
Epigenetic Modification of Mesenchymal Stromal Cells Derived from Bone Marrow and Embryonal Tumors to Facilitate Immunotherapeutic Approaches in Pediatric Malignancies


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Isolation of Tumor-Derived Mesenchymal Stromal Cells
2.2. CFSE Proliferation Assays
2.3. Cytokine Profiling
2.4. Flow Cytometry
2.5. MTS Proliferation Assays
2.6. BATDA Cytotoxicity Assay
2.7. Statistical Analysis
3. Results
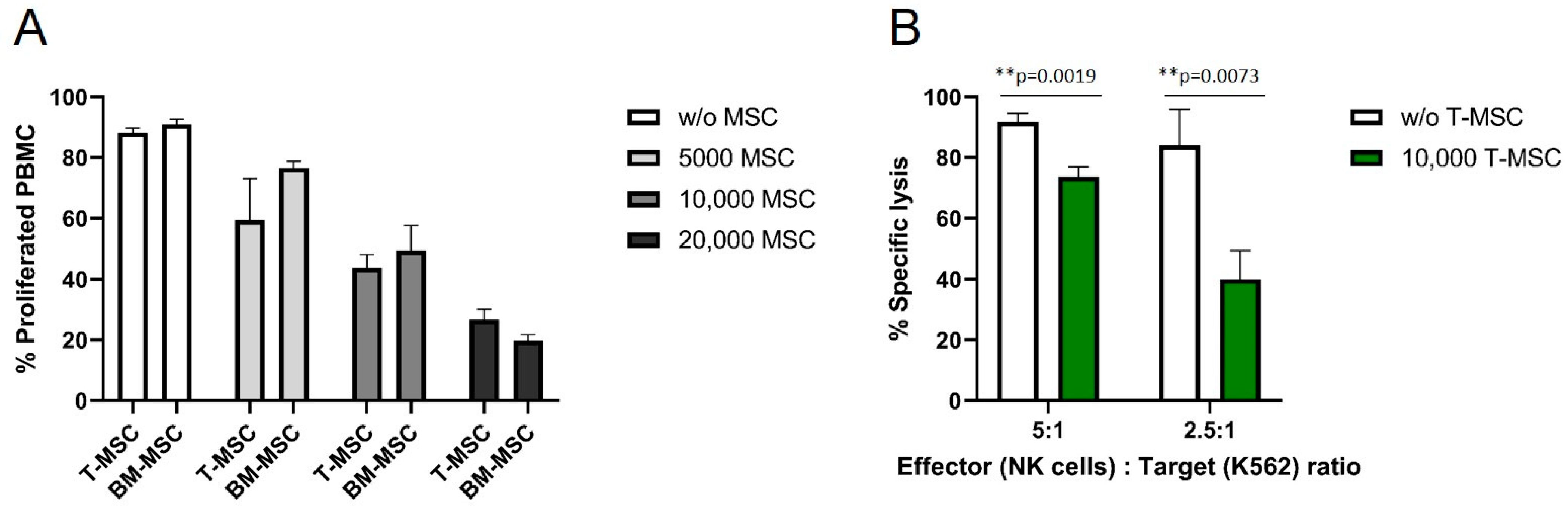
3.1. T-MSC Show Immunomodulatory Effects on Immune Effector Cells
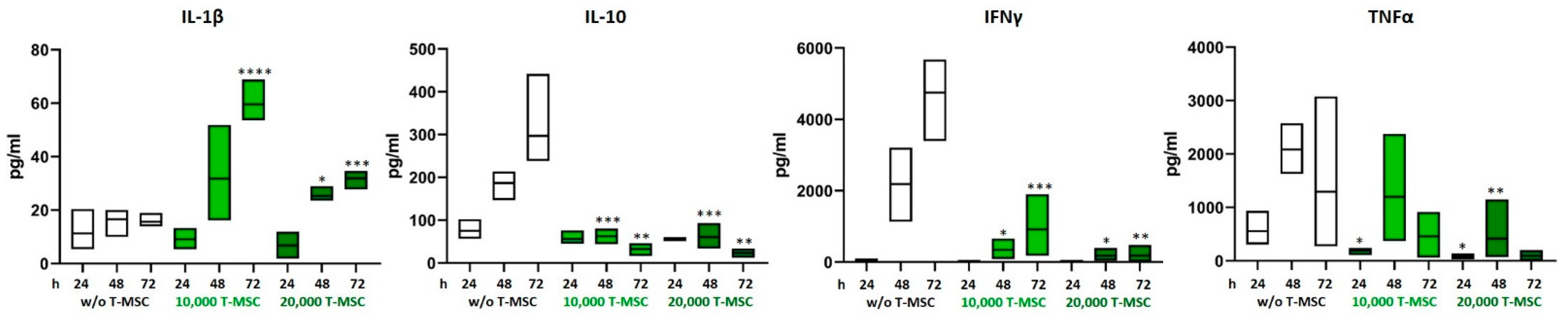
3.2. T-MSC Alter the Cytokine Profile of PBMC
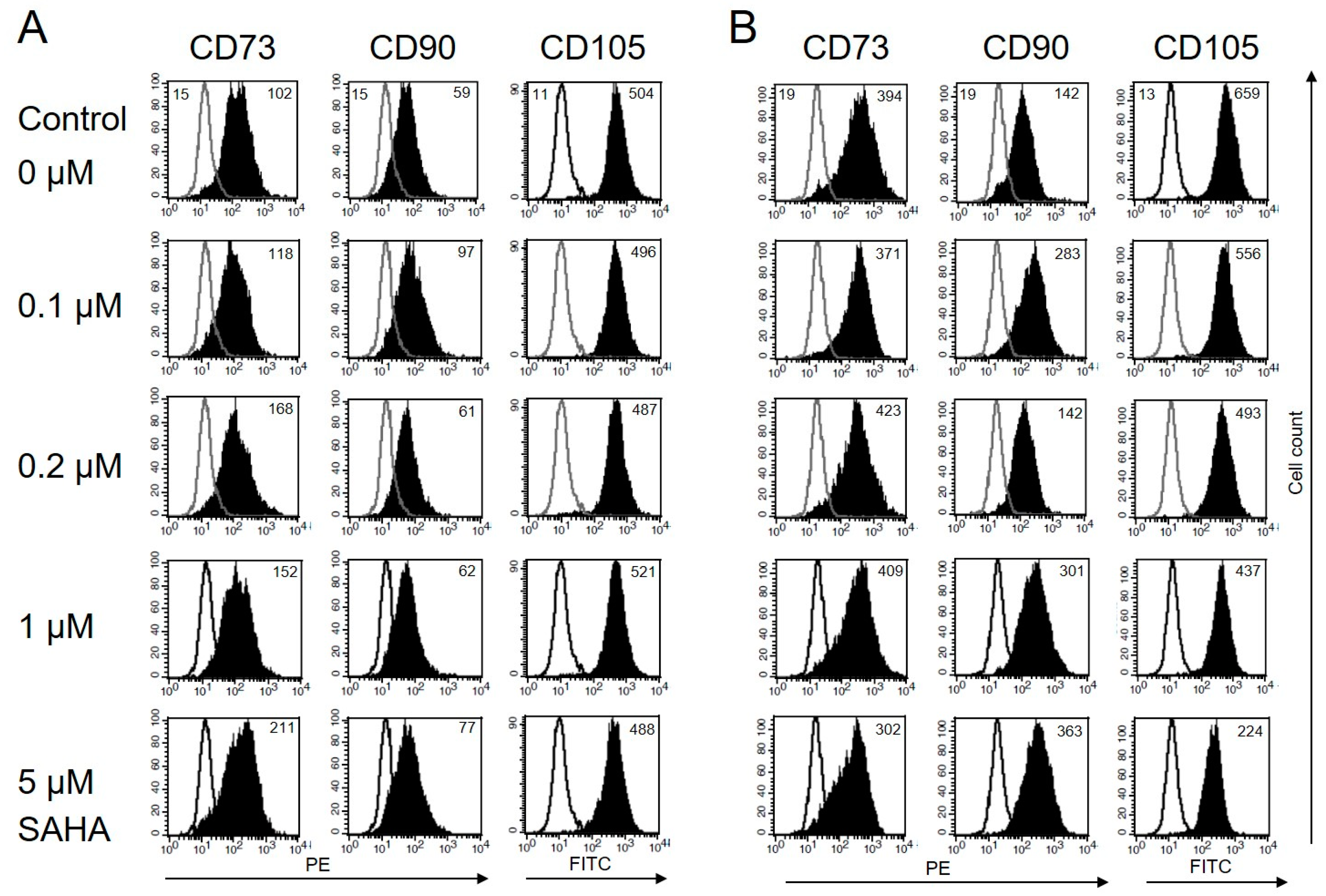
3.3. Immunophenotype of MSC Is Not Affected by Low Concentrations of SAHA
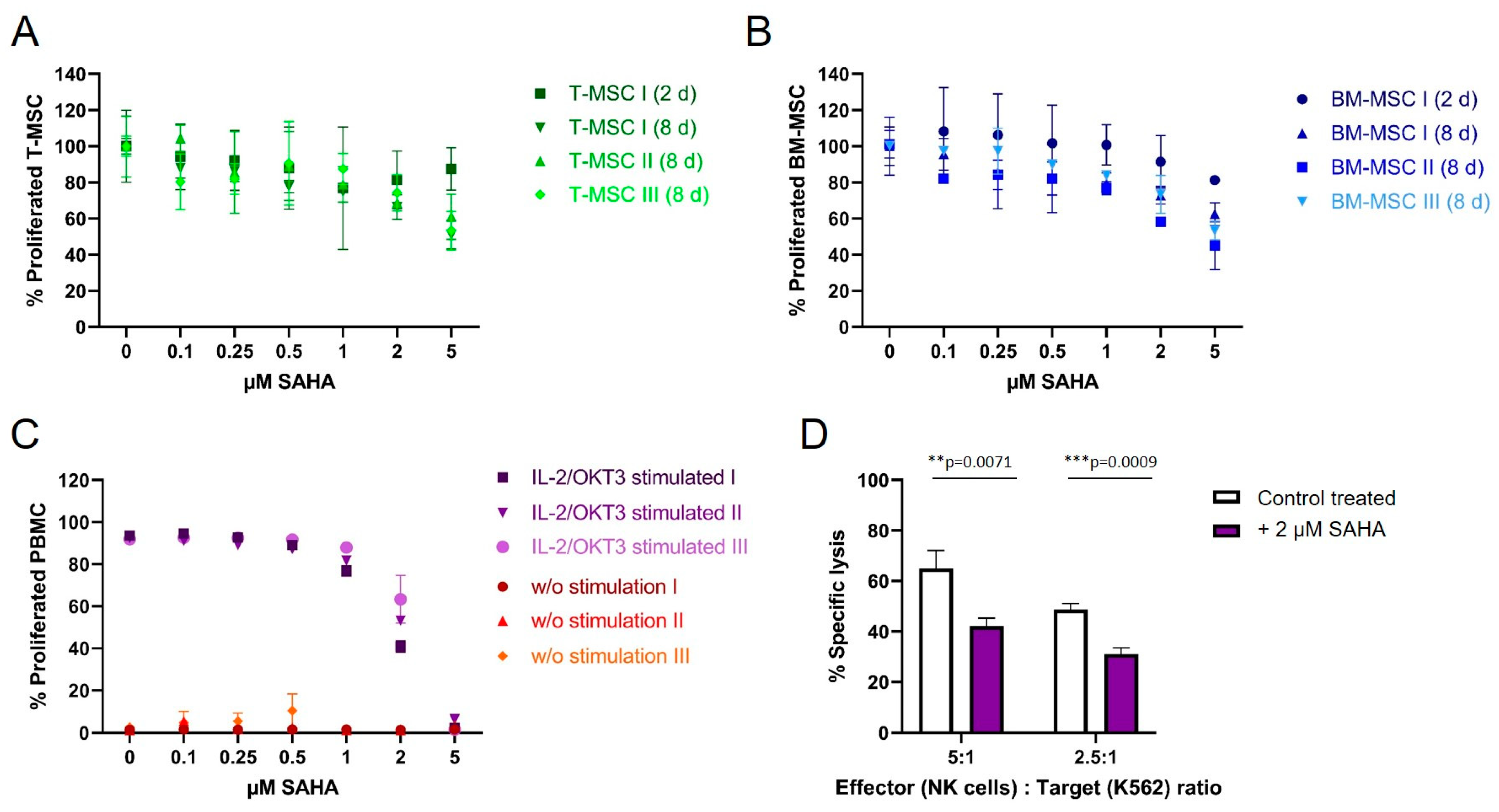
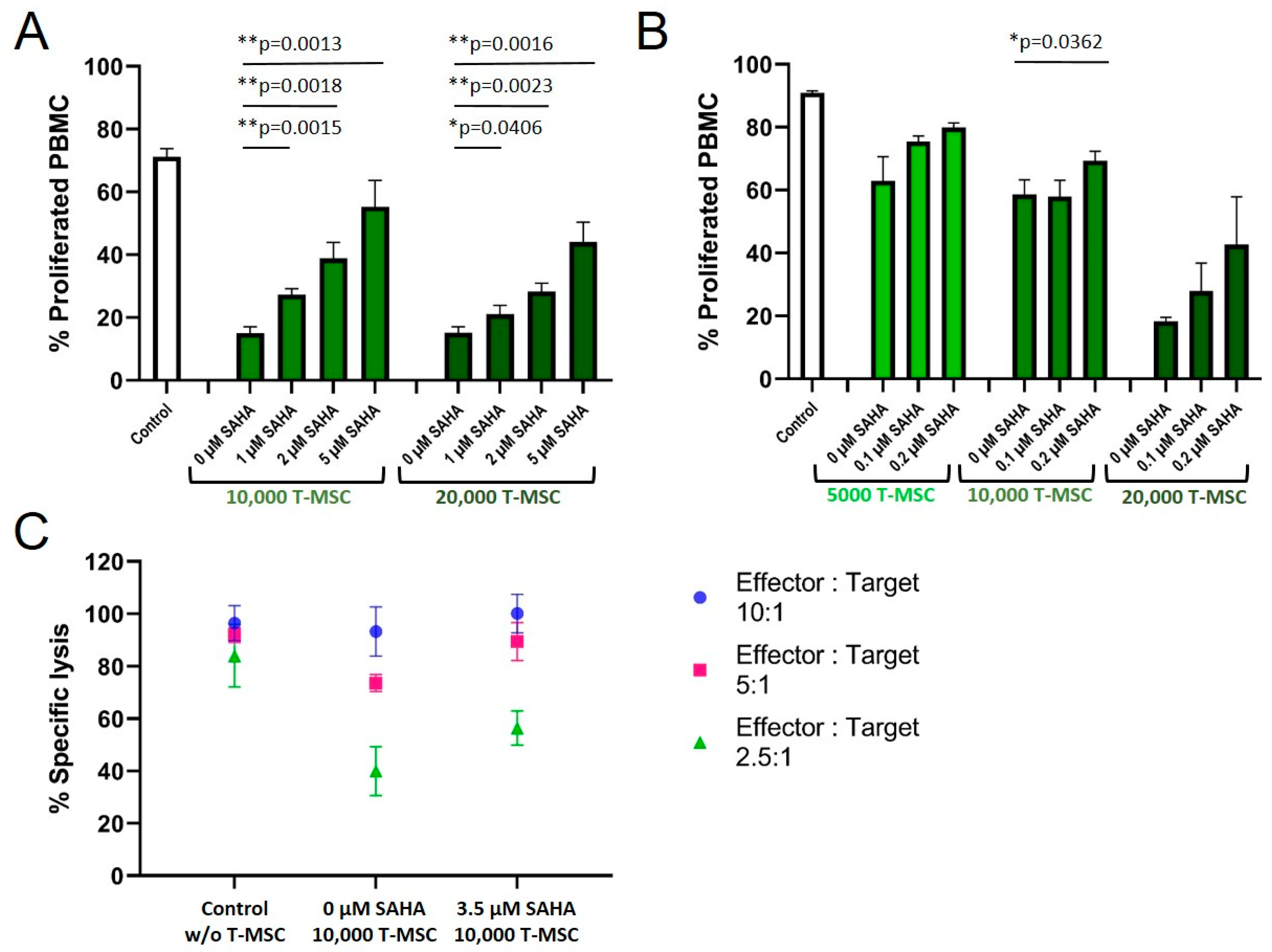
3.4. SAHA Exerts an Antiproliferative Effect on PBMC and MSC
3.5. SAHA Treatment Impairs the Immunomodulatory Effects of MSC
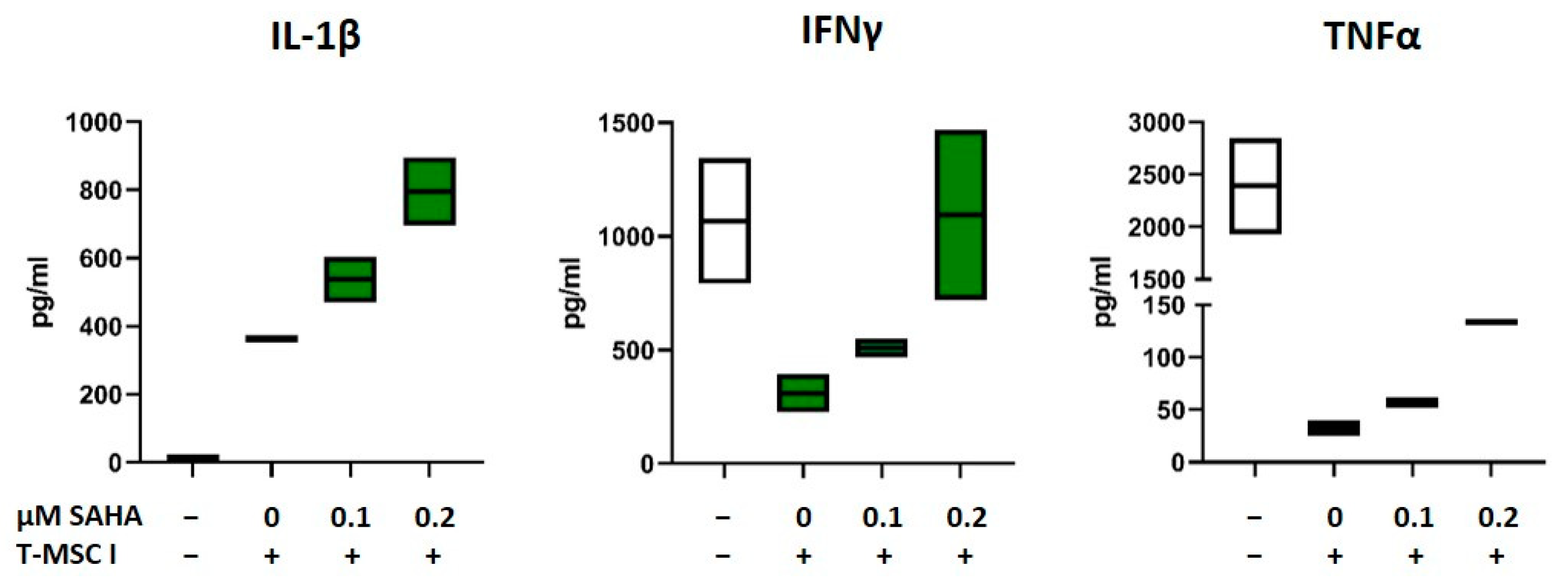
3.6. SAHA-Treated MSC Alter the Cytokine Profile of PBMC
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
List of Abbreviations
| AML | Acute myeloid leukemia |
| BM-MSC | Bone marrow-derived mesenchymal stromal cell |
| CAR | Chimeric antigen receptor |
| CLL | Chronic lymphocytic leukemia |
| FDA | United States Food and Drug Administration |
| HDACi | Histone deacetylase inhibitors |
| HPC | Hematopoietic progenitor cells |
| IFNγ | Interferon-gamma |
| IL | Interleukin |
| MDS | Myelodysplastic syndrome |
| MDSC | Myeloid-derived suppressor cell |
| MFI | Mean fluorescence intensity |
| MHC | Major histocompatibility complex |
| MSC | Mesenchymal stromal cells |
| NOS | Nitric oxide synthase |
| PBMC | Peripheral blood mononuclear cell |
| SAHA | Suberoylanilide hydroxamic acid |
| T-MSC | Tumor-derived mesenchymal stromal cell |
| TNFα | Tumor necrosis factor alpha |
References
- Ding, L.; Zhang, W.; Yang, L.; Pelicano, H.; Zhou, K.; Yin, R.; Huang, R.; Zeng, J. Targeting the autophagy in bone marrow stromal cells overcomes resistance to vorinostat in chronic lymphocytic leukemia. OncoTargets Ther. 2018, 11, 5151–5170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naveiras, O.; Daley, G.Q. Stem cells and their niche: A matter of fate. Cell Mol. Life Sci. 2006, 63, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Nauta, A.J.; Fibbe, W.E. Immunomodulatory properties of mesenchymal stromal cells. Blood 2007, 110, 3499–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef]
- Spaggiari, G.M.; Capobianco, A.; Becchetti, S.; Mingari, M.C.; Moretta, L. Mesenchymal stem cell-natural killer cell interactions: Evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood 2006, 107, 1484–1490. [Google Scholar] [CrossRef]
- Sotiropoulou, P.A.; Perez, S.A.; Gritzapis, A.D.; Baxevanis, C.N.; Papamichail, M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells 2006, 24, 74–85. [Google Scholar] [CrossRef]
- Rodriguez-Fuentes, D.E.; Fernandez-Garza, L.E.; Samia-Meza, J.A.; Barrera-Barrera, S.A.; Caplan, A.I.; Barrera-Saldana, H.A. Mesenchymal Stem Cells Current Clinical Applications: A Systematic Review. Arch. Med. Res. 2021, 52, 93–101. [Google Scholar] [CrossRef]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef] [Green Version]
- Krampera, M.; Le Blanc, K. Mesenchymal stromal cells: Putative microenvironmental modulators become cell therapy. Cell Stem Cell 2021, 28, 1708–1725. [Google Scholar] [CrossRef]
- Munk Pedersen, I.; Reed, J. Microenvironmental interactions and survival of CLL B-cells. Leuk. Lymphoma 2004, 45, 2365–2372. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Flores-Figueroa, E.; Montesinos, J.J.; Flores-Guzman, P.; Gutierrez-Espindola, G.; Arana-Trejo, R.M.; Castillo-Medina, S.; Perez-Cabrera, A.; Hernandez-Estevez, E.; Arriaga, L.; Mayani, H. Functional analysis of myelodysplastic syndromes-derived mesenchymal stem cells. Leuk. Res. 2008, 32, 1407–1416. [Google Scholar] [CrossRef]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef]
- Johann, P.D.; Vaegler, M.; Gieseke, F.; Mang, P.; Armeanu-Ebinger, S.; Kluba, T.; Handgretinger, R.; Muller, I. Tumour stromal cells derived from paediatric malignancies display MSC-like properties and impair NK cell cytotoxicity. BMC Cancer 2010, 10, 501. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [Green Version]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef] [Green Version]
- Johann, P.D.; Muller, I. Multipotent Mesenchymal Stromal Cells: Possible Culprits in Solid Tumors? Stem Cells Int. 2015, 2015, 914632. [Google Scholar] [CrossRef] [Green Version]
- San Jose-Eneriz, E.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Petruccelli, L.A.; Dupere-Richer, D.; Pettersson, F.; Retrouvey, H.; Skoulikas, S.; Miller, W.H., Jr. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS ONE 2011, 6, e20987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richon, V.M.; Garcia-Vargas, J.; Hardwick, J.S. Development of vorinostat: Current applications and future perspectives for cancer therapy. Cancer Lett. 2009, 280, 201–210. [Google Scholar] [CrossRef]
- Condorelli, F.; Gnemmi, I.; Vallario, A.; Genazzani, A.A.; Canonico, P.L. Inhibitors of histone deacetylase (HDAC) restore the p53 pathway in neuroblastoma cells. Br. J. Pharmacol. 2008, 153, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Peart, M.J.; Smyth, G.K.; van Laar, R.K.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 3697–3702. [Google Scholar] [CrossRef] [Green Version]
- Leoni, F.; Zaliani, A.; Bertolini, G.; Porro, G.; Pagani, P.; Pozzi, P.; Dona, G.; Fossati, G.; Sozzani, S.; Azam, T.; et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc. Natl. Acad. Sci. USA 2002, 99, 2995–3000. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.; Ou, Z.; Yang, Z.; Zhong, J.; Zhu, Y.; Tian, J.; Wu, J.; Deng, H.; Lin, X.; Peng, Y.; et al. A Pan-Histone Deacetylase Inhibitor Enhances the Antitumor Activity of B7-H3-Specific CAR T Cells in Solid Tumors. Clin. Cancer Res. 2021, 27, 3757–3771. [Google Scholar] [CrossRef]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Cornel, A.M.; Dunnebach, E.; Hofman, D.A.; Das, S.; Sengupta, S.; van den Ham, F.; Wienke, J.; Strijker, J.G.M.; van den Beemt, D.; Essing, A.H.W.; et al. Epigenetic modulation of neuroblastoma enhances T cell and NK cell immunogenicity by inducing a tumor-cell lineage switch. J. Immunother. Cancer 2022, 10, e005002. [Google Scholar] [CrossRef]
- Pasvolsky, O.; Daher, M.; Alatrash, G.; Marin, D.; Daver, N.; Ravandi, F.; Rezvani, K.; Shpall, E.; Kebriaei, P. CARving the Path to Allogeneic CAR T Cell Therapy in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 800110. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Wang, L.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Mineishi, S.; Rizvi, S.; Shike, H.; Bayerl, M.; Schell, T.D.; et al. Bone marrow CD8 T cells express high frequency of PD-1 and exhibit reduced anti-leukemia response in newly diagnosed AML patients. Blood Cancer J. 2018, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Muller, I.; Kordowich, S.; Holzwarth, C.; Spano, C.; Isensee, G.; Staiber, A.; Viebahn, S.; Gieseke, F.; Langer, H.; Gawaz, M.P.; et al. Animal serum-free culture conditions for isolation and expansion of multipotent mesenchymal stromal cells from human BM. Cytotherapy 2006, 8, 437–444. [Google Scholar] [CrossRef]
- Lang, P.; Pfeiffer, M.; Handgretinger, R.; Schumm, M.; Demirdelen, B.; Stanojevic, S.; Klingebiel, T.; Kohl, U.; Kuci, S.; Niethammer, D. Clinical scale isolation of T cell-depleted CD56+ donor lymphocytes in children. Bone Marrow Transplant. 2002, 29, 497–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaggiari, G.M.; Capobianco, A.; Abdelrazik, H.; Becchetti, F.; Mingari, M.C.; Moretta, L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: Role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood 2008, 111, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Gieseke, F.; Bohringer, J.; Bussolari, R.; Dominici, M.; Handgretinger, R.; Muller, I. Human multipotent mesenchymal stromal cells use galectin-1 to inhibit immune effector cells. Blood 2010, 116, 3770–3779. [Google Scholar] [CrossRef]
- Spoo, A.C.; Lubbert, M.; Wierda, W.G.; Burger, J.A. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood 2007, 109, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, A.; Poorfathollah, A.A.; Aghaiipour, M.; Rezaei, M.; Nikoo-ghoftar, M.; Abdi, M.; Gharib, A.; Amini, A. Diagnostic value of CD117 in differential diagnosis of acute leukemias. Tumour Biol. 2014, 35, 6763–6768. [Google Scholar] [CrossRef]
- Geyh, S.; Oz, S.; Cadeddu, R.P.; Frobel, J.; Bruckner, B.; Kundgen, A.; Fenk, R.; Bruns, I.; Zilkens, C.; Hermsen, D.; et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia 2013, 27, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.C.; Basu, S.K.; Zhao, X.; Chien, S.; Fang, M.; Oehler, V.G.; Appelbaum, F.R.; Becker, P.S. Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer J. 2015, 5, e302. [Google Scholar] [CrossRef]
- Poon, Z.; Dighe, N.; Venkatesan, S.S.; Cheung, A.M.S.; Fan, X.; Bari, S.; Hota, M.; Ghosh, S.; Hwang, W.Y.K. Bone marrow MSCs in MDS: Contribution towards dysfunctional hematopoiesis and potential targets for disease response to hypomethylating therapy. Leukemia 2019, 33, 1487–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.A.; Advani, A.; Fernandez, L.; Van Der Jagt, R.; Brandwein, J.; Kambhampati, S.; Kassis, J.; Davis, M.; Bonfils, C.; Dubay, M.; et al. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br. J. Haematol. 2009, 147, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.; Maeda, Y.; Hotary, K.; Liu, C.; Reznikov, L.L.; Dinarello, C.A.; Ferrara, J.L. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc. Natl. Acad. Sci. USA 2004, 101, 3921–3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieseke, F.; Kruchen, A.; Tzaribachev, N.; Bentzien, F.; Dominici, M.; Muller, I. Proinflammatory stimuli induce galectin-9 in human mesenchymal stromal cells to suppress T-cell proliferation. Eur. J. Immunol. 2013, 43, 2741–2749. [Google Scholar] [CrossRef] [Green Version]
- Godavarthy, P.S.; Kumar, R.; Herkt, S.C.; Pereira, R.S.; Hayduk, N.; Weissenberger, E.S.; Aggoune, D.; Manavski, Y.; Lucas, T.; Pan, K.T.; et al. The vascular bone marrow niche influences outcome in chronic myeloid leukemia via the E-selectin-SCL/TAL1-CD44 axis. Haematologica 2020, 105, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Passaro, D.; Di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 324–341.e326. [Google Scholar] [CrossRef] [Green Version]
- Javidi-Sharifi, N.; Martinez, J.; English, I.; Joshi, S.K.; Scopim-Ribeiro, R.; Viola, S.K.; Edwards, D.K.t.; Agarwal, A.; Lopez, C.; Jorgens, D.; et al. FGF2-FGFR1 signaling regulates release of Leukemia-Protective exosomes from bone marrow stromal cells. Elife 2019, 8, e40033. [Google Scholar] [CrossRef]
- Chaurasia, P.; Gajzer, D.C.; Schaniel, C.; D’Souza, S.; Hoffman, R. Epigenetic reprogramming induces the expansion of cord blood stem cells. J. Clin. Investig. 2014, 124, 2378–2395. [Google Scholar] [CrossRef]
- Huang, X.; Guo, B.; Liu, S.; Wan, J.; Broxmeyer, H.E. Neutralizing negative epigenetic regulation by HDAC5 enhances human haematopoietic stem cell homing and engraftment. Nat. Commun. 2018, 9, 2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, S.H.; Tufa, D.; Woods, R.; George, T.D.; Shank, T.; Yingst, A.; Lake, J.; Cobb, L.; Jones, D.; Jones, K.; et al. SAHA Enhances Differentiation of CD34+CD45+ Hematopoietic Stem and Progenitor Cells from Pluripotent Stem Cells Concomitant with an Increase in Hemogenic Endothelium. Stem Cells Transl. Med. 2022, 11, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ren, Y.; Weng, S.; Xu, H.; Li, L.; Han, X. A New Trend in Cancer Treatment: The Combination of Epigenetics and Immunotherapy. Front. Immunol. 2022, 13, 809761. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yan, Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer 2020, 6, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, L.; Alvarez-Errico, D.; Esteller, M. The Contribution of Epigenetics to Cancer Immunotherapy. Trends Immunol. 2020, 41, 676–691. [Google Scholar] [CrossRef]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Amoozgar, Z.; Huang, J.; Saleh, M.H.; Xing, D.; Orsulic, S.; Goldberg, M.S. Decitabine Enhances Lymphocyte Migration and Function and Synergizes with CTLA-4 Blockade in a Murine Ovarian Cancer Model. Cancer Immunol. Res. 2015, 3, 1030–1041. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PBMC Co-Cultivation | IL-1β [pg/mL] | IL-2 [pg/mL] | IL-4 [pg/mL] | IL-10 [pg/mL] | IL-12p70 [pg/mL] | IL-15 [pg/mL] | IL-17 [pg/mL] | IFNγ [pg/mL] | TNFα [pg/mL] | |
|---|---|---|---|---|---|---|---|---|---|---|
| w/o T-MSC | 24 h | 11.2 ± 6.7 | 1039 ± 94 | 3.1 ± 1.4 | 75.1 ± 22.7 | 5.3 ± 4.1 | oor < | 302 ± 121 | 74.4 ± 42.2 | 556 ± 272 |
| 10,000 T-MSC | 9.1 ± 4.0 | 747 ± 166 | 2.4 ± 0.9 | 56.1 ± 13.9 | oor < | 0.52 ± 0 | 504 ± 209 | oor < | 187 ± 63 | |
| 20,000 T-MSC | 6.8 ± 5.0 | 859 ± 179 | 2.9 ± 0.6 | 55.2 3.7 | oor < | 0.63 ± 0 | 524 ± 189 | oor < | 79.2 ± 51.5 | |
| w/o T-MSC | 48 h | 16.6 ± 4.7 | 375 ± 146 | 4.2 ± 1.4 | 187 ± 29.6 | 6.9 ± 4.0 | oor < | 1269 ± 903 | 2187 ± 1146 | 2086 ± 454 |
| 10,000 T-MSC | 31.8 ± 16.9 | 407 ± 141 | 12.9 ± 12.5 | 62.1 ± 18.2 | 7.1 ± 7.8 | 0.5 ± 0.08 | 2490 ± 1188 | 344 ± 255 | 1197 ± 991 | |
| 20,000 T-MSC | 25.3 ± 2.4 | 432 ± 149 | 11.7 ± 9.0 | 60.6 ± 27.7 | oor < | 1.4 ± 0.2 | 3818 ± 4133 | 178 ± 160 | 416 ± 505 | |
| w/o T-MSC | 72 h | 15.6 ± 2.3 | 179 ± 41 | 3.7 ± 3.1 | 297 ± 96.9 | 3.8 ± 2.8 | oor < | 7684 ± 5882 | 4754 ± 968 | 1294 ± 1322 |
| 10,000 T-MSC | 59.6 ± 6.7 | 236 ± 58 | 9.6 ± 1.8 | 32.2 ± 15.7 | 1.2 ± 0 | 1.7 ± 0.2 | 3057 ± 1834 | 917 ± 773 | 457 ± 460 | |
| 20,000 T-MSC | 31.9 ± 2.9 | 322 ± 129 | 4.7 ± 1.6 | 23.4 ± 11.5 | oor < | 3.2 ± 0.6 | 1344 ± 949 | 350 ± 180 | 128 ± 109 |
| PBMC Co-Cultivation | Treatment of T-MSC | IL-1β [pg/mL] | IL-2 [pg/mL] | IL-10 [pg/mL] | IL-17 [pg/mL] | IFNγ [pg/mL] | TNFα [pg/mL] | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T-MSC I | T-MSC II | T-MSC I | T-MSC II | T-MSC I | T-MSC II | T-MSC I | T-MSC II | T-MSC I | T-MSC II | T-MSC I | T-MSC II | ||
| w/o T-MSC | 14.3 ± 3.4 | 1135 ± 222 | 232 ± 26.3 | 79.1 ± 12.7 | 741 ± 446 | 2178 ± 453 | |||||||
| 20,000 T-MSC | 0 µM SAHA | 686 ± 382 | 335 ± 282 | 839 ± 398 | 978 ± 85.4 | 288 ± 125 | 198 ± 64.0 | 185 ± 64.3 | 140 ± 52.9 | 869 ± 690 | 124 ± 124 | 120 ± 102 | 15.2 ± 14.0 |
| 0.1 µM SAHA | 496 ± 81.4 | 411 ± 409 | 801 ± 303 | 987 ± 162 | 281 ± 85.8 | 170 ± 51 | 161 ± 118 | 260 ± 168 | 875 ± 436 | 323 ± 201 | 77.4 ± 25.3 | 20.0 ± 12.7 | |
| 0.2 µM SAHA | 736 ± 111 | 506 ± 524 | 816 ± 270 | 844 ± 118 | 305 ± 142 | 186 ± 102 | 118 ± 71.9 | 198 ± 133 | 1090 ± 306 | 427 ± 284 | 125 ± 10.5 | 15.9 ± 1.1 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruchen, A.; Johann, P.-D.; Rekowski, L.; Müller, I. Epigenetic Modification of Mesenchymal Stromal Cells Derived from Bone Marrow and Embryonal Tumors to Facilitate Immunotherapeutic Approaches in Pediatric Malignancies. Curr. Issues Mol. Biol. 2023, 45, 2121-2135. https://doi.org/10.3390/cimb45030136
Kruchen A, Johann P-D, Rekowski L, Müller I. Epigenetic Modification of Mesenchymal Stromal Cells Derived from Bone Marrow and Embryonal Tumors to Facilitate Immunotherapeutic Approaches in Pediatric Malignancies. Current Issues in Molecular Biology. 2023; 45(3):2121-2135. https://doi.org/10.3390/cimb45030136
Chicago/Turabian StyleKruchen, Anne, Pascal-David Johann, Laura Rekowski, and Ingo Müller. 2023. "Epigenetic Modification of Mesenchymal Stromal Cells Derived from Bone Marrow and Embryonal Tumors to Facilitate Immunotherapeutic Approaches in Pediatric Malignancies" Current Issues in Molecular Biology 45, no. 3: 2121-2135. https://doi.org/10.3390/cimb45030136