
Molecular Research in Pancreatic Cancer: Small Molecule Inhibitors, Their Mechanistic Pathways and Beyond

Abstract
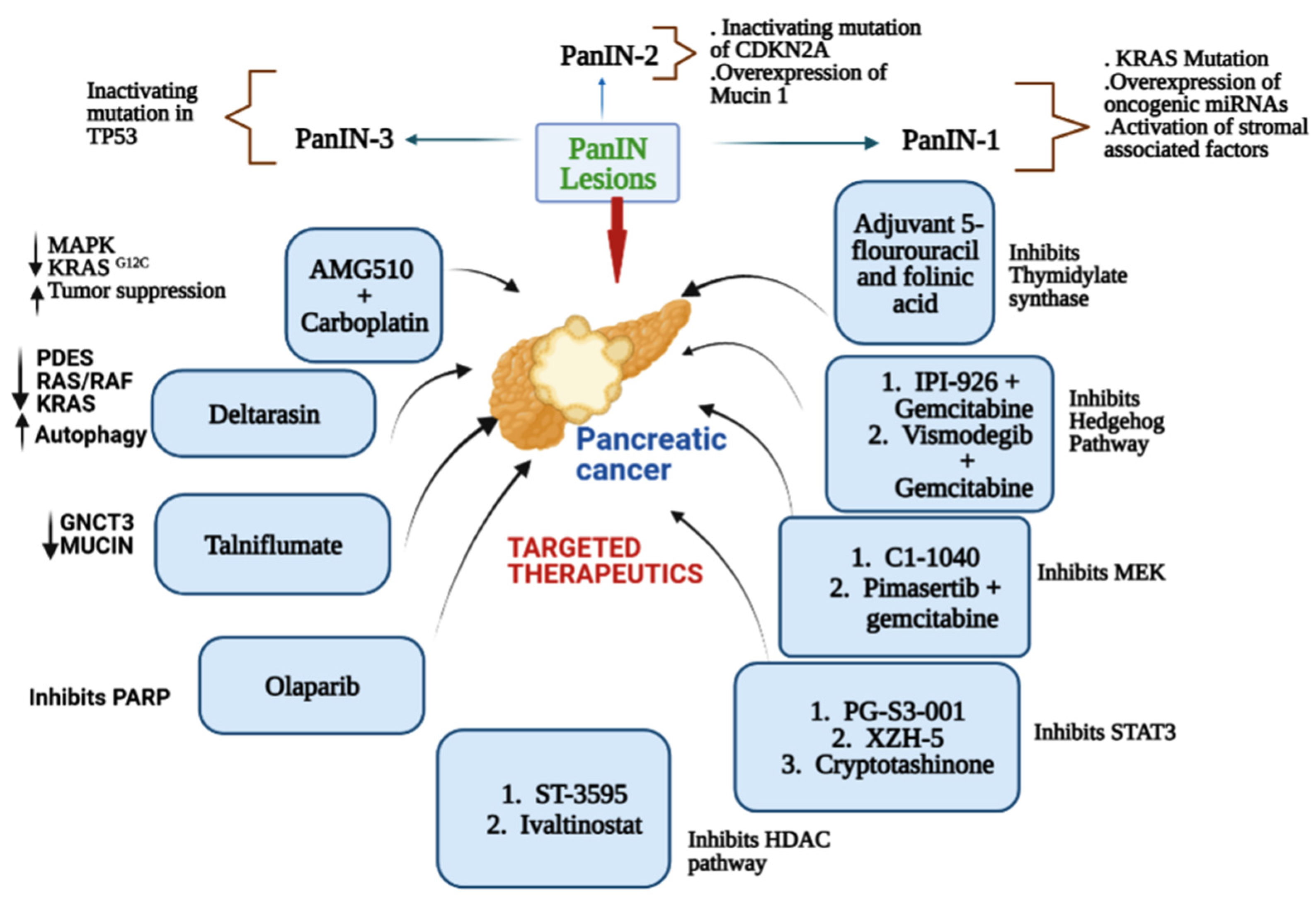
:1. Introduction
2. Major Drivers of Pancreatic Cancer
3. KRAS Inhibitors in Pancreatic Cancer
3.1. AMG 510 (Lumakras or Sotorasib)
3.2. MRTX849 (Adagrasib)
3.3. KRAS Inhibitor 11 [2-[4-(2-Methyl-3,5-Diphenylpyrazolo [1,5-a]Pyrimidin-7-yl)Piperazin-1-yl]Ethanol]
3.4. Deltarasin
3.5. Talniflumate
3.6. MDC-1016
3.7. Simvastatin
3.8. Avicin G
3.9. Prostratin
3.10. Lupeol
3.11. MRTX1133
4. RTK Inhibitor in Pancreatic Cancer
4.1. Pazopanib
4.2. Vandetanib
4.3. Lapatinib
4.4. Erlotinib
4.5. Axitinib
4.6. PD173074
4.7. Bemcentinib
4.8. Imatinib Mesylate
4.9. Sunitinib
4.10. Sorafenib
4.11. Losartan
4.12. Galunisertib
4.13. BMS-754807
5. Mixed (Combined) Inhibitors of Pancreatic Cancer
5.1. Thymidylate Synthase
5.1.1. Adjuvant 5-Fluorouracil and Folinic Acid
5.1.2. Gemcitabine + Capecitabine
5.2. Hedgehog Signaling
5.2.1. IPI-926 (Saridegib) + Gemcitabine
5.2.2. IPI-269609
5.2.3. Vismodegib + Gemcitabine (GDC-0449)
5.3. Mitogen-Activated Protein Kinase Kinase (MEK)
5.3.1. CI-1040 (Synonym: PD184352)
5.3.2. Pimasertib + Gemcitabine
5.4. Src Kinase
5.4.1. Dasatinib + Gemcitabine
5.4.2. Saracatinib (AZD0530) + Gemcitabine
5.5. Poly(ADP-Ribose) Polymerase Inhibitors (PARP)
5.5.1. Veliparib (Synonym: ABT-888)
5.5.2. Olaparib (AZD2281; KU0059436)
5.6. Transcription-3 (STAT3)
5.6.1. PG-S3-001
5.6.2. XZH-5
5.6.3. Cryptotanshinone
5.7. Hydroxamate-Based Histone Deacetylase (HDAC)
5.7.1. ST-3595
5.7.2. Ivaltinostat (CG200745)
5.7.3. MGCD0103 (Mocetinostat)
5.8. Bcl-2/Bcl-xL
5.8.1. UMI-77
5.8.2. TW-37
5.8.3. ABT-737
6. Other Promising Inhibitors of Pancreatic Cancer
6.1. Topoisomerase I
Nab-Paclitaxel + Gemcitabine
6.2. Protein Phosphatase 2A
LB-100
6.3. Proteasome
Marizomib + Vorinostat
6.4. mTOR
Everolimus
6.5. Thioredoxin-1 (Trx-1)
PX-12
6.6. Polo-Like Kinase 1 (PLK1) Phosphoinositide 3-Kinase (PI3K)
Rigosertib + Gemcitabine
6.7. Ataxia Telangiectasia and Rad3-Related Protein (ATR)
Ceralasertib (Synonym: AZD6738)
6.8. BET Bromodomain
JQ1
6.9. MDM2 Inhibitor
MI-319 + Cisplatin
6.10. Nuclear Factor Kappa Beta (NFκβ)
Spongiatrol
6.11. Gamma Secretase
MRK-003
7. NRF2 Role in KRAS-Driven Pancreatic Cancer
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Gordon-Dseagu, V.L.; Devesa, S.S.; Goggins, M.; Stolzenberg-Solomon, R. Pancreatic cancer incidence trends: Evidence from the Surveillance, Epidemiology and End Results (SEER) population-based data. Int. J. Epidemiol. 2018, 47, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Kota, J.; Hancock, J.; Kwon, J.; Korc, M. Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Lett. 2017, 391, 38–49. [Google Scholar] [CrossRef]
- Chang, D.K.; Merrett, N.D.; Biankin, A.V. Improving outcomes for operable pancreatic cancer: Is access to safer surgery the problem? J. Gastroenterol. Hepatol. 2008, 23, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Dunn, J.A.; Stocken, D.D.; Almond, J.; Link, K.; Beger, H.; Bassi, C.; Falconi, M.; Pederzoli, P.; Dervenis, C.; et al. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: A randomised controlled trial. Lancet 2001, 358, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Chin, V.; Nagrial, A.; Sjoquist, K.; O’Connor, C.A.; Chantrill, L.; Biankin, A.V.; Scholten, R.J.; Yip, D. Chemotherapy and radiotherapy for advanced pancreatic cancer. Cochrane Database Syst. Rev. 2018, 3, CD011044. [Google Scholar] [CrossRef]
- Krishnan, S.; Chadha, A.S.; Suh, Y.; Chen, H.C.; Rao, A.; Das, P.; Minsky, B.D.; Mahmood, U.; Delclos, M.E.; Sawakuchi, G.O.; et al. Focal Radiation Therapy Dose Escalation Improves Overall Survival in Locally Advanced Pancreatic Cancer Patients Receiving Induction Chemotherapy and Consolidative Chemoradiation. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 755–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Are, C.; Chowdhury, S.; Ahmad, H.; Ravipati, A.; Song, T.; Shrikandhe, S.; Smith, L. Predictive global trends in the incidence and mortality of pancreatic cancer based on geographic location, socio-economic status, and demographic shift. J. Surg. Oncol. 2016, 114, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [Green Version]
- Ferro, R.; Falasca, M. Emerging role of the KRAS-PDK1 axis in pancreatic cancer. World J. Gastroenterol. 2014, 20, 10752–10757. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar]
- Yonemori, K.; Kurahara, H.; Maemura, K.; Natsugoe, S. MicroRNA in pancreatic cancer. J. Hum. Genet. 2017, 62, 33–40. [Google Scholar] [CrossRef]
- Khan, S.; Ansarullah; Kumar, D.; Jaggi, M.; Chauhan, S.C. Targeting microRNAs in pancreatic cancer: Microplayers in the big game. Cancer Res. 2013, 73, 6541–6547. [Google Scholar] [CrossRef] [Green Version]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, T.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R.; et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Tinder, T.L.; Subramani, D.B.; Basu, G.D.; Bradley, J.M.; Schettini, J.; Million, A.; Skaar, T.; Mukherjee, P. MUC1 enhances tumor progression and contributes toward immunosuppression in a mouse model of spontaneous pancreatic adenocarcinoma. J. Immunol. 2008, 181, 3116–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanfredini, S.; Thapa, A.; O’Neill, E. RAS in pancreatic cancer. Biochem. Soc. Trans. 2019, 47, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordelas, L.; Rebmann, V.; Ludwig, A.K.; Radtke, S.; Ruesing, J.; Doeppner, T.R.; Epple, M.; Horn, P.A.; Beelen, D.W.; Giebel, B. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 2014, 28, 970–973. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorde Khvalevsky, E.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [Green Version]
- Mosolits, S.; Ullenhag, G.; Mellstedt, H. Therapeutic vaccination in patients with gastrointestinal malignancies. A review of immunological and clinical results. Ann. Oncol. 2005, 16, 847–862. [Google Scholar] [CrossRef]
- Quandt, J.; Schlude, C.; Bartoschek, M.; Will, R.; Cid-Arregui, A.; Schölch, S.; Reissfelder, C.; Weitz, J.; Schneider, M.; Wiemann, S.; et al. Long-peptide vaccination with driver gene mutations in p53 and Kras induces cancer mutation-specific effector as well as regulatory T cell responses. Oncoimmunology 2018, 7, e1500671. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, M.J.; Pagba, C.V.; Prakash, P.; Naji, A.K.; Van Der Hoeven, D.; Liang, H.; Gupta, A.K.; Zhou, Y.; Cho, K.J.; Hancock, J.F.; et al. Discovery of High-Affinity Noncovalent Allosteric KRAS Inhibitors That Disrupt Effector Binding. ACS Omega 2019, 4, 2921–2930. [Google Scholar] [CrossRef] [Green Version]
- Leung, E.L.; Luo, L.X.; Li, Y.; Liu, Z.Q.; Li, L.L.; Shi, D.F.; Xie, Y.; Huang, M.; Lu, L.L.; Duan, F.G.; et al. Identification of a new inhibitor of KRAS-PDEδ interaction targeting KRAS mutant nonsmall cell lung cancer. Int. J. Cancer 2019, 145, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Janakiram, N.B.; Madka, V.; Kumar, G.; Scott, E.J.; Pathuri, G.; Bryant, T.; Kutche, H.; Zhang, Y.; Biddick, L.; et al. Small-Molecule Inhibition of GCNT3 Disrupts Mucin Biosynthesis and Malignant Cellular Behaviors in Pancreatic Cancer. Cancer Res. 2016, 76, 1965–1974. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, G.G.; Bartels, L.E.; Xie, G.; Papayannis, I.; Alston, N.; Vrankova, K.; Ouyang, N.; Rigas, B. A novel Ras inhibitor (MDC-1016) reduces human pancreatic tumor growth in mice. Neoplasia 2013, 15, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Gbelcová, H.; Rimpelová, S.; Knejzlík, Z.; Šáchová, J.; Kolář, M.; Strnad, H.; Repiská, V.; D’Acunto, W.C.; Ruml, T.; Vítek, L. Isoprenoids responsible for protein prenylation modulate the biological effects of statins on pancreatic cancer cells. Lipids Health Dis. 2017, 16, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido, C.M.; Henkels, K.M.; Rehl, K.M.; Liang, H.; Zhou, Y.; Gutterman, J.U.; Cho, K.J. Avicin G is a potent sphingomyelinase inhibitor and blocks oncogenic K- and H-Ras signaling. Sci. Rep. 2020, 10, 9120. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- Sturm, S.; Gil, R.R.; Chai, H.-B.; Ngassapa, O.D.; Santisuk, T.; Reutrakul, V.; Howe, A.; Moss, M.; Besterman, J.M.; Yang, S.-L.; et al. Lupane Derivatives from Lophopetalum wallichii with Farnesyl Protein Transferase Inhibitory Activity. J. Nat. Prod. 1996, 59, 658–663. [Google Scholar] [CrossRef]
- Román, M.; Baraibar, I.; López, I.; Nadal, E.; Rolfo, C.; Vicent, S.; Gil-Bazo, I. KRAS oncogene in non-small cell lung cancer: Clinical perspectives on the treatment of an old target. Mol. Cancer 2018, 17, 33. [Google Scholar] [CrossRef] [Green Version]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e517. [Google Scholar] [CrossRef] [Green Version]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguel-García, A.; Orero, T.; Matutes, E.; Carbonell, F.; Miguel-Sosa, A.; Linares, M.; Tarín, F.; Herrera, M.; García-Talavera, J.; Carbonell-Ramón, F. bcl-2 expression in plasma cells from neoplastic gammopathies and reactive plasmacytosis: A comparative study. Haematologica 1998, 83, 298–304. [Google Scholar]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug. Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gysin, S.; Salt, M.; Young, A.; McCormick, F. Therapeutic strategies for targeting ras proteins. Genes Cancer 2011, 2, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, G.; Schultz-Fademrecht, C.; Küchler, P.; Murarka, S.; Ismail, S.; Triola, G.; Nussbaumer, P.; Wittinghofer, A.; Waldmann, H. Structure guided design and kinetic analysis of highly potent benzimidazole inhibitors targeting the PDEδ prenyl binding site. J. Med. Chem. 2014, 57, 5435–5448. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Li, Y.; Li, L.L.; Fan, X.X.; Wang, Y.W.; Wei, C.L.; Liu, L.; Leung, E.L.; Yao, X.J. Identification of a New Potent Inhibitor Targeting KRAS in Non-small Cell Lung Cancer Cells. Front. Pharm. 2017, 8, 823. [Google Scholar] [CrossRef] [Green Version]
- Wallis, N.; Oberman, F.; Shurrush, K.; Germain, N.; Greenwald, G.; Gershon, T.; Pearl, T.; Abis, G.; Singh, V.; Singh, A.; et al. Small molecule inhibitor of Igf2bp1 represses Kras and a pro-oncogenic phenotype in cancer cells. RNA Biol. 2022, 19, 26–43. [Google Scholar] [CrossRef]
- Evelyn, C.R.; Duan, X.; Biesiada, J.; Seibel, W.L.; Meller, J.; Zheng, Y. Rational design of small molecule inhibitors targeting the Ras GEF, SOS1. Chem. Biol. 2014, 21, 1618–1628. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Kumar, S.; Momi, N.; Sasson, A.R.; Batra, S.K. Mucins in pancreatic cancer and its microenvironment. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geltz, N.R.; Augustine, J.A. The p85 and p110 subunits of phosphatidylinositol 3-kinase-alpha are substrates, in vitro, for a constitutively associated protein tyrosine kinase in platelets. Blood 1998, 91, 930–939. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, M.-Y.; Kang, H.-M.; Han, D.C.; Son, K.-H.; Yang, D.C.; Sung, N.-D.; Lee, C.W.; Kim, H.M.; Kwon, B.-M. Anti-tumor activity of the farnesyl-protein transferase inhibitors arteminolides, isolated from Artemisa. Bioorg. Med. Chem. 2003, 11, 4545–4549. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Agnihotri, N. Piperlongumine, a piper alkaloid targets Ras/PI3K/Akt/mTOR signaling axis to inhibit tumor cell growth and proliferation in DMH/DSS induced experimental colon cancer. Biomed. Pharm. 2019, 109, 1462–1477. [Google Scholar] [CrossRef] [PubMed]
- Yaqoob, A.; Li, W.M.; Liu, V.; Wang, C.; Mackedenski, S.; Tackaberry, L.E.; Massicotte, H.B.; Egger, K.N.; Reimer, K.; Lee, C.H. Grifolin, neogrifolin and confluentin from the terricolous polypore Albatrellus flettii suppress KRAS expression in human colon cancer cells. PLoS ONE 2020, 15, e0231948. [Google Scholar] [CrossRef]
- Kim, C.-K.; Wang, D.; Bokesch, H.R.; Fuller, R.W.; Smith, E.; Henrich, C.J.; Durrant, D.E.; Morrison, D.K.; Bewley, C.A.; Gustafson, K.R. Swinhopeptolides A and B: Cyclic Depsipeptides from the Sponge Theonella swinhoei That Inhibit Ras/Raf Interaction. J. Nat. Prod. 2020, 83, 1288–1294. [Google Scholar] [CrossRef]
- Kumar, V.B.; Yuan, T.C.; Liou, J.W.; Yang, C.J.; Sung, P.J.; Weng, C.F. Antroquinonol inhibits NSCLC proliferation by altering PI3K/mTOR proteins and miRNA expression profiles. Mutat. Res. 2011, 707, 42–52. [Google Scholar] [CrossRef]
- Samantha, B.K.; Noah, C.; Nune, M.; Rina, S.; Jill, H.; Jason, S.; Liz, Q.; Natalie, V.B.; Jared, B.B.; Nikhil, J.; et al. Efficacy of a small molecule inhibitor of KRASG12D in immunocompetent models of pancreatic cancer. Cancer Discov. 2023, 13, 298–311. [Google Scholar] [CrossRef]
- American Institute of Cancer Research. Pancreatic Cancer. 2020. Available online: www.aicr.org/cancer-survival/cancer/pancreatic-cancer (accessed on 31 December 2022).
- Yang, H.H.; Liu, J.W.; Lee, J.H.; Harn, H.J.; Chiou, T.W. Pancreatic Adenocarcinoma Therapeutics Targeting RTK and TGF Beta Receptor. Int. J. Mol. Sci. 2021, 22, 8125. [Google Scholar] [CrossRef] [PubMed]
- Itakura, J.; Ishiwata, T.; Shen, B.; Kornmann, M.; Korc, M. Concomitant over-expression of vascular endothelial growth factor and its receptors in pancreatic cancer. Int. J. Cancer 2000, 85, 27–34. [Google Scholar] [CrossRef]
- Hakam, A.; Fang, Q.; Karl, R.; Coppola, D. Coexpression of IGF-1R and c-Src proteins in human pancreatic ductal adenocarcinoma. Dig. Dis. Sci. 2003, 48, 1972–1978. [Google Scholar] [CrossRef]
- Phan, A.T.; Halperin, D.M.; Chan, J.A.; Fogelman, D.R.; Hess, K.R.; Malinowski, P.; Regan, E.; Ng, C.S.; Yao, J.C.; Kulke, M.H. Pazopanib and depot octreotide in advanced, well-differentiated neuroendocrine tumours: A multicentre, single-group, phase 2 study. Lancet Oncol. 2015, 16, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Middleton, G.; Palmer, D.H.; Greenhalf, W.; Ghaneh, P.; Jackson, R.; Cox, T.; Evans, A.; Shaw, V.E.; Wadsley, J.; Valle, J.W.; et al. Vandetanib plus gemcitabine versus placebo plus gemcitabine in locally advanced or metastatic pancreatic carcinoma (ViP): A prospective, randomised, double-blind, multicentre phase 2 trial. Lancet Oncol. 2017, 18, 486–499. [Google Scholar] [CrossRef]
- Wu, Z.; Gabrielson, A.; Hwang, J.J.; Pishvaian, M.J.; Weiner, L.M.; Zhuang, T.; Ley, L.; Marshall, J.L.; He, A.R. Phase II study of lapatinib and capecitabine in second-line treatment for metastatic pancreatic cancer. Cancer Chemother. Pharm. 2015, 76, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Renouf, D.J.; Tang, P.A.; Hedley, D.; Chen, E.; Kamel-Reid, S.; Tsao, M.S.; Tran-Thanh, D.; Gill, S.; Dhani, N.; Au, H.J.; et al. A phase II study of erlotinib in gemcitabine refractory advanced pancreatic cancer. Eur. J. Cancer 2014, 50, 1909–1915. [Google Scholar] [CrossRef] [PubMed]
- Ioka, T.; Okusaka, T.; Ohkawa, S.; Boku, N.; Sawaki, A.; Fujii, Y.; Kamei, Y.; Takahashi, S.; Namazu, K.; Umeyama, Y.; et al. Efficacy and safety of axitinib in combination with gemcitabine in advanced pancreatic cancer: Subgroup analyses by region, including Japan, from the global randomized Phase III trial. Jpn. J. Clin. Oncol. 2015, 45, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Büchler, P.; Reber, H.A.; Roth, M.M.; Shiroishi, M.; Friess, H.; Hines, O.J. Target therapy using a small molecule inhibitor against angiogenic receptors in pancreatic cancer. Neoplasia 2007, 9, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, K.F.; Du, W.; Sorrelle, N.B.; Wnuk-Lipinska, K.; Topalovski, M.; Toombs, J.E.; Cruz, V.H.; Yabuuchi, S.; Rajeshkumar, N.V.; Maitra, A.; et al. Small-Molecule Inhibition of Axl Targets Tumor Immune Suppression and Enhances Chemotherapy in Pancreatic Cancer. Cancer Res. 2018, 78, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Moss, R.A.; Moore, D.; Mulcahy, M.F.; Nahum, K.; Saraiya, B.; Eddy, S.; Kleber, M.; Poplin, E.A. A Multi-institutional Phase 2 Study of Imatinib Mesylate and Gemcitabine for First-Line Treatment of Advanced Pancreatic Cancer. Gastrointest. Cancer Res. 2012, 5, 77–83. [Google Scholar]
- Martínez-Bosch, N.; Guerrero, P.E.; Moreno, M.; José, A.; Iglesias, M.; Munné-Collado, J.; Anta, H.; Gibert, J.; Orozco, C.A.; Vinaixa, J.; et al. The pancreatic niche inhibits the effectiveness of sunitinib treatment of pancreatic cancer. Oncotarget 2016, 7, 48265–48279. [Google Scholar] [CrossRef] [Green Version]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef] [Green Version]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, A.; Gilabert, M.; François, E.; Dahan, L.; Perrier, H.; Lamy, R.; Re, D.; Largillier, R.; Gasmi, M.; Tchiknavorian, X.; et al. BAYPAN study: A double-blind phase III randomized trial comparing gemcitabine plus sorafenib and gemcitabine plus placebo in patients with advanced pancreatic cancer. Ann. Oncol. 2012, 23, 2799–2805. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carboni, J.M.; Wittman, M.; Yang, Z.; Lee, F.; Greer, A.; Hurlburt, W.; Hillerman, S.; Cao, C.; Cantor, G.H.; Dell-John, J.; et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009, 8, 3341–3349. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, A.C.; Wang, Y.J.; Wang, X.; Wei, D.Q. Irinotecan and vandetanib create synergies for treatment of pancreatic cancer patients with concomitant TP53 and KRAS mutations. Brief Bioinform. 2021, 22, bbaa149. [Google Scholar] [CrossRef] [PubMed]
- Komoto, M.; Nakata, B.; Nishii, T.; Kawajiri, H.; Shinto, O.; Amano, R.; Yamada, N.; Yashiro, M.; Hirakawa, K. In vitro and in vivo evidence that a combination of lapatinib plus S-1 is a promising treatment for pancreatic cancer. Cancer Sci. 2010, 101, 468–473. [Google Scholar] [CrossRef]
- El-Rayes, B.F.; Ali, S.; Ali, I.F.; Philip, P.A.; Abbruzzese, J.; Sarkar, F.H. Retraction: Potentiation of the Effect of Erlotinib by Genistein in Pancreatic Cancer: The Role of Akt and Nuclear Factor-κB. Cancer Res. 2018, 78, 5472. [Google Scholar] [CrossRef] [Green Version]
- Canu, B.; Fioravanti, A.; Orlandi, P.; Di Desidero, T.; Alì, G.; Fontanini, G.; Di Paolo, A.; Del Tacca, M.; Danesi, R.; Bocci, G. Irinotecan synergistically enhances the antiproliferative and proapoptotic effects of axitinib in vitro and improves its anticancer activity in vivo. Neoplasia 2011, 13, 217–229. [Google Scholar] [CrossRef]
- Peran, I.; Vietsch, E.E.; Yan, G.; Riegel, A.T.; Wellstein, A. Distinct Response of Circulating microRNAs to the Treatment of Pancreatic Cancer Xenografts with FGFR and ALK Kinase Inhibitors. Cancers 2022, 14, 1517. [Google Scholar] [CrossRef]
- Takayama, Y.; Kokuryo, T.; Yokoyama, Y.; Nagino, M.; Nimura, Y.; Senga, T.; Hamaguchi, M. MEK inhibitor enhances the inhibitory effect of imatinib on pancreatic cancer cell growth. Cancer Lett. 2008, 264, 241–249. [Google Scholar] [CrossRef]
- Cuneo, K.C.; Geng, L.; Fu, A.; Orton, D.; Hallahan, D.E.; Chakravarthy, A.B. SU11248 (sunitinib) sensitizes pancreatic cancer to the cytotoxic effects of ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 873–879. [Google Scholar] [CrossRef]
- Huang, S.; Sinicrope, F.A. Sorafenib inhibits STAT3 activation to enhance TRAIL-mediated apoptosis in human pancreatic cancer cells. Mol. Cancer Ther. 2010, 9, 742–750. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.A.; Rivera, L.B.; Carbon, J.G.; Toombs, J.E.; Chang, C.L.; Bradshaw, A.D.; Brekken, R.A. Losartan slows pancreatic tumor progression and extends survival of SPARC-null mice by abrogating aberrant TGFβ activation. PLoS ONE 2012, 7, e31384. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, N.; Zhang, C.; Ruan, W.; Schwarz, M.A.; Schwarz, R.E. BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol. Cancer Ther. 2012, 11, 2644–2653. [Google Scholar] [CrossRef] [Green Version]
- Neoptolemos, J.P.; Stocken, D.D.; Tudur Smith, C.; Bassi, C.; Ghaneh, P.; Owen, E.; Moore, M.; Padbury, R.; Doi, R.; Smith, D.; et al. Adjuvant 5-fluorouracil and folinic acid vs observation for pancreatic cancer: Composite data from the ESPAC-1 and -3(v1) trials. Br. J. Cancer 2009, 100, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, G.; Fendrich, V.; McGovern, K.; Bedja, D.; Bisht, S.; Alvarez, H.; Koorstra, J.B.; Habbe, N.; Karikari, C.; Mullendore, M.; et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol. Cancer Ther. 2008, 7, 2725–2735. [Google Scholar] [CrossRef] [Green Version]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, P.M.; Adjei, A.A.; Varterasian, M.; Gadgeel, S.; Reid, J.; Mitchell, D.Y.; Hanson, L.; DeLuca, P.; Bruzek, L.; Piens, J.; et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 5281–5293. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Hidalgo, M.; Canon, J.L.; Macarulla, T.; Bazin, I.; Poddubskaya, E.; Manojlovic, N.; Radenkovic, D.; Verslype, C.; Raymond, E.; et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int. J. Cancer 2018, 143, 2053–2064. [Google Scholar] [CrossRef]
- Evans, T.R.J.; Van Cutsem, E.; Moore, M.J.; Bazin, I.S.; Rosemurgy, A.; Bodoky, G.; Deplanque, G.; Harrison, M.; Melichar, B.; Pezet, D.; et al. Phase 2 placebo-controlled, double-blind trial of dasatinib added to gemcitabine for patients with locally-advanced pancreatic cancer. Ann. Oncol. 2017, 28, 354–361. [Google Scholar] [CrossRef]
- Renouf, D.J.; Moore, M.J.; Hedley, D.; Gill, S.; Jonker, D.; Chen, E.; Walde, D.; Goel, R.; Southwood, B.; Gauthier, I.; et al. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Invest New Drugs 2012, 30, 779–786. [Google Scholar] [CrossRef]
- Lowery, M.A.; Kelsen, D.P.; Capanu, M.; Smith, S.C.; Lee, J.W.; Stadler, Z.K.; Moore, M.J.; Kindler, H.L.; Golan, T.; Segal, A.; et al. Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur. J. Cancer 2018, 89, 19–26. [Google Scholar] [CrossRef]
- Deeks, E.D. Olaparib: First global approval. Drugs 2015, 75, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Arpin, C.C.; Mac, S.; Jiang, Y.; Cheng, H.; Grimard, M.; Page, B.D.; Kamocka, M.M.; Haftchenary, S.; Su, H.; Ball, D.P.; et al. Applying Small Molecule Signal Transducer and Activator of Transcription-3 (STAT3) Protein Inhibitors as Pancreatic Cancer Therapeutics. Mol. Cancer Ther. 2016, 15, 794–805. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Liu, Y.; Jin, Z.; Hu, Q.; Lin, L.; Jou, D.; Yang, J.; Xu, Z.; Wang, H.; Li, C.; et al. XZH-5 inhibits STAT3 phosphorylation and enhances the cytotoxicity of chemotherapeutic drugs in human breast and pancreatic cancer cells. PLoS ONE 2012, 7, e46624. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.S.; Kim, H.N.; Shin, K.D.; Yoon, Y.J.; Kim, S.J.; Han, D.C.; Kwon, B.M. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer Res. 2009, 69, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Yang, B.; Chen, Z.; Cheng, R. Cryptotanshinone suppresses the proliferation and induces the apoptosis of pancreatic cancer cells via the STAT3 signaling pathway. Mol. Med. Rep. 2015, 12, 7782–7788. [Google Scholar] [CrossRef] [Green Version]
- Minjie, S.; Defei, H.; Zhimin, H.; Weiding, W.; Yuhua, Z. Targeting pancreatic cancer cells by a novel hydroxamate-based histone deacetylase (HDAC) inhibitor ST-3595. Tumour Biol. 2015, 36, 9015–9022. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, S.B.; Kim, S.A.; Kwon, S.K.; Cha, H.; Lee, D.Y.; Ro, S.; Cho, J.M.; Song, S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017, 7, 41615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfils, C.; Kalita, A.; Dubay, M.; Siu, L.L.; Carducci, M.A.; Reid, G.; Martell, R.E.; Besterman, J.M.; Li, Z. Evaluation of the pharmacodynamic effects of MGCD0103 from preclinical models to human using a novel HDAC enzyme assay. Clin. Cancer Res. 2008, 14, 3441–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, V.; Richard, N.; Brady, H.; Maier, A.; Kelter, G.; Heise, C. Histone deacetylase inhibitor MGCD0103 synergizes with gemcitabine in human pancreatic cells. Cancer Sci. 2011, 102, 1201–1207. [Google Scholar] [CrossRef]
- Abulwerdi, F.; Liao, C.; Liu, M.; Azmi, A.S.; Aboukameel, A.; Mady, A.S.; Gulappa, T.; Cierpicki, T.; Owens, S.; Zhang, T.; et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2014, 13, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Azmi, A.S.; Ahmad, A.; Banerjee, S.; Wang, S.; Sarkar, F.H.; Mohammad, R.M. TW-37, a small-molecule inhibitor of Bcl-2, inhibits cell growth and induces apoptosis in pancreatic cancer: Involvement of Notch-1 signaling pathway. Cancer Res. 2009, 69, 2757–2765. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Song, W.; Aboukameel, A.; Mohammad, M.; Wang, G.; Banerjee, S.; Kong, D.; Wang, S.; Sarkar, F.H.; Mohammad, R.M. TW-37, a small-molecule inhibitor of Bcl-2, inhibits cell growth and invasion in pancreatic cancer. Int. J. Cancer 2008, 123, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Kasai, S.; Sasaki, T.; Watanabe, A.; Nishiya, M.; Yasuhira, S.; Shibazaki, M.; Maesawa, C. Bcl-2/Bcl-x(L) inhibitor ABT-737 sensitizes pancreatic ductal adenocarcinoma to paclitaxel-induced cell death. Oncol. Lett. 2017, 14, 903–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddia, L.; D’Arca, D.; Ferrari, S.; Marraccini, C.; Severi, L.; Ponterini, G.; Assaraf, Y.G.; Marverti, G.; Costi, M.P. Inside the biochemical pathways of thymidylate synthase perturbed by anticancer drugs: Novel strategies to overcome cancer chemoresistance. Drug Resist. Updat. 2015, 23, 20–54. [Google Scholar] [CrossRef]
- Showalter, S.L.; Showalter, T.N.; Witkiewicz, A.; Havens, R.; Kennedy, E.P.; Hucl, T.; Kern, S.E.; Yeo, C.J.; Brody, J.R. Evaluating the drug-target relationship between thymidylate synthase expression and tumor response to 5-fluorouracil. Is it time to move forward? Cancer Biol. Ther. 2008, 7, 986–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avallone, A.; Di Gennaro, E.; Bruzzese, F.; Laus, G.; Delrio, P.; Caraglia, M.; Pepe, S.; Comella, P.; Budillon, A. Synergistic antitumour effect of raltitrexed and 5-fluorouracil plus folinic acid combination in human cancer cells. Anticancer. Drugs 2007, 18, 781–791. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Chung, V.; Mansfield, A.S.; Braiteh, F.; Richards, D.; Durivage, H.; Ungerleider, R.S.; Johnson, F.; Kovach, J.S. Safety, Tolerability, and Preliminary Activity of LB-100, an Inhibitor of Protein Phosphatase 2A, in Patients with Relapsed Solid Tumors: An Open-Label, Dose Escalation, First-in-Human, Phase I Trial. Clin. Cancer Res. 2017, 23, 3277–3284. [Google Scholar] [CrossRef] [Green Version]
- Millward, M.; Price, T.; Townsend, A.; Sweeney, C.; Spencer, A.; Sukumaran, S.; Longenecker, A.; Lee, L.; Lay, A.; Sharma, G.; et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest New Drugs 2012, 30, 2303–2317. [Google Scholar] [CrossRef]
- Vandamme, T.; Beyens, M.; De Beeck, K.O.; Dogan, F.; Van Koetsveld, P.M.; Pauwels, P.; Mortier, G.; Vangestel, C.; De Herder, W.; Van Camp, G.; et al. Long-term acquired everolimus resistance in pancreatic neuroendocrine tumours can be overcome with novel PI3K-AKT-mTOR inhibitors. Br. J. Cancer 2016, 114, 650–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, R.K.; Abbruzzese, J.; Dragovich, T.; Kirkpatrick, L.; Guillen, J.M.; Baker, A.F.; Pestano, L.A.; Green, S.; Von Hoff, D.D. A randomized phase II study of PX-12, an inhibitor of thioredoxin in patients with advanced cancer of the pancreas following progression after a gemcitabine-containing combination. Cancer Chemother Pharm. 2011, 67, 503–509. [Google Scholar] [CrossRef]
- O’Neil, B.H.; Scott, A.J.; Ma, W.W.; Cohen, S.J.; Aisner, D.L.; Menter, A.R.; Tejani, M.A.; Cho, J.K.; Granfortuna, J.; Coveler, L.; et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 1923–1929. [Google Scholar] [CrossRef] [PubMed]
- Wallez, Y.; Dunlop, C.R.; Johnson, T.I.; Koh, S.B.; Fornari, C.; Yates, J.W.T.; Bernaldo de Quiros Fernandez, S.; Lau, A.; Richards, F.M.; Jodrell, D.I. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol. Cancer Ther. 2018, 17, 1670–1682. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.; Raza, S.S.; Knab, L.M.; Chow, C.R.; Kwok, B.; Bentrem, D.J.; Popovic, R.; Ebine, K.; Licht, J.D.; Munshi, H.G. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci. Rep. 2015, 5, 9489. [Google Scholar] [CrossRef] [Green Version]
- Azmi, A.S.; Aboukameel, A.; Banerjee, S.; Wang, Z.; Mohammad, M.; Wu, J.; Wang, S.; Yang, D.; Philip, P.A.; Sarkar, F.H.; et al. MDM2 inhibitor MI-319 in combination with cisplatin is an effective treatment for pancreatic cancer independent of p53 function. Eur. J. Cancer 2010, 46, 1122–1131. [Google Scholar] [CrossRef] [Green Version]
- Guzmán, E.; Maher, M.; Temkin, A.; Pitts, T.; Wright, A. Spongiatriol inhibits nuclear factor kappa B activation and induces apoptosis in pancreatic cancer cells. Mar. Drugs 2013, 11, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Mizuma, M.; Rasheed, Z.A.; Yabuuchi, S.; Omura, N.; Campbell, N.R.; De Wilde, R.F.; De Oliveira, E.; Zhang, Q.; Puig, O.; Matsui, W.; et al. The gamma secretase inhibitor MRK-003 attenuates pancreatic cancer growth in preclinical models. Mol. Cancer Ther. 2012, 11, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, V. Gemcitabine: Progress in the treatment of pancreatic cancer. Oncology 2001, 60, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Neoptolemos, J.P.; Stocken, D.D.; Friess, H.; Bassi, C.; Dunn, J.A.; Hickey, H.; Beger, H.; Fernandez-Cruz, L.; Dervenis, C.; Lacaine, F.; et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N. Engl. J. Med. 2004, 350, 1200–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girardi, D.; Barrichello, A.; Fernandes, G.; Pereira, A. Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives. Cells 2019, 8, 153. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.R.; Lescarbeau, A.; Grogan, M.J.; Tan, E.; Lin, G.; Austad, B.C.; Yu, L.C.; Behnke, M.L.; Nair, S.J.; Hagel, M.; et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418. [Google Scholar] [CrossRef]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef] [Green Version]
- Karaca, M.; Dutta, R.; Ozsoy, Y.; Mahato, R.I. Micelle Mixtures for Coadministration of Gemcitabine and GDC-0449 To Treat Pancreatic Cancer. Mol. Pharm. 2016, 13, 1822–1832. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: Molecular mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef] [Green Version]
- Allen, L.F.; Sebolt-Leopold, J.; Meyer, M.B. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Semin. Oncol. 2003, 30, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Vena, F.; Li Causi, E.; Rodriguez-Justo, M.; Goodstal, S.; Hagemann, T.; Hartley, J.A.; Hochhauser, D. The MEK1/2 Inhibitor Pimasertib Enhances Gemcitabine Efficacy in Pancreatic Cancer Models by Altering Ribonucleotide Reductase Subunit-1 (RRM1). Clin. Cancer Res. 2015, 21, 5563–5577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, A.; Li, Z.R.; Bepler, G. RRM1-induced metastasis suppression through PTEN-regulated pathways. Oncogene 2003, 22, 2135–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akita, H.; Zheng, Z.; Takeda, Y.; Kim, C.; Kittaka, N.; Kobayashi, S.; Marubashi, S.; Takemasa, I.; Nagano, H.; Dono, K.; et al. Significance of RRM1 and ERCC1 expression in resectable pancreatic adenocarcinoma. Oncogene 2009, 28, 2903–2909. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Wei, J.; Su, G.H.; Lin, J. Dasatinib can enhance paclitaxel and gemcitabine inhibitory activity in human pancreatic cancer cells. Cancer Biol. Ther. 2019, 20, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Iyer, R.; Fountzilas, C. Poly(ADP-Ribose) Polymerase Inhibitors in Pancreatic Cancer: A New Treatment Paradigms and Future Implications. Cancers 2019, 11, 1980. [Google Scholar] [CrossRef] [Green Version]
- Pahuja, S.; Appleman, L.J.; Belani, C.P.; Chen, A.; Chu, E.; Beumer, J.H.; Puhalla, S. Preliminary activity of veliparib (V) in BRCA2-mutated metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2015, 33, 170, (Meeting abstract: Genitourinary Cancers Symposium). [Google Scholar] [CrossRef]
- Yarchoan, M.; Myzak, M.C.; Johnson, B.A., 3rd; De Jesus-Acosta, A.; Le, D.T.; Jaffee, E.M.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget 2017, 8, 44073–44081. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, W.; Li, Y. Discovery of novel inhibitors of signal transducer and activator of transcription 3 (STAT3) signaling pathway by virtual screening. Eur. J. Med. Chem. 2013, 62, 301–310. [Google Scholar] [CrossRef]
- Wu, Y.H.; Wu, Y.R.; Li, B.; Yan, Z.Y. Cryptotanshinone: A review of its pharmacology activities and molecular mechanisms. Fitoterapia 2020, 145, 104633. [Google Scholar] [CrossRef]
- Jo, J.H.; Jung, D.E.; Lee, H.S.; Park, S.B.; Chung, M.J.; Park, J.Y.; Bang, S.; Park, S.W.; Cho, S.; Song, S.Y. A phase I/II study of ivaltinostat combined with gemcitabine and erlotinib in patients with untreated locally advanced or metastatic pancreatic adenocarcinoma. Int. J. Cancer 2022, 151, 1565–1577. [Google Scholar] [CrossRef] [PubMed]
- Grassadonia, A.; Cioffi, P.; Simiele, F.; Iezzi, L.; Zilli, M.; Natoli, C. Role of Hydroxamate-Based Histone Deacetylase Inhibitors (Hb-HDACIs) in the Treatment of Solid Malignancies. Cancers 2013, 5, 919–942. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; He, J.; Zhao, J.; Yun, W.; Xie, C.; Taub, J.W.; Azmi, A.; Mohammad, R.M.; Dong, Y.; Kong, W.; et al. Class I and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PLoS ONE 2012, 7, e52095. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Dai, N.; Tang, Z.; Fang, H. Small-molecule Mcl-1 inhibitors: Emerging anti-tumor agents. Eur. J. Med. Chem. 2018, 146, 471–482. [Google Scholar] [CrossRef]
- Azmi, A.S.; Mohammad, R.M. Non-peptidic small molecule inhibitors against Bcl-2 for cancer therapy. J. Cell. Physiol. 2009, 218, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Parsels, L.A.; Karnak, D.; Davis, M.A.; Parsels, J.D.; Marsh, A.C.; Zhao, L.; Maybaum, J.; Lawrence, T.S.; Sun, Y.; et al. Inhibition of protein phosphatase 2A radiosensitizes pancreatic cancers by modulating CDC25C/CDK1 and homologous recombination repair. Clin. Cancer Res. 2013, 19, 4422–4432. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Guo, Y.; Wu, H.; Xiong, J.; Peng, T. Everolimus regulates the activity of gemcitabine-resistant pancreatic cancer cells by targeting the Warburg effect via PI3K/AKT/mTOR signaling. Mol. Med. 2021, 27, 38. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.W.; Messersmith, W.A.; Dy, G.K.; Weekes, C.D.; Whitworth, A.; Ren, C.; Maniar, M.; Wilhelm, F.; Eckhardt, S.G.; Adjei, A.A.; et al. Phase I study of Rigosertib, an inhibitor of the phosphatidylinositol 3-kinase and Polo-like kinase 1 pathways, combined with gemcitabine in patients with solid tumors and pancreatic cancer. Clin. Cancer Res. 2012, 18, 2048–2055. [Google Scholar] [CrossRef] [Green Version]
- Wilson, Z.; Odedra, R.; Wallez, Y.; Wijnhoven, P.W.G.; Hughes, A.M.; Gerrard, J.; Jones, G.N.; Bargh-Dawson, H.; Brown, E.; Young, L.A.; et al. ATR Inhibitor AZD6738 (Ceralasertib) Exerts Antitumor Activity as a Monotherapy and in Combination with Chemotherapy and the PARP Inhibitor Olaparib. Cancer Res. 2022, 82, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2020, 207, 107450. [Google Scholar] [CrossRef]
- Jette, N.R.; Radhamani, S.; Ye, R.; Yu, Y.; Arthur, G.; Goutam, S.; Bismar, T.A.; Kumar, M.; Bose, P.; Yip, S.; et al. ATM-deficient lung, prostate and pancreatic cancer cells are acutely sensitive to the combination of olaparib and the ATR inhibitor AZD6738. Genome Instab. Dis. 2020, 1, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.L.; Miller, A.L.; Kreitzburg, K.M.; Council, L.N.; Gamblin, T.L.; Christein, J.D.; Heslin, M.J.; Arnoletti, J.P.; Richardson, J.H.; Chen, D.; et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene 2016, 35, 833–845. [Google Scholar] [CrossRef]
- Leal, A.S.; Williams, C.R.; Royce, D.B.; Pioli, P.A.; Sporn, M.B.; Liby, K.T. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017, 394, 76–87. [Google Scholar] [CrossRef]
- Wang, X.; Hamann, M.T. Marine natural products in the discovery and development of potential pancreatic cancer therapeutics. Adv. Cancer Res. 2019, 144, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Suman, M.; Debanjan, G.; Pavan, P.; William, B.; Ming, Y.; Theresa, M.; Serguei, V.; Dwright, V.N.; Frank, M. Undermining Glutaminolysis bolsters Chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancer. Cancer Res. 2020, 80, 1630–1643. [Google Scholar]
- Shin, H.; Ryotaro, M.; Yu, T.; Keiko, T.; Masayuki, Y.; Atsushi, M. Nrf2 Activation Sensitizes K-Ras Mutant Pancreatic Cancer to glutaminase Inhibition. Int. J. Mol. Sci. 2021, 22, 1870. [Google Scholar]
- Suman, M.; Matthew, G.; Frank, M. The metabolic landscape of RAS-driven cancers from biology to therapy. Nat. Cancer 2022, 2, 271–283. [Google Scholar]
- Rushika, M.; Nabeel, B. Pancreatic cancer metabolism- breaking it down to build it back up. Cancer Discov. 2015, 5, 1247–1261. [Google Scholar]
- Xiaolung, W.; Shelley, A.; James, F.B.; Vickie, B.; David, M.B.; Andrew, C.; Joshua, R.D.; Jay, B.F.; John, P.F.; Matthew, A.M.; et al. Identification of MRTX1133, a noncovalent, potent and selective KRASG12D Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef]


{kind=link}
| Inhibitors | Structure | Target | Ref |
|---|---|---|---|
| AMG 510 (Lumakras or Sotorasib) |  | KRASG12C inhibitor | [29] |
| MRTX849 (Adagrasib) |  | KRASG12C inhibitor | [30] |
| KRAS inhibitor 11 † |  | Inhibits MAPK/RAF signaling, | [31] |
| Deltarasin |  | Downregulates RAS/RAF signaling pathway. | [32] |
| Talniflumate + Ggefitinib |  | Inhibition of 2 β-1,6 N-acetylglucosaminyltransferase (GCNT3) | [33] |
| MDC-1016 |  | Ras inhibitor | [34] |
| Natural Product Inhibitors | |||
| Simvastatin |  | GFP-K-Ras protein trafficking | [35] |
| Avicin G |  | KRASG12V and HRASG12V | [36] |
| Prostratin |  | KRAS | [37] |
| Lupeol |  | KRASG12V | [38] |
| Inhibitor | Structure | Target | Ref |
|---|---|---|---|
|  | VEGF receptors 1, 2, and 3 | [62] |
|  | VEGFR2, RET, and EGFR | [63] |
Capecitabine |  | EGFR and HER-2 | [64] |
Gemcitabine |  | EGFR | [65] |
Gemcitabine |  | VEGFR1, VEGFR2, VEGFR3 and PDGFRβ | [66] |
|  | VEGF-RII and FGF-RI | [67] |
|  | Axl | [68,69] |
|  | PDGFR, c-Kit, v-Abl | [70] |
|  | PDGFR, FLT3, IRE1α, Kit | [71] |
|  | EGFR | [72] |
|  | VEGFR | [73] |
|  | VEGFR-2/PDGFR-beta | [74] |
|  | Angiotensin Receptor | [75] |
|  | TGF-β type I receptor (TβRI) | [76] |
|  | insulin-like growth factor-1R/IR | [77] |
| Thymidylate Synthase | ||
|---|---|---|
| Inhibitor | Structure | Ref |
| Adjuvant 5-fluorouracil + folinic acid |  | [88] |
| Gemcitabine + Capecitabine |  | [89] |
| Hedgehog signaling | ||
| Inhibitor | Structure | Ref |
| IPI-926 (Saridegib) + gemcitabine |  | [90] |
| IPI-269609 |  | [91] |
| Vismodegib (GDC-0449) + Gemcitabine |  | [92] |
| Mitogen-activated protein kinase kinase(MEK) | ||
| Inhibitor | Structure | Ref |
| CI-1040 (Synonyms: PD 184352) |  | [93] |
| Pimasertib + gemcitabine |  | [94] |
| Src kinase | ||
| Inhibitor | Structure | Ref |
| Dasatinib + Gemcitabine |  | [95] |
| Saracatinib (AZD0530) + Gemcitabine |  | [96] |
| Poly(ADP-ribose) polymerase inhibitors (PARP) | ||
| Inhibitor | Structure | Ref |
| Veliparib (Synonyms: ABT-888) |  | [97] |
| Olaparib (AZD2281; KU0059436) |  | [98] |
| Transcription-3 (STAT3) | ||
| Inhibitor | Structure | Ref |
| PG-S3-001 |  | [99] |
| XZH-5 |  | [100] |
| Cryptotanshinone |  | [101,102] |
| hydroxamate-based histone deacetylase (HDAC) | ||
| Inhibitor | Structure | Ref |
| ST-3595 |  | [103] |
| Ivaltinostat (CG200745) |  | [104] |
| MGCD0103 (Mocetinostat) |  | [105,106] |
| Bcl-2/Bcl-xL | ||
| Inhibitor | Structure | Ref |
| UMI-77 |  | [107] |
| TW-37 |  | [108,109] |
| ABT-737 |  | [110] |
| Inhibitor | Structure | Target | Ref |
|---|---|---|---|
| nab-Paclitaxel + gemcitabine |  | Topoisomerase I | [114] |
| LB-100 |  | Protein Phosphatase 2A | [115] |
| Marizomib + Vorinostat |  | Proteasome | [116] |
| Everolimus |  | mTOR | [117] |
| PX-12 |  | Thioredoxin-1 (Trx-1) | [118] |
| Rigosertib + Gemcitabine |  | polo-like kinase 1 (PLK1) phosphoinositide 3-kinase (PI3K) | [119] |
| Ceralasertib (Synonyms: AZD6738) |  | Ataxia telangiectasia and Rad3-related protein (ATR) | [120] |
| JQ1 |  | BET bromodomain | [121] |
| MI-319+ Cisplatin |  | MDM2 inhibitor | [122] |
| Spongiatriol |  | Nuclear Factor Kappa B Nuclear Factor Kappa B | [123] |
| MRK-003 |  | Gamma Secretase | [124] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shetu, S.A.; James, N.; Rivera, G.; Bandyopadhyay, D. Molecular Research in Pancreatic Cancer: Small Molecule Inhibitors, Their Mechanistic Pathways and Beyond. Curr. Issues Mol. Biol. 2023, 45, 1914-1949. https://doi.org/10.3390/cimb45030124
Shetu SA, James N, Rivera G, Bandyopadhyay D. Molecular Research in Pancreatic Cancer: Small Molecule Inhibitors, Their Mechanistic Pathways and Beyond. Current Issues in Molecular Biology. 2023; 45(3):1914-1949. https://doi.org/10.3390/cimb45030124
Chicago/Turabian StyleShetu, Shaila A., Nneoma James, Gildardo Rivera, and Debasish Bandyopadhyay. 2023. "Molecular Research in Pancreatic Cancer: Small Molecule Inhibitors, Their Mechanistic Pathways and Beyond" Current Issues in Molecular Biology 45, no. 3: 1914-1949. https://doi.org/10.3390/cimb45030124