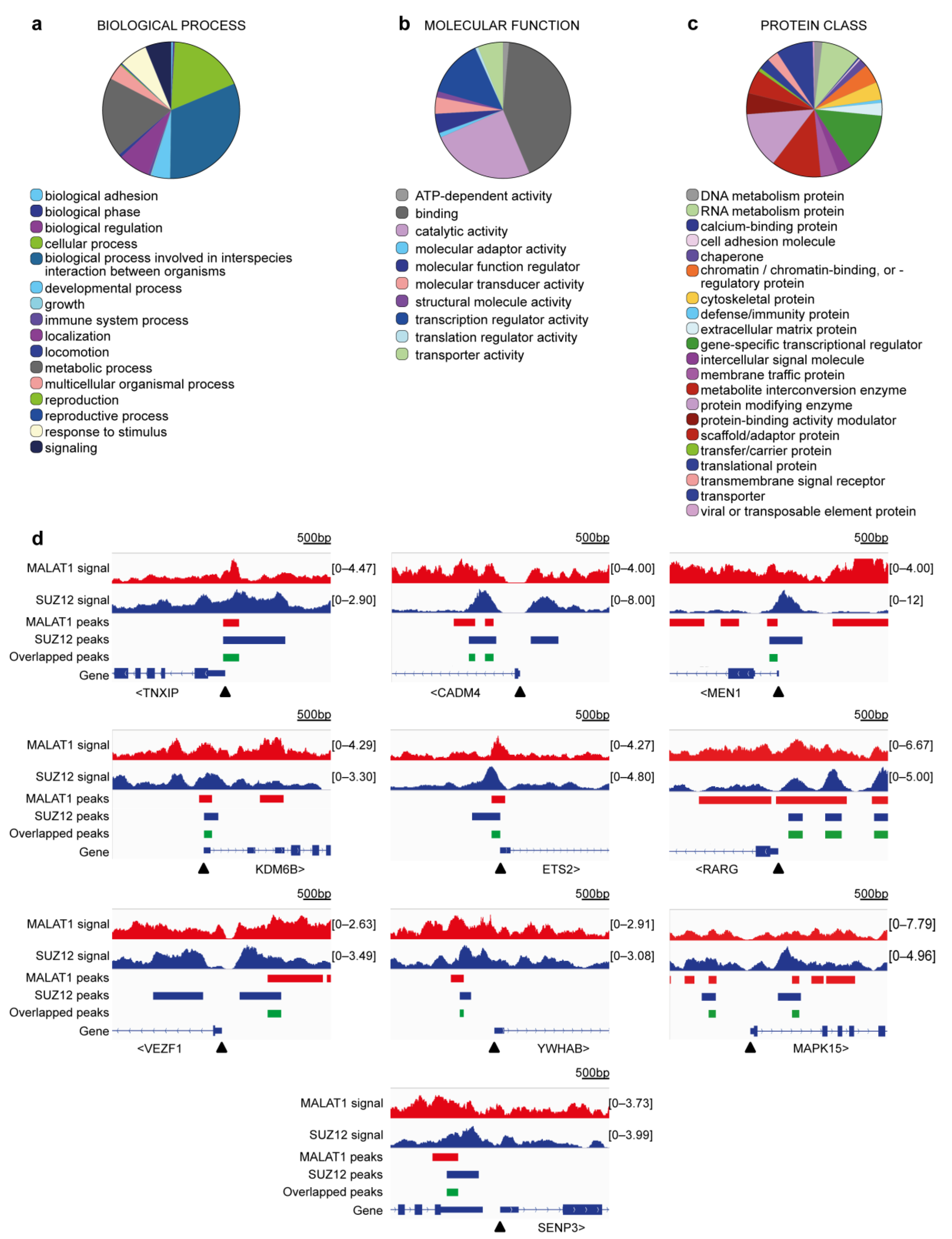
2.3. Detection of Overlapped Peaks and Gene Ontology Analyses
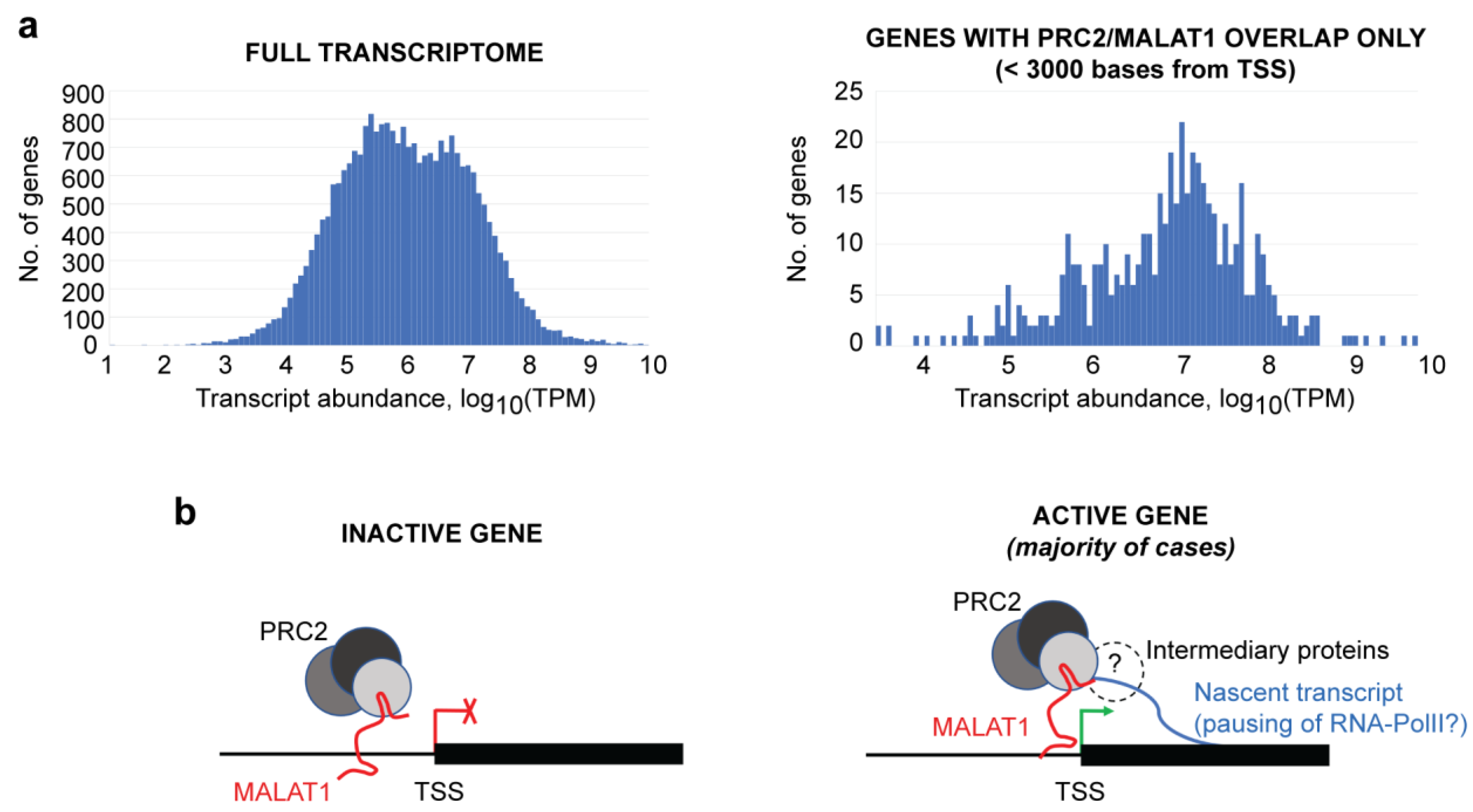
2.4. RNA-Seq Analyses
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, R.; Bustos, F.J.; Saez, M.; Rojas, A.; Allende, M.L.; van Wijnen, A.J.; van Zundert, B.; Montecino, M. Polycomb PRC2 complex mediates epigenetic silencing of a critical osteogenic master regulator in the hippocampus. Biochim. Biophys. Acta 2016, 1859, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.; Blum, R.; Kesner, B.; Aeby, E.; Garant, J.M.; Szanto, A.; Lee, J.T. Motif-driven interactions between RNA and PRC2 are rheostats that regulate transcription elongation. Nat. Struct. Mol. Biol. 2021, 28, 103–117. [Google Scholar] [CrossRef]
- Henriquez, B.; Bustos, F.J.; Aguilar, R.; Becerra, A.; Simon, F.; Montecino, M.; van Zundert, B. Ezh1 and Ezh2 differentially regulate PSD-95 gene transcription in developing hippocampal neurons. Mol. Cell Neurosci. 2013, 57, 130–143. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Adibfar, S.; Elveny, M.; Kashikova, H.S.; Mikhailova, M.V.; Farhangnia, P.; Vakili-Samiani, S.; Tarokhian, H.; Jadidi-Niaragh, F. The molecular mechanisms and therapeutic potential of EZH2 in breast cancer. Life Sci. 2021, 286, 120047. [Google Scholar] [CrossRef]
- Laugesen, A.; Hojfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef]
- Aguilar, R.; Spencer, K.B.; Kesner, B.; Rizvi, N.F.; Badmalia, M.D.; Mrozowich, T.; Mortison, J.D.; Rivera, C.; Smith, G.F.; Burchard, J.; et al. Targeting Xist with compounds that disrupt RNA structure and X inactivation. Nature 2022, 604, 160–166. [Google Scholar] [CrossRef]
- Ji, P.; Diederichs, S.; Wang, W.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef]
- Gutschner, T.; Hammerle, M.; Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. 2013, 91, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Goyal, B.; Yadav, S.R.M.; Awasthee, N.; Gupta, S.; Kunnumakkara, A.B.; Gupta, S.C. Diagnostic, prognostic, and therapeutic significance of long non-coding RNA MALAT1 in cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188502. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Bai, Z.; Liu, Y.; Gao, Y.; Zhou, J.; Zhang, Y.; Li, J. Exosomes Derived from Tumor Cells Initiate Breast Cancer Cell Metastasis and Chemoresistance through a MALAT1-Dependent Mechanism. J. Oncol. 2022, 2022, 5483523. [Google Scholar] [CrossRef] [PubMed]
- Abulwerdi, F.A.; Xu, W.; Ageeli, A.A.; Yonkunas, M.J.; Arun, G.; Nam, H.; Schneekloth, J.S., Jr.; Dayie, T.K.; Spector, D.; Baird, N.; et al. Selective Small-Molecule Targeting of a Triple Helix Encoded by the Long Noncoding RNA, MALAT1. ACS Chem. Biol. 2019, 14, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Lino Cardenas, C.L.; Kessinger, C.W.; Cheng, Y.; MacDonald, C.; MacGillivray, T.; Ghoshhajra, B.; Huleihel, L.; Nuri, S.; Yeri, A.S.; Jaffer, F.A.; et al. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat. Commun. 2018, 9, 1009. [Google Scholar] [CrossRef] [PubMed]
- Warda, A.S.; Kretschmer, J.; Hackert, P.; Lenz, C.; Urlaub, H.; Hobartner, C.; Sloan, K.E.; Bohnsack, M.T. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017, 18, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Achour, C.; Aguilo, F. Long non-coding RNA and Polycomb: An intricate partnership in cancer biology. Front. Biosci. 2018, 23, 2106–2132. [Google Scholar] [CrossRef]
- Ye, M.; Xie, L.; Zhang, J.; Liu, B.; Liu, X.; He, J.; Ma, D.; Dong, K. Determination of long non-coding RNAs associated with EZH2 in neuroblastoma by RIP-seq, RNA-seq and ChIP-seq. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef]
- Huang, J.; Fang, J.; Chen, Q.; Chen, J.; Shen, J. Epigenetic silencing of E-cadherin gene induced by lncRNA MALAT-1 in acute myeloid leukaemia. J. Clin. Lab. Anal. 2022, 36, e24556. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Ooi, H.S.; Wu, J.; Chen, J.; Zhang, X.; Tan, S.; Yu, Q.; Li, Y.Y.; Kang, Y.; Li, H.; et al. MALAT1 long ncRNA promotes gastric cancer metastasis by suppressing PCDH10. Oncotarget 2016, 7, 12693–12703. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, X.; Wang, H.; Wang, L.; Liu, T.; Du, L.; Yang, Y.; Wang, C. MALAT1 Is Associated with Poor Response to Oxaliplatin-Based Chemotherapy in Colorectal Cancer Patients and Promotes Chemoresistance through EZH2. Mol. Cancer Ther 2017, 16, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Zovoilis, A.; Cifuentes-Rojas, C.; Chu, H.P.; Hernandez, A.J.; Lee, J.T. Destabilization of B2 RNA by EZH2 Activates the Stress Response. Cell 2016, 167, 1788–1802.e1713. [Google Scholar] [CrossRef]
- Fan, Y.; Shen, B.; Tan, M.; Mu, X.; Qin, Y.; Zhang, F.; Liu, Y. TGF-beta-induced upregulation of malat1 promotes bladder cancer metastasis by associating with suz12. Clin. Cancer Res. 2014, 20, 1531–1541. [Google Scholar] [CrossRef]
- Orlando, V.; Paro, R. Mapping Polycomb-repressed domains in the bithorax complex using in vivo formaldehyde cross-linked chromatin. Cell 1993, 75, 1187–1198. [Google Scholar] [CrossRef]
- El Said, N.H.; Della Valle, F.; Liu, P.; Paytuvi-Gallart, A.; Adroub, S.; Gimenez, J.; Orlando, V. Malat-1-PRC2-EZH1 interaction supports adaptive oxidative stress dependent epigenome remodeling in skeletal myotubes. Cell Death Dis. 2021, 12, 850. [Google Scholar] [CrossRef]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Sirokman, K.; McDonel, P.; Shishkin, A.A.; Surka, C.; Russell, P.; Grossman, S.R.; Chow, A.Y.; Guttman, M.; Lander, E.S. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell 2014, 159, 188–199. [Google Scholar] [CrossRef]
- Lu, J.Y.; Shao, W.; Chang, L.; Yin, Y.; Li, T.; Zhang, H.; Hong, Y.; Percharde, M.; Guo, L.; Wu, Z.; et al. Genomic Repeats Categorize Genes with Distinct Functions for Orchestrated Regulation. Cell Rep. 2020, 30, 3296–3311.e5. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Soule, H.D.; Vazguez, J.; Long, A.; Albert, S.; Brennan, M. A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer Inst. 1973, 51, 1409–1416. [Google Scholar] [CrossRef]
- GSM1411209: MCF7 MALAT1 CO1 CHART-seq. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM1411209 (accessed on 1 July 2021).
- GSM1411210: MCF7 MALAT1 CO2 CHART-seq. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM1411210 (accessed on 1 July 2021).
- GSM1411212: MCF7 Input for MALAT1 CHART-seq. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM1411212 (accessed on 1 July 2021).
- GSM2828862: ChIP-seq from MCF-7. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM2828862 (accessed on 1 July 2021).
- GSM2828863: ChIP-seq from MCF-7. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM2828863 (accessed on 1 July 2021).
- GSM2423179: ChIP-seq from MCF-7. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM2423179 (accessed on 1 July 2021).
- ENCODE Transcription Factor and Histone ChIP-Seq Processing Pipeline. Available online: https://github.com/ENCODE-DCC/chip-seq-pipeline2 (accessed on 1 July 2021).
- ENCODE Transcription Factor and Histone ChIP-Seq Processing Pipeline: Input JSON. Available online: https://github.com/ENCODE-DCC/chip-seq-pipeline2/blob/master/docs/input.md (accessed on 1 July 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Picard. Available online: http://broadinstitute.github.io/picard/ (accessed on 1 July 2021).
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Dieudonné, F.X.; O’Connor, P.B.; Gubler-Jaquier, P.; Yasrebi, H.; Conne, B.; Nikolaev, S.; Antonarakis, S.; Baranov, P.V.; Curran, J. GSM1915041: MCF7 Total RNA-seq, from GSE74232: The Effect of Heterogeneous Transcription Start Sites (TSS) on the Translatome: Implications for the Mammalian Cellular Phenotype. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM1915041 (accessed on 1 November 2022).
- Dieudonne, F.X.; O’Connor, P.B.; Gubler-Jaquier, P.; Yasrebi, H.; Conne, B.; Nikolaev, S.; Antonarakis, S.; Baranov, P.V.; Curran, J. The effect of heterogeneous Transcription Start Sites (TSS) on the translatome: Implications for the mammalian cellular phenotype. BMC Genom. 2015, 16, 986. [Google Scholar] [CrossRef]
- GSM1172885: MCF7, RNA-Seq, from GSE48216: Modeling Precision Treatment of Breast Cancer. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM1172885 (accessed on 1 November 2022).
- Costello, J.C.; Heiser, L.M.; Georgii, E.; Gonen, M.; Menden, M.P.; Wang, N.J.; Bansal, M.; Ammad-ud-din, M.; Hintsanen, P.; Khan, S.A.; et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 2014, 32, 1202–1212. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Teinturier, R.; Abou Ziki, R.; Kassem, L.; Luo, Y.; Malbeteau, L.; Gherardi, S.; Corbo, L.; Bertolino, P.; Bachelot, T.; Treilleux, I.; et al. Reduced menin expression leads to decreased ERalpha expression and is correlated with the occurrence of human luminal B-like and ER-negative breast cancer subtypes. Breast Cancer Res. Treat. 2021, 190, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Takihara, Y.; Matsuda, Y.; Hara, J. Role of the beta isoform of 14-3-3 proteins in cellular proliferation and oncogenic transformation. Carcinogenesis 2000, 21, 2073–2077. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.D.; Lee, Y.J.; Liu, K.; Li, G.; Lin, F.T.; Lin, W.C. E2F1 sumoylation as a protective cellular mechanism in oxidative stress response. Proc. Natl. Acad. Sci. USA 2020, 117, 14958–14969. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.; Tham, K.M.; Gill, D.J.; Bard-Chapeau, E.A.; Bard, F.A. ERK8 is a negative regulator of O-GalNAc glycosylation and cell migration. eLife 2014, 3, e01828. [Google Scholar] [CrossRef]
- Saito, M.; Goto, A.; Abe, N.; Saito, K.; Maeda, D.; Ohtake, T.; Murakami, Y.; Takenoshita, S. Decreased expression of CADM1 and CADM4 are associated with advanced stage breast cancer. Oncol. Lett. 2018, 15, 2401–2406. [Google Scholar] [CrossRef]
- Chen, Y.; Ning, J.; Cao, W.; Wang, S.; Du, T.; Jiang, J.; Feng, X.; Zhang, B. Research Progress of TXNIP as a Tumor Suppressor Gene Participating in the Metabolic Reprogramming and Oxidative Stress of Cancer Cells in Various Cancers. Front. Oncol. 2020, 10, 568574. [Google Scholar] [CrossRef]
- Xu, D.; Dwyer, J.; Li, H.; Duan, W.; Liu, J.P. Ets2 maintains hTERT gene expression and breast cancer cell proliferation by interacting with c-Myc. J. Biol. Chem. 2008, 283, 23567–23580. [Google Scholar] [CrossRef]
- Mendes-Pereira, A.M.; Sims, D.; Dexter, T.; Fenwick, K.; Assiotis, I.; Kozarewa, I.; Mitsopoulos, C.; Hakas, J.; Zvelebil, M.; Lord, C.J.; et al. Genome-wide functional screen identifies a compendium of genes affecting sensitivity to tamoxifen. Proc. Natl. Acad. Sci. USA 2012, 109, 2730–2735. [Google Scholar] [CrossRef]
- Yin, R.; Guo, L.; Gu, J.; Li, C.; Zhang, W. Over expressing miR-19b-1 suppress breast cancer growth by inhibiting tumor microenvironment induced angiogenesis. Int. J. Biochem. Cell Biol. 2018, 97, 43–51. [Google Scholar] [CrossRef]
- Xun, J.; Gao, R.; Wang, B.; Li, Y.; Ma, Y.; Guan, J.; Zhang, Q. Histone demethylase KDM6B inhibits breast cancer metastasis by regulating Wnt/beta-catenin signaling. FEBS Open Bio. 2021, 11, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, C.; Zheng, L.; Goodrich, K.J.; Cech, T.R. Promiscuous RNA binding by Polycomb repressive complex 2. Nat. Struct Mol. Biol. 2013, 20, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Rojas, C.; Hernandez, A.J.; Sarma, K.; Lee, J.T. Regulatory interactions between RNA and polycomb repressive complex 2. Mol. Cell 2014, 55, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Bolanos, B.; Gong, L.; Liu, W.; Goodrich, K.J.; Yang, X.; Chen, S.; Gooding, A.R.; Maegley, K.A.; Gajiwala, K.S.; et al. Conserved RNA-binding specificity of polycomb repressive complex 2 is achieved by dispersed amino acid patches in EZH2. eLife 2017, 6, e31558. [Google Scholar] [CrossRef]
- Wang, X.; Goodrich, K.J.; Gooding, A.R.; Naeem, H.; Archer, S.; Paucek, R.D.; Youmans, D.T.; Cech, T.R.; Davidovich, C. Targeting of Polycomb Repressive Complex 2 to RNA by Short Repeats of Consecutive Guanines. Mol. Cell 2017, 65, 1056–1067.E5. [Google Scholar] [CrossRef]
- Mousavi, K.; Zare, H.; Wang, A.H.; Sartorelli, V. Polycomb protein Ezh1 promotes RNA polymerase II elongation. Mol. Cell 2012, 45, 255–262. [Google Scholar] [CrossRef]
- Comsa, S.; Cimpean, A.M.; Raica, M. The Story of MCF-7 Breast Cancer Cell Line: 40 years of Experience in Research. Anticancer Res. 2015, 35, 3147–3154. [Google Scholar]
- Wang, Y.; Xie, Y.; Li, L.; He, Y.; Zheng, D.; Yu, P.; Yu, L.; Tang, L.; Wang, Y.; Wang, Z. EZH2 RIP-seq Identifies Tissue-specific Long Non-coding RNAs. Curr. Gene Ther. 2018, 18, 275–285. [Google Scholar] [CrossRef]
- Wang, Z.; Katsaros, D.; Biglia, N.; Shen, Y.; Fu, Y.; Loo, L.W.M.; Jia, W.; Obata, Y.; Yu, H. High expression of long non-coding RNA MALAT1 in breast cancer is associated with poor relapse-free survival. Breast Cancer Res. Treat. 2018, 171, 261–271. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef]
- Bodega, B.; Marasca, F.; Ranzani, V.; Cherubini, A.; Della Valle, F.; Neguembor, M.V.; Wassef, M.; Zippo, A.; Lanzuolo, C.; Pagani, M.; et al. A cytosolic Ezh1 isoform modulates a PRC2-Ezh1 epigenetic adaptive response in postmitotic cells. Nat. Struct. Mol. Biol. 2017, 24, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Quinodoz, S.A.; Bhat, P.; Chovanec, P.; Jachowicz, J.W.; Ollikainen, N.; Detmar, E.; Soehalim, E.; Guttman, M. SPRITE: A genome-wide method for mapping higher-order 3D interactions in the nucleus using combinatorial split-and-pool barcoding. Nat. Protoc. 2022, 17, 36–75. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Full Gene Name | Related Functions | Ref. |
|---|---|---|---|
| MEN1 | Menin | Part of CCKR signaling. Chromatin binding (component of a MLL/SET1 histone methyltransferase (HMT) complex). Negative regulator of cell cycle. Negative regulation of proliferation. Inactivation favors tumorigenesis of mammary cells. | [53] |
| YWHAB | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein beta | Part of EGF, FGF, and CCKR signaling. Stimulates cell growth. | [54] |
| SENP3 | SUMO specific peptidase 3 | Involved in processing of sumoylated proteins. High levels are associated with poor survival in breast cancer. | [55] |
| MAPK15 | Mitogen-activated protein kinase 15 | Part of multiple signaling pathways, including: PDGF, TGF-beta, EGF, FGF, IFG. Associated with apoptosis. Regulates autophagy, ciliogenesis, protein trafficking/secretion and genome integrity. Downregulation activates cell motility in breast cancer. | [56] |
| CADM4 | Cell adhesion molecule 4 | Cell–cell adhesion. Associated to wound healing. Associated with cell growth. Low expression associated with advanced breast cancer stages. | [57] |
| TXNIP | Thioredoxin interacting protein | Oxidative stress mediator. Transcriptional repressor. Tumor suppressor. Low expression in breast cancer. | [58] |
| ETS2 | ETS proto-oncogene 2, transcription factor | Transcription factor. Regulator of telomerase for breast cancer cell survival. Promotes hTERT expression. | [59] |
| RARG | Retinoic acid receptor gamma | Receptor for retinoic acid. Downregulated in tumors. Silencing causes tamoxifen resistance in breast cancer. | [60] |
| VEZF1 | Vascular endothelial zinc finger 1 | Regulator of angiogenesis in cancer. | [61] |
| KDM6B | Lysine demethylase 6B | Demethylates lysine 27 of histone H3. Regulates HOX expression. Inhibits metastasis in breast cancer cells | [62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arratia, F.; Fierro, C.; Blanco, A.; Fuentes, S.; Nahuelquen, D.; Montecino, M.; Rojas, A.; Aguilar, R. Selective Concurrence of the Long Non-Coding RNA MALAT1 and the Polycomb Repressive Complex 2 to Promoter Regions of Active Genes in MCF7 Breast Cancer Cells. Curr. Issues Mol. Biol. 2023, 45, 4735-4748. https://doi.org/10.3390/cimb45060301
Arratia F, Fierro C, Blanco A, Fuentes S, Nahuelquen D, Montecino M, Rojas A, Aguilar R. Selective Concurrence of the Long Non-Coding RNA MALAT1 and the Polycomb Repressive Complex 2 to Promoter Regions of Active Genes in MCF7 Breast Cancer Cells. Current Issues in Molecular Biology. 2023; 45(6):4735-4748. https://doi.org/10.3390/cimb45060301
Chicago/Turabian StyleArratia, Felipe, Cristopher Fierro, Alejandro Blanco, Sebastian Fuentes, Daniela Nahuelquen, Martin Montecino, Adriana Rojas, and Rodrigo Aguilar. 2023. "Selective Concurrence of the Long Non-Coding RNA MALAT1 and the Polycomb Repressive Complex 2 to Promoter Regions of Active Genes in MCF7 Breast Cancer Cells" Current Issues in Molecular Biology 45, no. 6: 4735-4748. https://doi.org/10.3390/cimb45060301