
Conflicts in Mitochondrial Phylogenomics of Branchiopoda, with the First Complete Mitogenome of Laevicaudata (Crustacea: Branchiopoda)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. PCR Amplification, Sequencing, Sequence Assembly, and Gene Annotation
2.3. Sequence Alignment and Substitutional Saturation Test
2.4. Analyses of Sequence Heterogeneity and Phylogenetic Signal Dissection
2.5. Phylogenetic Analysis
2.5.1. Phylogenetic Analyses under Site-Homogeneous Models
2.5.2. Phylogenetic Analyses under Site-Heterogeneous Models
3. Results
3.1. Characteristics of L. grossipedia Mitogenome
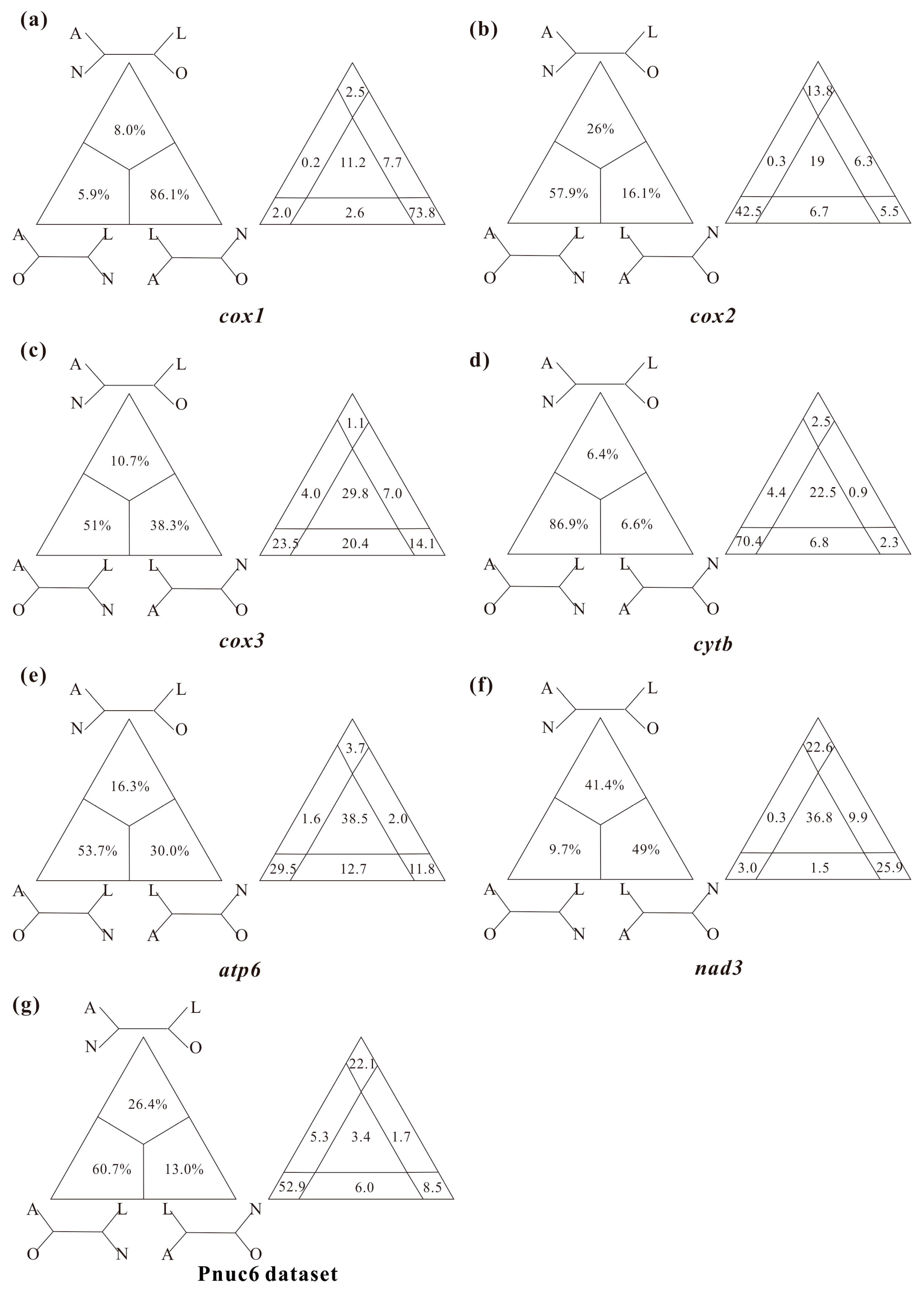
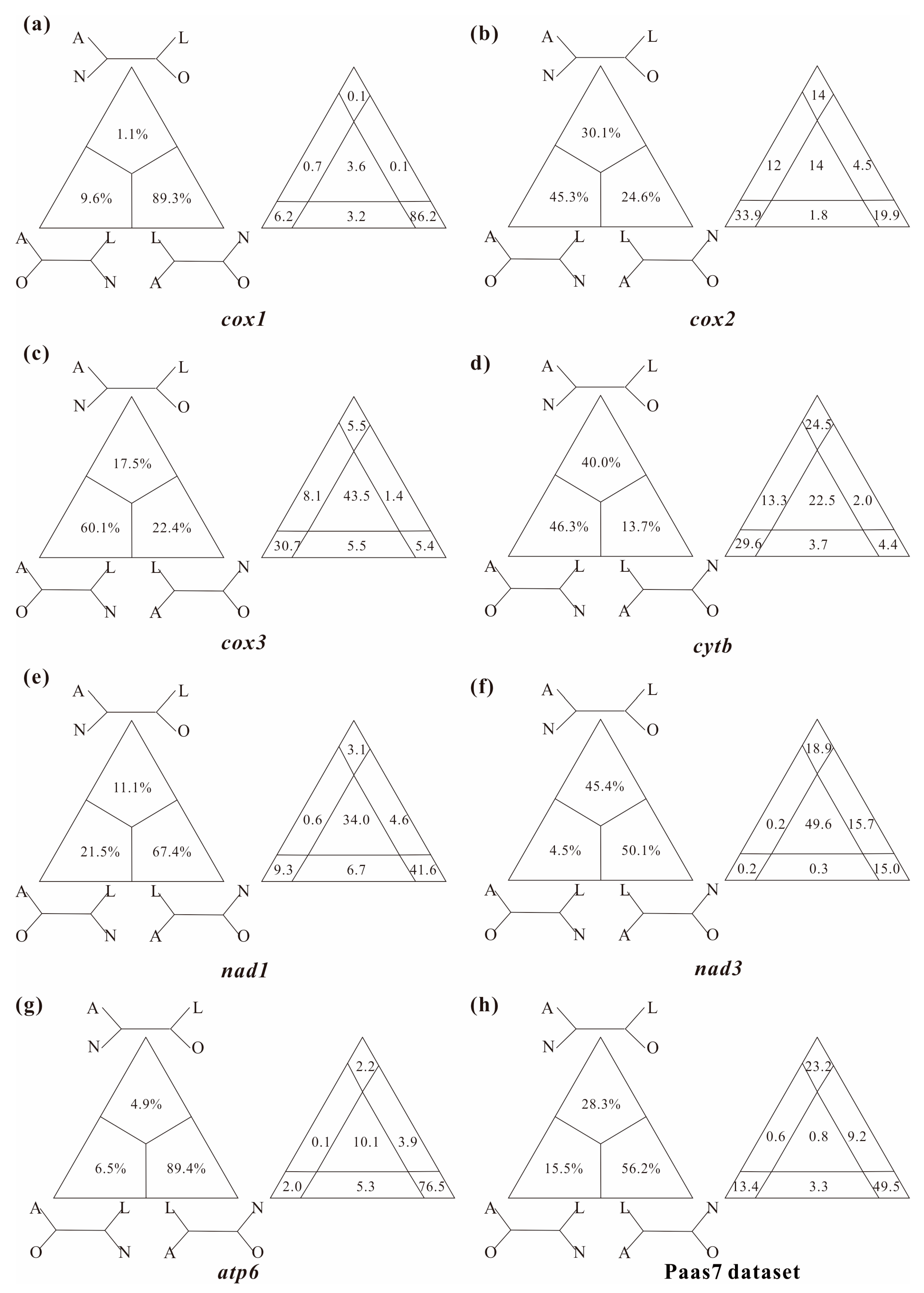
3.2. Levels of Substitutional Saturation and Heterogeneous Sequence Divergence within Branchiopod Mitogenomes
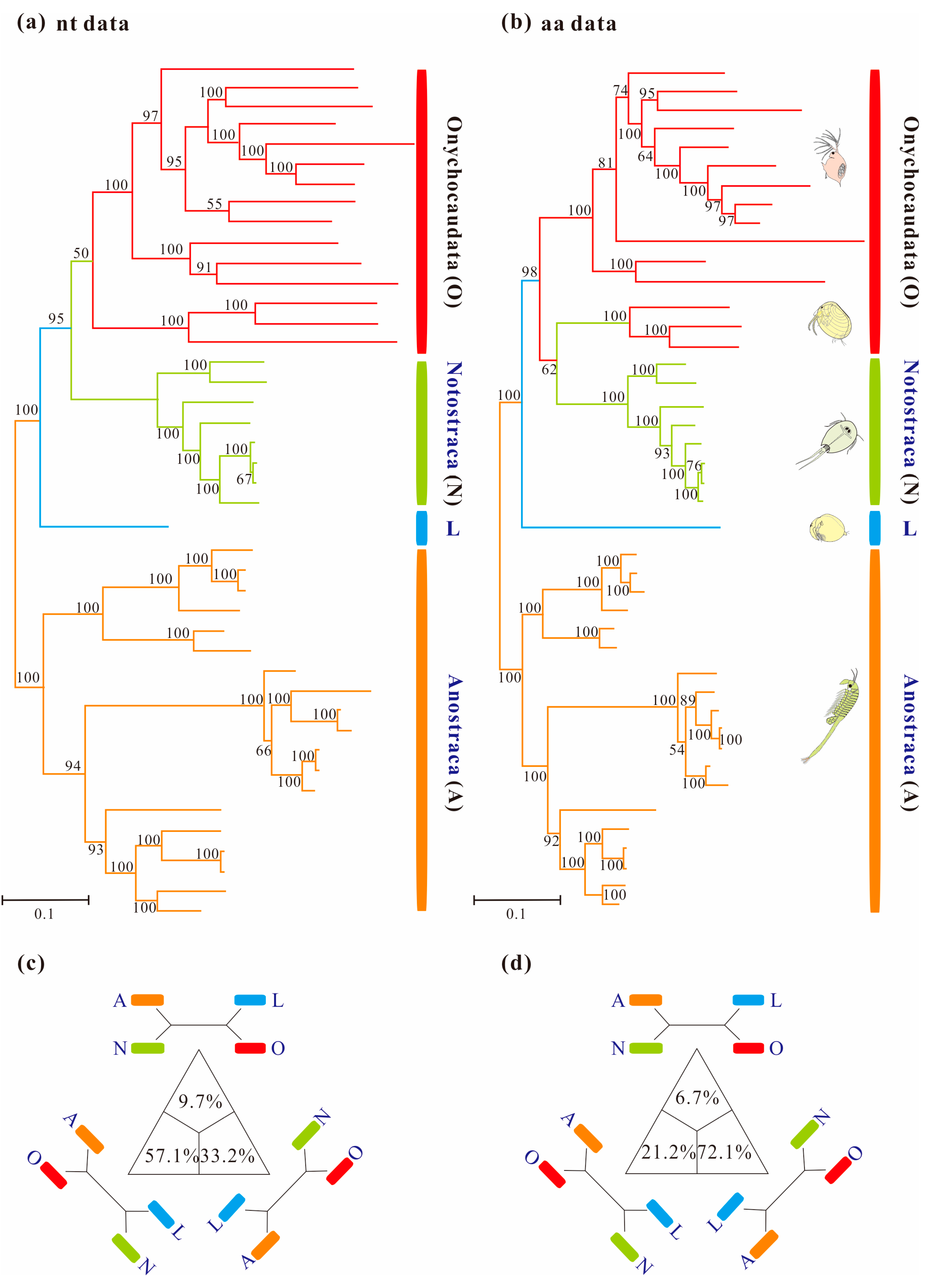
3.3. Phylogenetic Analyses Using Standard Homogeneous Models
3.4. Reducing Compositional Heterogeneity in Sequence Data
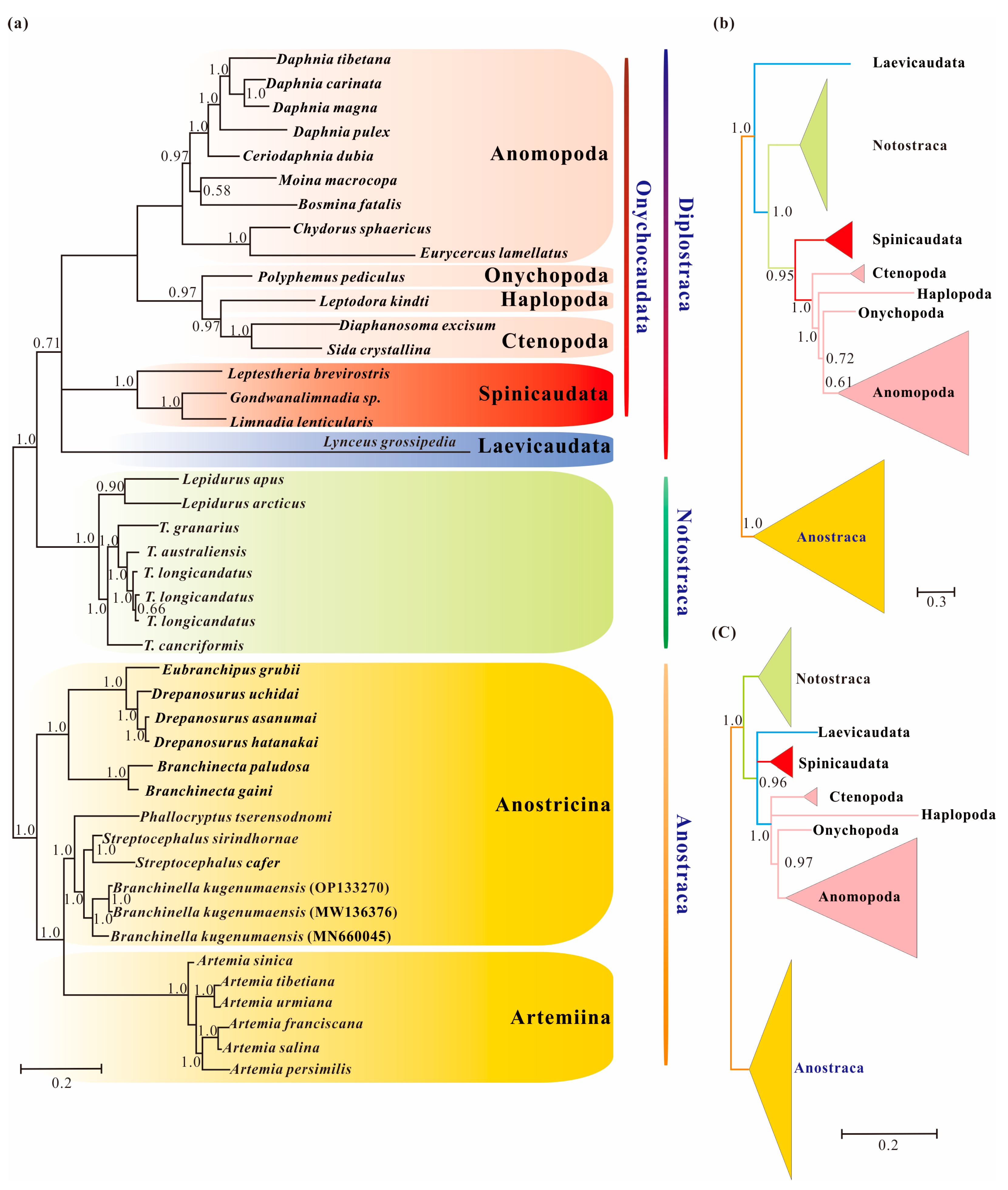
3.5. Phylogenetic Results under Heterogeneous Model
4. Discussion
4.1. Pervasiveness of Phylogenetic Conflicts
4.2. Heterogeneity and Tree Topology
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cameron, S.L.; Lambkin, C.L.; Barker, S.C.; Whiting, M.F. A mitochondrial genome phylogeny of Diptera: Whole genome sequence data accurately resolve relationships over broad timescales with high precision. Syst. Entomol. 2007, 32, 40–59. [Google Scholar] [CrossRef]
- Harrison, G.L.; McLenachan, P.A.; Phillips, M.J.; Slack, K.E.; Cooper, A.; Penny, D. Four new avian mitochondrial genomes help get to basic evolutionary questions in the late Cretaceous. Mol. Biol. Evol. 2004, 21, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Penny, D. The root of the mammalian tree inferred from whole mitochondrial genomes. Mol. Phylogenet. Evol. 2003, 28, 171–185. [Google Scholar] [CrossRef]
- Cook, C.E.; Yue, Q.; Akam, M. Mitochondrial genomes suggest that hexapods and crustaceans are mutually paraphyletic. Proc. Biol. Sci. 2005, 272, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Rota-Stabelli, O.; Kayal, E.; Gleeson, D.; Daub, J.; Boore, J.L.; Telford, M.J.; Pisani, D.; Blaxter, M.; Lavrov, D.V. Ecdysozoan mitogenomics: Evidence for a common origin of the legged invertebrates, the Panarthropoda. Genome Biol. Evol. 2010, 2, 425–440. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Vila, R. What is the phylogenetic signal limit from mitogenomes? The reconciliation between mitochondrial and nuclear data in the Insecta class phylogeny. BMC Evol. Biol. 2011, 11, 315. [Google Scholar] [CrossRef] [Green Version]
- Delsuc, F.; Brinkmann, H.; Chourrout, D.; Philippe, H. Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 2006, 439, 965–968. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ezpeleta, N.; Brinkmann, H.; Roure, B.; Lartillot, N.; Lang, B.F.; Philippe, H. Detecting and Overcoming Systematic Errors in Genome-Scale Phylogenies. Syst. Biol. 2007, 56, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Philippe, H.; Brinkmann, H.; Lavrov, D.V.; Littlewood, D.T.J.; Manuel, M.; Wörheide, G.; Baurain, D. Resolving difficult phylogenetic questions: Why more sequences are not enough. PLoS Biol. 2011, 9, e1000602. [Google Scholar] [CrossRef] [Green Version]
- Foster, P.G. Modeling compositional heterogeneity. Syst. Biol. 2004, 53, 485–495. [Google Scholar] [CrossRef]
- Lartillot, N.; Philippe, H. Improvement of molecular phylogenetic inference and the phylogeny of Bilateria. Philos. Trans. R. Soc. B 2008, 363, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.W.; Jermiin, L.S. Tracing the decay of the historical signal in biological sequence data. Syst. Biol. 2004, 53, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Pagel, M.; Meade, A. A phylogenetic mixture model for detecting pattern-heterogeneity in gene sequence or character-state data. Syst. Biol. 2004, 53, 571–581. [Google Scholar] [CrossRef]
- Martin, J.W.; Belk, D. Review of the clam shrimp family Lynceidae Stebbing, 1902 (Branchiopoda: Conchostraca), in the Americas. J. Crustac. Biol. 1988, 8, 451–482. [Google Scholar] [CrossRef]
- Timms, B.V. A revision of the Australian species of Lynceus Müller, 1776 (Crustacea: Branchiopoda: Laevicaudata: Lynceidae). Zootaxa 2013, 3702, 501–533. [Google Scholar] [CrossRef]
- Rogers, D.C.; Olesen, J. Laevicaudata catalogus (Crustacea: Branchiopoda): An overview of diversity and terminology. Arthropod Syst. Phylogeny 2016, 74, 221–240. [Google Scholar]
- Sigvardt, Z.M.S.; Rogers, D.C.; De los Ríos, P.; Palero, F.; Olesen, J. First molecular phylogeny of Laevicaudata (Crustacea: Branchiopoda) with description of a new species of Lynceus from Chile and an updated key to species in the Americas. Invertebr. Syst. 2019, 33, 597–617. [Google Scholar] [CrossRef]
- Olesen, J.; Martin, J.W. Laevicaudata. In Atlas of Crustacean Larvae; Martin, J.W., Olesen, J., Høeg, J.T., Eds.; Johns Hopkins University Press: Baltimore, MD, USA, 2014; pp. 47–50. [Google Scholar]
- Pessacq, P.; Epele, L.B.; Rogers, D.C. A new species of Lynceus (Crustacea: Branchiopoda: Laevicaudata) from Patagonia, with comments on laevicaudatan systematics. Zootaxa 2011, 3043, 25–32. [Google Scholar] [CrossRef]
- Rogers, D.C.; Olesen, J.; Martin, J.W. A new possibly parthenogenic species of Lynceus from Canada (Crustacea: Branchiopoda: Laevicaudata), with key to the Nearctic female Laevicaudata. Sci. Pap. Nat. Hist. Mus. Univ. Kansas 2015, 47, 1–9. [Google Scholar]
- Olesen, J.; Pöllabauer, C.; Sigvardt, Z.M.S.; Rogers, D.C. A new species of Lynceus Müller, 1776 from New Caledonia (Crustacea: Branchiopoda: Laevicaudata) from dolines, with remarks on zoogeography. Eur. J. Taxon. 2016, 224, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Shu, S.; Sigvardt, Z.M.S.; Chen, X.; Olesen, J.; Rogers, D.C.; Sanoamuang, L. Lynceus amplopedia sp. nov., a new laevicaudatan clam shrimp with asymmetrically modified thoracopods from Yunnan, China (Crustacea: Branchiopoda). Zool. Stud. 2019, 58, 28. [Google Scholar] [CrossRef]
- Sigvardt, Z.M.S.; Shu, S.; Alonso, M.; Ventura, M.; Sanoamuang, L.; Rogers, D.C.; Palero, F.; Olesen, J. A new Northeast Asian Lynceus (Crustacea: Branchiopoda: Laevicaudata) with uniquely modified thoracopods and an evaluation of DNA barcoding for clam shrimp species identification. Nauplius 2020, 28, e2020013. [Google Scholar] [CrossRef]
- Sigvardt, Z.M.S.; Olesen, J.; Rogers, D.C.; Timms, B.; Mlambo, M.; Rabet, N.; Palero, F. Multilocus phylogenetics of smooth clam shrimps (Branchiopoda, Laevicaudata). Zool. Scr. 2021, 50, 795–811. [Google Scholar] [CrossRef]
- Richter, S.; Olesen, J.; Wheeler, W.C. Phylogeny of Branchiopoda (Crustacea) based on a combined analysis of morphological data and six molecular loci. Cladistics 2007, 23, 301–336. [Google Scholar] [CrossRef]
- Olesen, J. Phylogeny of Branchiopoda (Crustacea)—Character evolution and contribution of uniquely preserved fossils. Arthropod Syst. Phylogeny 2009, 67, 3–39. [Google Scholar]
- Olesen, J.; Richter, S. Onychocaudata (Branchiopoda: Diplostraca), a new high-level taxon in branchiopod systematics. J. Crustac. Biol. 2013, 33, 62–65. [Google Scholar] [CrossRef] [Green Version]
- Schwentner, M.; Richter, S.; Rogers, D.C.; Giribet, G. Tetraconatan phylogeny with special focus on Malacostraca and Branchiopoda: Highlighting the strength of taxon-specific matrices in phylogenomics. Proc. Biol. Sci. 2018, 285, 20181524. [Google Scholar] [CrossRef] [Green Version]
- Uozumi, T.; Ishiwata, K.; Grygier, M.J.; Sanoamuang, L.O.; Su, Z. Three nuclear protein-coding genes corroborate a recent phylogenomic model of the Branchiopoda (Crustacea) and provide estimates of the divergence times of the major branchiopodan taxa. Genes Genet. Syst. 2021, 96, 13–24. [Google Scholar] [CrossRef]
- Olesen, J. Monophyly and phylogeny of Branchiopoda, with focus on morphology and homologies of branchiopod phyllopodous limbs. J. Crustac. Biol. 2007, 27, 165–183. [Google Scholar] [CrossRef] [Green Version]
- Lozano-Fernandez, J.; Giacomelli, M.; Fleming, J.F.; Chen, A.; Vinther, J.; Thomsen, P.F.; Glenner, H.; Palero, F.; Legg, D.A.; Iliffe, T.M.; et al. Pancrustacean evolution illuminated by taxon-rich genomic-scale data sets with an expanded remipede sampling. Genome Biol. Evol. 2019, 11, 2055–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchetti, A.; Forni, G.; Skaist, A.M.; Wheelan, S.J.; Mantovani, B. Mitochondrial genome diversity and evolution in Branchiopoda (Crustacea). Zool. Lett. 2019, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, F.; Luchetti, A.; Mantovani, B. Exploring mitogenome evolution in Branchiopoda (Crustacea) lineages reveals gene order rearrangements in Cladocera. Sci. Rep. 2022, 12, 4931. [Google Scholar] [CrossRef] [PubMed]
- Kitano, T.; Sato, H.; Takahashi, N.; Igarashi, S.; Hatanaka, Y.; Igarashi, K.; Umetsu, K. Complete mitochondrial genomes of three fairy shrimps from snowmelt pools in Japan. BMC Zool. 2022, 7, 11. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, J. Comparative Mitogenomic Analyses and New Insights into the Phylogeny of Thamnocephalidae (Branchiopoda: Anostraca). Genes 2022, 13, 1765. [Google Scholar] [CrossRef]
- Xu, S.; Han, B.; Martínez, A.; Schwentner, M.; Fontaneto, D.; Dumont, H.J.; Kotov, A.A. Mitogenomics of Cladocera (Branchiopoda): Marked gene order rearrangements and independent predation roots. Mol. Phylogenet. Evol. 2021, 164, 107275. [Google Scholar] [CrossRef]
- Sun, X. Divergence across the mitogenomes of Branchinella kugenumaensis (Anostraca: Thamnocephalidae) with implications for species delimitation. Mitochondrial DNA Part B 2021, 6, 631–633. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, J. Characterization of the complete mitochondrial genome of Chinese Triops granarius and implications for species delimitation. Int. J. Biol. Macromol. 2019, 135, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- BLAST. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 28 September 2022).
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE6: New tools for microbial genomics, phylogenetics, and molecular evolution. J. Hered. 2017, 108, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Wu, D.; Goremykin, V.; Xiao, J.; Xu, Y.; Garg, S.; Zhang, C.; Martin, W.F.; Zhu, R. Phylogenetic analyses with systematic taxon sampling show that mitochondria branch within Alphaproteobacteria. Nat. Ecol. Evol. 2020, 4, 1213–1219. [Google Scholar] [CrossRef]
- Stone, M. Cross-validatory choice and assessment of statistical predictions. J. R. Stat. Soc. Ser. B Methodol. 1974, 36, 111–147. [Google Scholar] [CrossRef]
- Nesnidal, M.P.; Helmkampf, M.; Bruchhaus, I.; Hausdorf, B. Compositional heterogeneity and phylogenomic inference of metazoan relationships. Mol. Biol. Evol. 2010, 27, 2095–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rota-Stabelli, O.; Pisani, D. Serine codon-usage bias in deep phylogenomics: Pancrustacean relationships as a case study. Syst. Biol. 2013, 62, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, F.; Jiang, P.; Wilson, J.-J.; Cai, W.; Li, H. Compositional heterogeneity in true bug mitochondrial phylogenomics. Mol. Phylogenet. Evol. 2018, 18, 135–144. [Google Scholar] [CrossRef]
- Phillips, M.J.; Delsuc, F.; Penny, D. Genome-scale phylogeny and the detection of systematic biases. Mol. Biol. Evol. 2004, 21, 1455–1458. [Google Scholar] [CrossRef] [Green Version]
- Brochier, C.; Philippe, H. Phylogeny: A nonhyperthermophilic ancestor for Bacteria. Nature 2002, 417, 244. [Google Scholar] [CrossRef]
- Regier, J.C.; Shultz, J.W.; Zwick, A.; Hussey, A.; Ball, B.; Wetzer, R.; Martin, J.W.; Cunningham, C.W. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature 2010, 463, 1079–1084. [Google Scholar] [CrossRef]
- Brinkmann, H.; Philippe, H. Archaea sister group of Bacteria? Indications from tree reconstruction artifacts in ancient phylogenies. Mol. Biol. Evol. 1999, 16, 817–825. [Google Scholar] [CrossRef]
- Ruiz-Trillo, I.; Riutort, M.; Littlewood, D.T.; Herniou, E.A.; Baguna, J. Acoel flatworms: Earliest extant bilaterian Metazoans, not members of Platyhelminthes. Science 1999, 283, 1919–1923. [Google Scholar] [CrossRef] [Green Version]
- Martijn, J.; Vosseberg, J.; Guy, L.; Offre, P.; Ettema, T.J.G. Deep mitochondrial origin outside the sampled alphaproteobacteria. Nature 2018, 557, 101–105. [Google Scholar] [CrossRef]
- Curole, J.; Kocher, T. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Nardi, F.; Spinsanti, G.; Boore, J.L.; Carapelli, A.; Dallai, R.; Frati, F. Hexapod origins: Monophyletic or paraphyletic? Science 2003, 299, 1887–1889. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L.; Miller, K.B.; D’Haese, C.A.; Whiting, M.F.; Barker, S.C. Mitochondrial genome data alone are not enough to unambiguously resolve the relationships of Entognatha, Insecta and Crustacea sensu lato (Athropoda). Cladistics 2004, 20, 534–557. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Song, H.; Cameron, S.L.; Whiting, M.F. Nonstationary evolution and compositional heterogeneity in beetle mitochondrial phylogenomics. Syst. Biol. 2009, 58, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Crease, T.J. The complete sequence of the mitochondrial genome of Daphnia pulex (Cladocera: Crustacea). Gene 1999, 233, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Cheng, R.; Cheng, D.; Zhang, H. The complete mitochondrial DNA genome of Chinese Daphnia carinata (Clasocera: Daphniidae). Mitochondrial DNA Part B 2016, 1, 323–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.; Deng, B.; Wang, Y.; Geng, X.; Li, J.; Zhang, X.; Peng, S.; Deng, D.; Zhang, H. Complete mitochondrial genome sequence of Daphnia magna (Crustacea: Cladocera) from Huaihe in China. J. Lake Sci. 2016, 28, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Chen, Y. The complete mitochondrial genome of the freshwater fairy shrimp Branchinella kugenumaensis Ishikawa 1894 (Crustacea: Anostraca: Thamnocephalidae). Mitochondrial DNA Part B 2020, 5, 1048–1049. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Lu, B.; Yang, J. The complete mitogenome of the fairy shrimp Phallocryptus tserensodnomi (Crustacea: Anostraca: Thamnocephalidae). Mitochondrial DNA Part A 2016, 27, 3113–3114. [Google Scholar] [CrossRef]
- Tladi, M.; Dalu, T.; Rogers, D.C.; Nyamukondiwa, C.; Emami-Khoyi, A.; Oliver, J.C.; Teske, P.R.; Wasserman, R.J. The complete mitogenome of an undescribed clam shrimp of the genus Gondwanalimnadia (Branchiopoda: Spinicaudata), from a temporary wetland in Central District, Botswana. Mitochondrial DNA Part B 2020, 5, 1238–1240. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, H.; Jermnak, U.; Yang, J. The complete mitogenome of the freshwater fairy shrimp Streptocephalus sirindhornae (Crustacea: Anostraca: Streptocephalidae). Mitochondrial DNA Part A 2016, 27, 3189–3191. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; Kim, J.; Ko, Y.; Kim, S.; Kang, S. The complete mitochondrial genome of the Antarctic fairy shrimp Branchinecta gaini Daday, 1910 (Branchiopoda, Anostraca, Branchinectidae). Biodivers. Data J. 2022, 10, e94051. [Google Scholar] [CrossRef]
- Perez, M.; Valverde, J.; Batuecas, B.; Amat, F.; Marco, R.; Garesse, R. Speciation in the Artemia genus: Mitochondrial DNA analysis of bisexual and parthenogenetic brine shrimps. J. Mol. Evol. 1994, 38, 156–168. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Luo, Q.; Sun, J.; Liu, F.; Wu, G.; Yu, J.; Wang, W. Mitochondrial genome sequences of Artemia tibetiana and Artemia urmiana: Assessing molecular changes for high plateau adaptation. Sci. China Life Sci. 2013, 56, 440–452. [Google Scholar] [CrossRef]
- Asem, A.; Li, W.; Wang, P.; Eimanifar, A.; Shen, C.; de Vos, S.; van Stappen, G. The complete mitochondrial genome of Artemia sinica Cai, 1989 (Crustacea: Anostraca) using next-generation sequencing. Mitochondrial DNA Part B 2019, 4, 746–747. [Google Scholar] [CrossRef] [Green Version]
- Deji, G.; Zhang, C.; Sui, L.; Han, X. The complete mitochondrial genome of Artemia salina Leach, 1819 (Crustacea: Anostraca). Mitochondrial DNA Part B 2021, 6, 3255–3256. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Tashi, L.; Sui, L.; Wang, G.; Deji, G.; Zhang, C. The complete mitochondrial genome of Artemia persimilis Piccinelli and Prosdocimi, 1968 (Crustacea: Anostraca). Mitochondrial DNA Part B 2022, 7, 464–465. [Google Scholar] [CrossRef] [PubMed]
- Bellec, L.; Debruyne, R.; Utge, J.; Rabet, N. The first complete mitochondrial genome of Limnadia lenticularis (Branchiopoda, Spinicaudata), with new insights on its phylogeography and on the taxonomy of the genus. Hydrobiologia 2019, 826, 145–158. [Google Scholar] [CrossRef]
- Emami-Khoyi, A.; Tladi, M.; Dalu, T.; Teske, P.R.; van Vuuren, B.J.; Rogers, D.C.; Nyamukondiwa, C.; Wasserman, R.J. The complete mitogenome of Leptestheria brevirostris Barnard, 1924, a rock pool clam shrimp (Branchiopoda: Spinicaudata) from Central District, Botswana. Mitochondrial DNA Part B 2021, 6, 608–610. [Google Scholar] [CrossRef]
- Umetsu, K.; Iwabuchi, N.; Yuasa, I.; Saitou, N.; Clark, P.F.; Boxshall, G.; Osawa, M.; Igarashi, K. Complete mitochondrial DNA sequence of a tadpole shrimp (Triops cancriformis) and analysis of museum samples. Electrophoresis 2002, 23, 4080–4084. [Google Scholar] [CrossRef]
- Gan, H.; Tan, M.; Austin, C.M. The complete mitogenome of the Australian tadpole shrimp Triops australiensis (Spencer and Hall, 1895) (Crustacea: Branchiopoda: Notostraca). Mitochondrial DNA 2016, 27, 2028–2029. [Google Scholar] [CrossRef] [PubMed]
- Horn, R.L.; Cowley, D.E. Evolutionary relationships within Triops (Branchiopoda: Notostraca) using complete mitochondrial genomes. J. Crustac. Biol. 2014, 34, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.; Hwang, U. Complete mitochondrial genome of the longtail tadpole shrimp Triops longicaudatus (Crustacea, Branchiopoda, Notostraca). Mitochondrial DNA 2010, 21, 170–172. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. Phylogenetic position of the Pentastomida and (pan)crustacean relationships. Proc. R. Soc. Lond. B 2004, 271, 537–544. [Google Scholar] [CrossRef]
- Koga, C.; Rouse, G.W. Mitogenomics and the phylogeny of mantis shrimp (Crustacea: Stomatopoda). Diversity 2021, 13, 647. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | GenBank Position no. | Size (nts) | Strand a | Start Codon | Stop Codon | Anticodon | IGN b |
|---|---|---|---|---|---|---|---|
| trnI | 1–64 | 64 | - | GAT | 46 | ||
| trnQ | 111–179 | 69 | - | TTG | 9 | ||
| trnM | 189–253 | 65 | + | CAT | 12 | ||
| nd2 | 266–1216 | 951 | + | ATA | TAG | −2 | |
| trnW | 1215–1277 | 63 | + | TCA | −1 | ||
| trnC | 1277–1339 | 63 | - | GCA | 0 | ||
| trnY | 1340–1405 | 66 | - | GTA | −5 | ||
| cox1 | 1401–2939 | 1539 | + | ATA | TAA | 2 | |
| trnL1-CUN | 2942–3006 | 65 | + | TAG | 1 | ||
| trnL2-UUR | 3008–3069 | 62 | + | TAA | 19 | ||
| cox2 | 3089–3770 | 682 | + | ATT | T | 1 | |
| trnK | 3771–3835 | 65 | + | CTT | 0 | ||
| trnD | 3836–3898 | 63 | + | GTC | 0 | ||
| atp8 | 3899–4060 | 162 | + | ATT | TAA | −4 | |
| atp6 | 4057–4720 | 664 | + | ATA | T | 0 | |
| cox3 | 4721–5508 | 788 | + | ATG | T | −1 | |
| trnG | 5509–5569 | 61 | + | TCC | 63 | ||
| nd3 | 5633–5990 | 358 | + | ATA | T | 3 | |
| trnA | 5991–6052 | 62 | + | TGC | 14 | ||
| trnR | 6067–6126 | 60 | + | TCG | −3 | ||
| trnN | 6124–6187 | 64 | + | GTT | 0 | ||
| trnS1-AGN | 6188–6244 | 57 | + | GCT | 0 | ||
| trnE | 6245–6307 | 63 | + | TTC | 0 | ||
| trnF | 6308–6369 | 62 | - | GAA | 0 | ||
| nd5 | 6370–8041 | 1672 | - | ATT | T | 0 | |
| trnH | 8042–8103 | 62 | - | GTG | 0 | ||
| nd4 | 8104–9403 | 1300 | - | ATG | T | −1 | |
| nd4L | 9403–9690 | 295 | - | ATG | TAA | 5 | |
| trnT | 9696–9757 | 62 | + | TGT | 0 | ||
| trnP | 9758–9820 | 63 | - | TGG | 2 | ||
| nd6 | 9823–10,303 | 481 | + | ATT | T | 0 | |
| cytb | 10,304–11,434 | 1131 | + | ATG | TAA | −2 | |
| trnS2-UCN | 11,433–11,499 | 67 | + | TGA | 4 | ||
| nd1 | 11,504–12,415 | 912 | - | ATT | TAA | 0 | |
| rrnL | 12,416–13,738 | 1323 | - | 0 | |||
| trnV | 13,739–13,806 | 68 | - | TAC | 0 | ||
| rrnS | 13,807–14,586 | 780 | - | 0 | |||
| Control region | 14,589–15,023 | 437 | + | 0 |
| Regions | Nucleotide Composition (%) | AT-Skew | GC-Skew | ||||
|---|---|---|---|---|---|---|---|
| T (U) | C | A | G | A + T | |||
| Whole genome | 36.8/31.9 | 15.3/22.4 | 38.1/26.5 | 9.9/19.2 | 75.0/59.5 | 0.02/−0.09 | −0.21/−0.08 |
| PCGs | 43.8/35.4 | 12.5/22.6 | 29.8/22.0 | 13.8/20.0 | 73.6/57.4 | −0.19/−0.23 | 0.05/−0.06 |
| 1st codon position | 35.4/29.0 | 12.6/20.5 | 32.0/25.6 | 20.0/25.1 | 67.4/54.4 | −0.05/−0.06 | 0.24/0.10 |
| 2nd codon position | 48.6/44.0 | 16.9/22.2 | 20.5/17.3 | 14.0/16.5 | 69.2/61.4 | −0.41/−0.43 | −0.10/−0.15 |
| 3rd codon position | 47.4/33.0 | 8.1/25.1 | 37.0/23.0 | 7.6/18.5 | 84.3/56.0 | −0.12/−0.19 | −0.03/−0.15 |
| rRNA | 39.3/27.3 | 7.6/16.3 | 37.5/34.8 | 15.7/21.53 | 76.8/62.1 | −0.02/0.12 | 0.35/0.14 |
| tRNA | 36.7/31.3 | 10.7/16.6 | 39.1/30.1 | 13.5/22.1 | 75.8/61.3 | 0.03/−0.02 | 0.12/0.14 |
| atp6 | 42.0/35.1 | 16.9/25.2 | 31.5/20.1 | 9.6/19.5 | 73.5/55.3 | −0.14/−0.27 | −0.28/−0.13 |
| atp8 | 45.6/33.3 | 12.2/28.8 | 38.1/23.1 | 4.1/14.7 | 83.7/56.4 | −0.09/−0.18 | −0.5/−0.32 |
| cox1 | 39.2/33.5 | 16.7/23.7 | 27.6/20.5 | 16.5/22.2 | 66.8/54.1 | −0.17/−0.24 | −0.01/−0.03 |
| cox2 | 38.2/30.8 | 16.6/24.7 | 32.9/22.9 | 12.3/21.6 | 71.1/53.7 | −0.07/−0.15 | −0.15/−0.07 |
| cox3 | 39.5/37.1 | 17.1/22.3 | 29.8/19.6 | 13.7/21.0 | 69.2/56.7 | −0.14/−0.31 | 0.11/−0.03 |
| cytb | 39.5/34.3 | 16.6/24.4 | 31.6/20.6 | 12.4/20.7 | 71.0/54.9 | −0.11/−0.25 | −0.14/−0.08 |
| nad1 | 48.7/36.4 | 8.5/18.6 | 26.2/24.1 | 16.6/20.8 | 74.9/60.6 | −0.30/−0.20 | 0.32/0.06 |
| nad2 | 46.3/40.0 | 11.4/27.1 | 31.8/17.6 | 10.4/15.4 | 78.2/57.6 | −0.19/−0.39 | −0.05/−0.28 |
| nad3 | 43.2/40.2 | 15.3/19.9 | 30.2/18.8 | 11.3/21.1 | 73.4/59.0 | −0.18/−0.36 | −0.15/0.03 |
| nad4 | 49.8/36.5 | 7.4/21.1 | 27.9/22.5 | 14.9/20.8 | 77.8/59.0 | −0.28/−0.24 | 0.34/0.02 |
| nad4L | 52.2/34.3 | 3.4/19.9 | 26.8/24.9 | 17.5/20.9 | 79.0/59.3 | −0.32/−0.16 | 0.67/0.02 |
| nad5 | 44.5/32.3 | 8.1/20.6 | 31.8/26.6 | 15.6/20.5 | 76.2/58.8 | −0.17/0.10 | 0.31/0.00 |
| nad6 | 45.8/39.1 | 13.1/25.7 | 34.2/20.1 | 6.9/15.1 | 80.0/59.2 | −0.15/−0.32 | −0.31/−0.26 |
| Gene | Number of Comparisons with Significant Heterogeneity | Proportion of Significant Heterogeneity (%) | AT% a | |||
|---|---|---|---|---|---|---|
| Max | Mean | Min | LG b | |||
| nd2 | 554 | 61.4 | 78.2 | 68.6 | 57.2 | 78.2 |
| cox1 | 588 | 65.1 | 66.8 | 61.7 | 54.1 | 66.8 |
| cox2 | 368 | 40.8 | 71.1 | 64.2 | 53.7 | 71.1 |
| atp8 | 123 | 13.6 | 83.7 | 70.2 | 47.8 | 83.7 |
| atp6 | 435 | 48.2 | 73.5 | 64.8 | 54.6 | 73.5 |
| cox3 | 597 | 66.1 | 69.2 | 62.3 | 51.2 | 69.2 |
| nd3 | 350 | 38.8 | 76.8 | 68.7 | 56.8 | 73.5 |
| nd5 | 710 | 78.6 | 76.2 | 66.5 | 54.8 | 76.2 |
| nd4 | 692 | 76.6 | 77.8 | 67.0 | 55.1 | 77.8 |
| nd4l | 318 | 35.2 | 79.0 | 68.8 | 59.3 | 79.0 |
| nd6 | 334 | 37.0 | 80.0 | 69.9 | 55.7 | 80.0 |
| cytb | 569 | 63.0 | 71.0 | 62.9 | 54.9 | 71.0 |
| nd1 | 598 | 66.2 | 74.9 | 65.6 | 56.9 | 74.9 |
| Gene/Datasets | Nucleotide Data Sets | Amino Acid Data Sets | ||||
|---|---|---|---|---|---|---|
| Z Score | p Score | NDT * | Z Score | p Score | NDT * | |
| nd2 | 6.42 | 0.00 | 31 | 3.50 | 0.00 | 18 |
| cox1 | 5.20 | 0.02 | 32 | 0.37 | 0.24 | 6 |
| cox2 | 4.82 | 0.03 | 18 | 0.01 | 0.42 | 0 |
| atp8 | 5.67 | 0.00 | 11 | 2.11 | 0.03 | 14 |
| atp6 | 4.67 | 0.00 | 25 | 0.26 | 0.30 | 4 |
| cox3 | 4.31 | 0.02 | 31 | −0.89 | 0.81 | 2 |
| nd3 | 6.05 | 0.01 | 20 | 0.41 | 0.29 | 3 |
| nd5 | 13.10 | 0.00 | 33 | 5.79 | 0.00 | 16 |
| nd4 | 6.81 | 0.00 | 39 | 4.34 | 0.00 | 10 |
| nd4l | 7.12 | 0.00 | 26 | 2.04 | 0.05 | 5 |
| nd6 | 5.67 | 0.01 | 24 | 2.15 | 0.05 | 5 |
| cytb | 6.59 | 0.00 | 26 | 0.92 | 0.19 | 3 |
| nd1 | 5.13 | 0.00 | 35 | 1.28 | 0.09 | 7 |
| 13 PCGs | 5.60 | 0.00 | 39 | 13.18 | 0.00 | 33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Cheng, J. Conflicts in Mitochondrial Phylogenomics of Branchiopoda, with the First Complete Mitogenome of Laevicaudata (Crustacea: Branchiopoda). Curr. Issues Mol. Biol. 2023, 45, 820-837. https://doi.org/10.3390/cimb45020054
Sun X, Cheng J. Conflicts in Mitochondrial Phylogenomics of Branchiopoda, with the First Complete Mitogenome of Laevicaudata (Crustacea: Branchiopoda). Current Issues in Molecular Biology. 2023; 45(2):820-837. https://doi.org/10.3390/cimb45020054
Chicago/Turabian StyleSun, Xiaoyan, and Jinhui Cheng. 2023. "Conflicts in Mitochondrial Phylogenomics of Branchiopoda, with the First Complete Mitogenome of Laevicaudata (Crustacea: Branchiopoda)" Current Issues in Molecular Biology 45, no. 2: 820-837. https://doi.org/10.3390/cimb45020054