
Semi-Site-Specific Primer PCR: A Simple but Reliable Genome-Walking Tool

Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic DNA
2.2. Primer
2.3. PCR Procedure
2.4. Agarose Electrophoresis and DNA Sequencing
3. Results
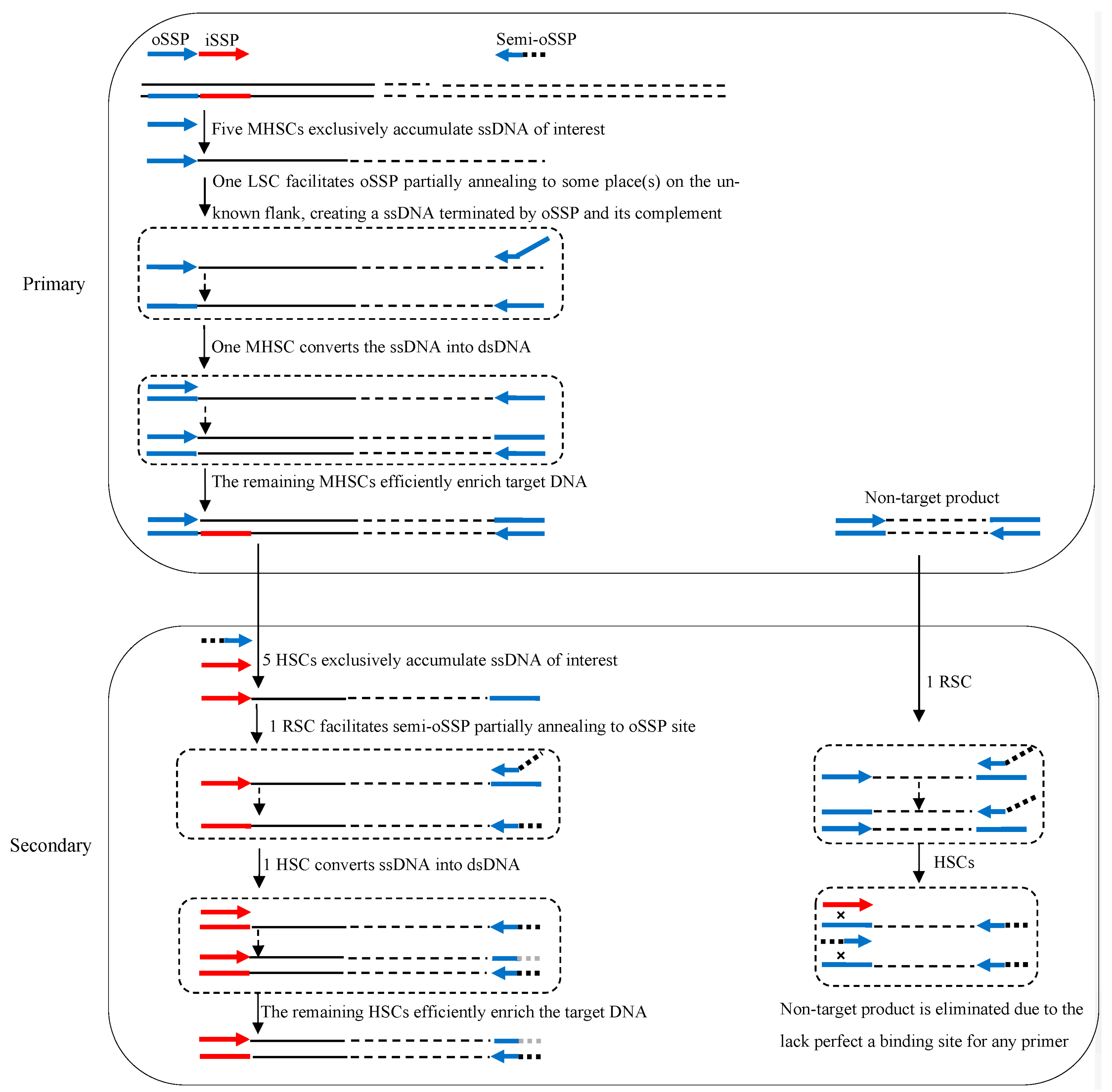
3.1. Principle of 3SP-PCR
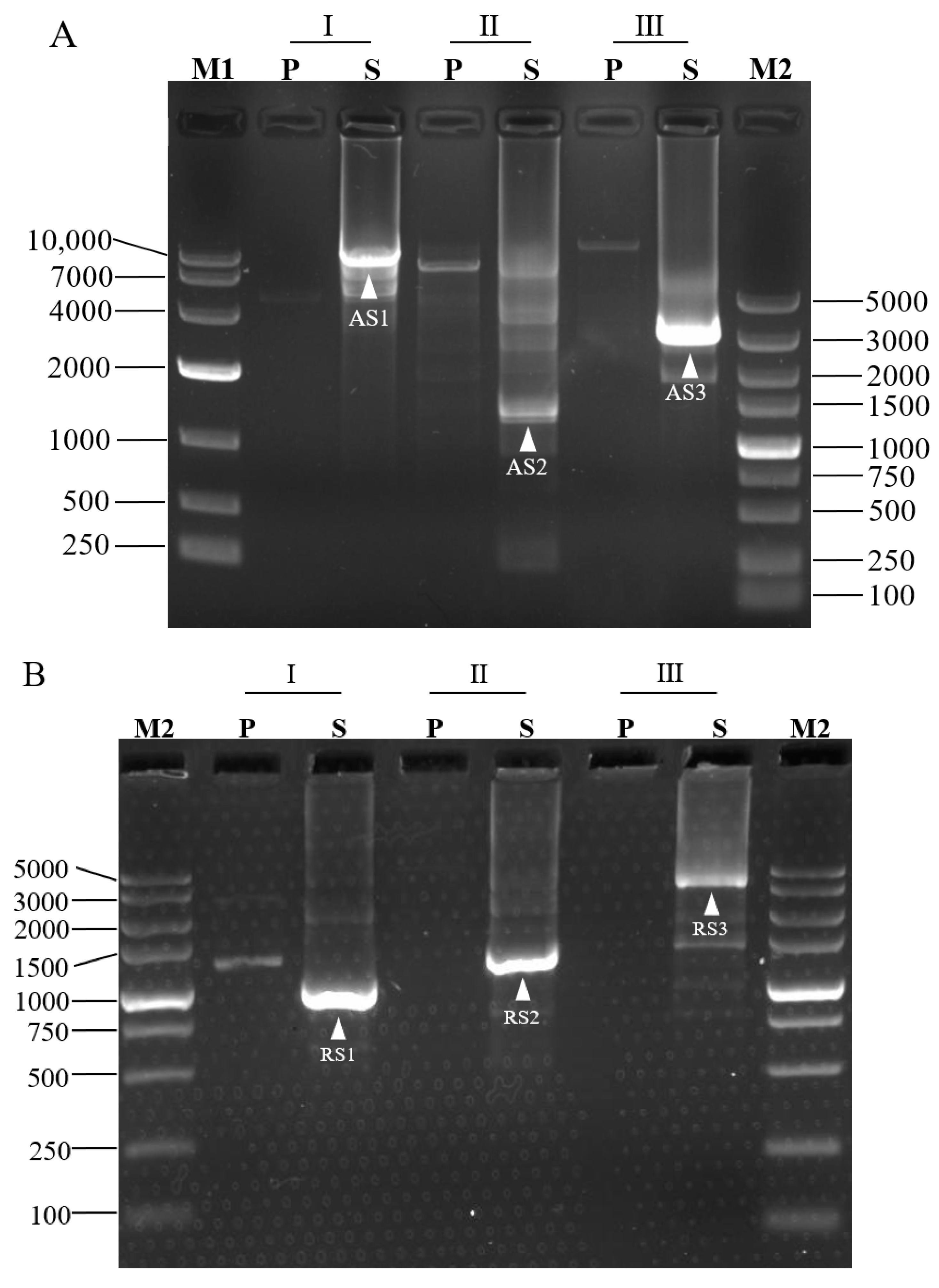
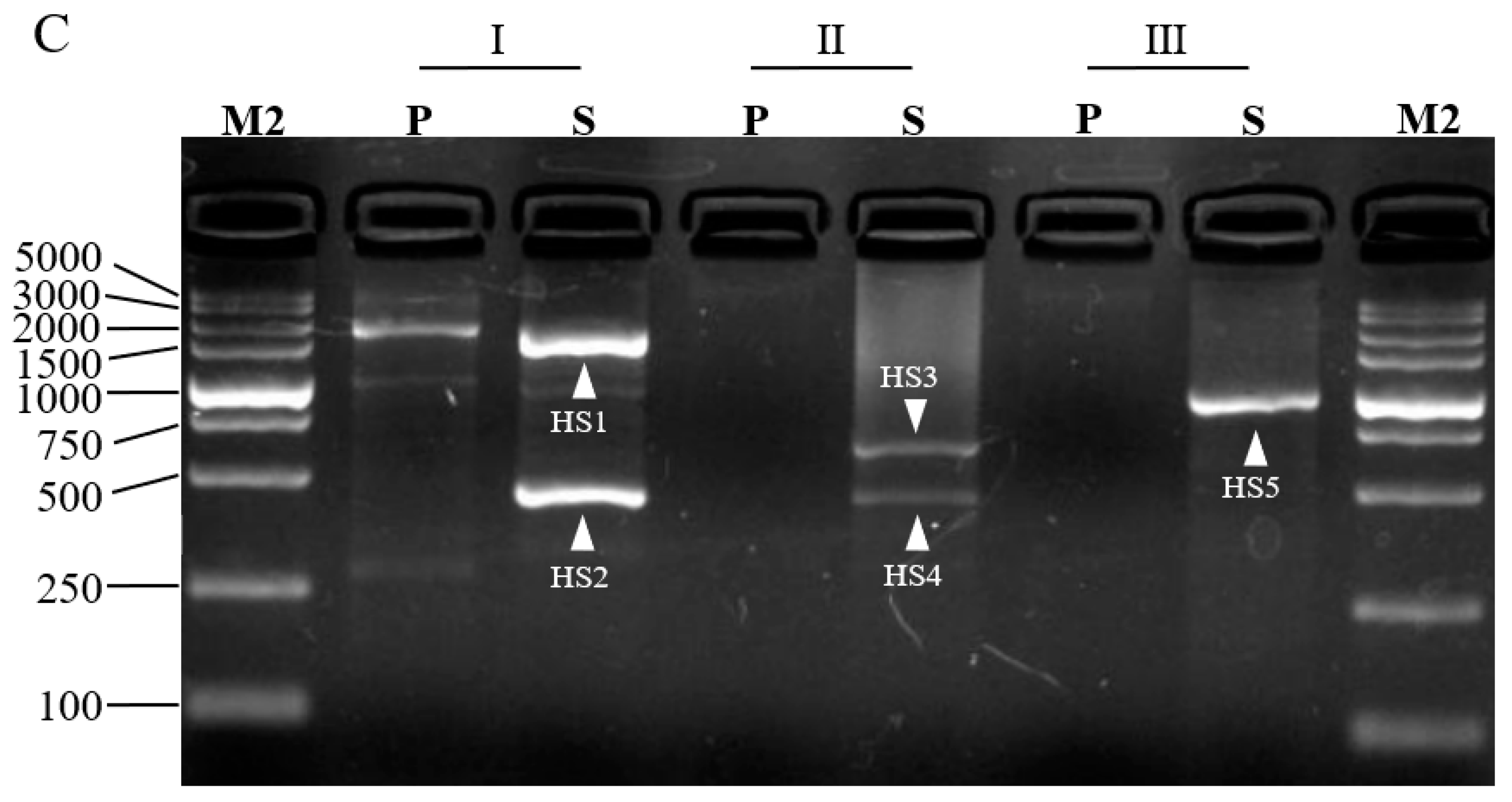
3.2. Validation of 3SP-PCR
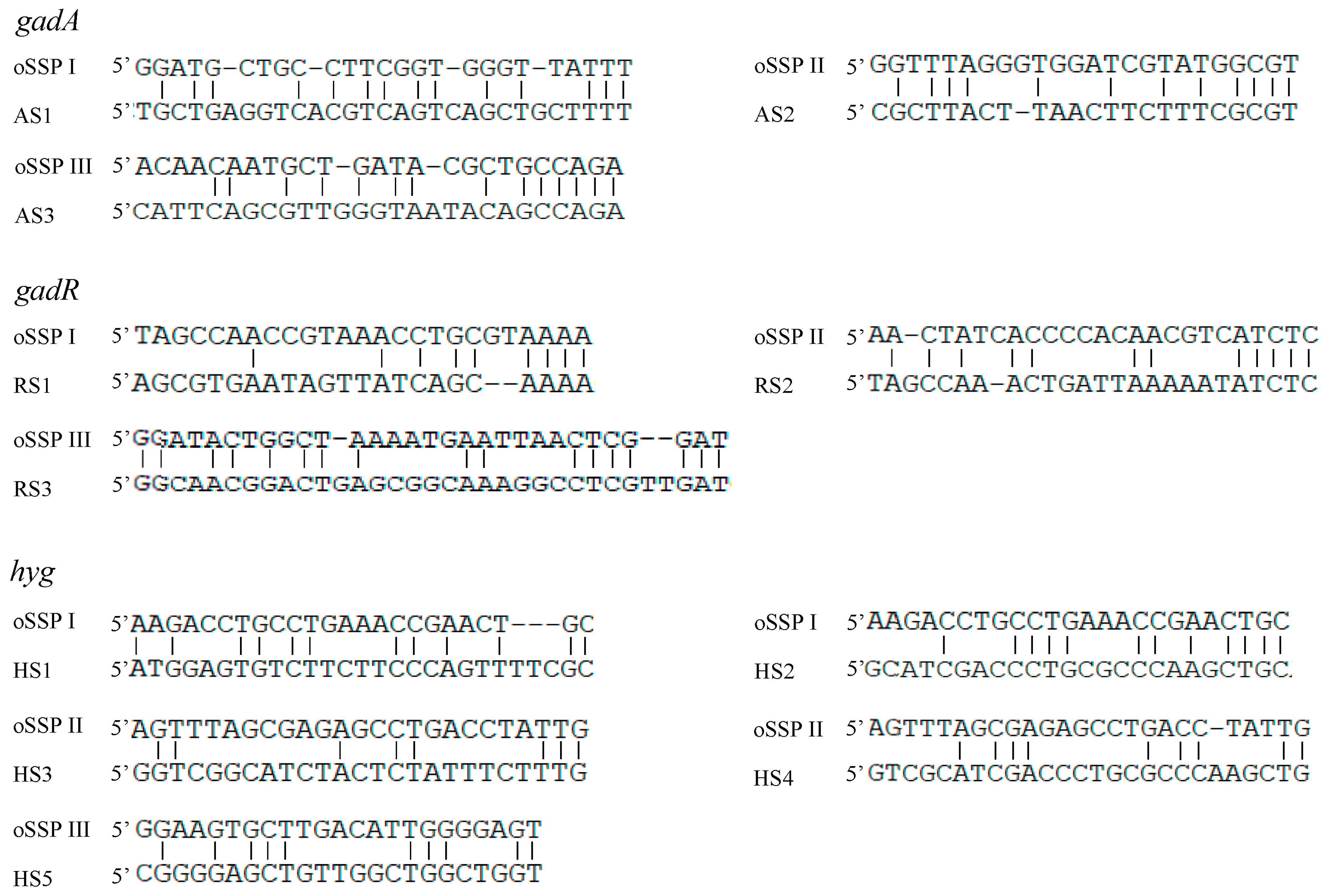
3.3. Partial Annealing Sites of Walking Primers
3.4. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kotik, M.; Stepanek, V.; Maresova, H.; Kyslik, P.; Archelas, A. Environmental DNA as a source of a novel epoxide hydrolase reacting with aliphatic terminal epoxides. J. Mol. Catal. 2009, 56, 288–293. [Google Scholar] [CrossRef]
- Leoni, C.; Volpicella, M.; De Leo, F.; Gallerani, R.; Ceci, L.R. Genome walking in eukaryotes. FEBS J. 2011, 278, 3953–3977. [Google Scholar] [CrossRef] [PubMed]
- Fraiture, M.A.; Papazova, N.; Roosens, N.H.C. DNA walking strategy to identify unauthorized genetically modified bacteria in microbial fermentation products. Int. J. Food Microbiol. 2021, 337, 108913. [Google Scholar] [CrossRef] [PubMed]
- Myrick, K.V.; Gelbart, W.M. Universal fast walking for direct and versatile determination of flanking sequence. Gene 2002, 284, 125–131. [Google Scholar] [CrossRef]
- Thirulogachandar, V.; Pandey, P.; Vaishnavi, C.S.; Reddy, M.K. An affinity-based genome walking method to find transgene integration loci in transgenic genome. Anal. Biochem. 2011, 416, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Pan, Q.; Hu, Z.; Qiu, J.; Yu, Z. Heterologous Expression and Characterization of Flavinadenine Dinucleotide Synthetase from Candida famata for Flavin Adenine Dinucleotide Production. Protein Peptide Lett. 2021, 28, 229–239. [Google Scholar] [CrossRef]
- Li, H.; Ding, D.; Cao, Y.; Yu, B.; Guo, L.; Liu, X. Partially Overlapping Primer-Based PCR for Genome Walking. PLoS ONE 2015, 10, e0120139. [Google Scholar] [CrossRef] [Green Version]
- Ochman, H.; Gerber, A.S.; Hartl, D.L. Genetic applications of an inverse polymerase chain reaction. Genetics 1988, 120, 621–623. [Google Scholar] [CrossRef]
- Uchiyama, T.; Watanabe, K. Improved inverse PCR scheme for metagenome walking. BioTechniques. 2006, 41, 183–188. [Google Scholar] [CrossRef]
- Yuanxin, Y.; Chengcai, A.; Li, L.; Jiayu, G.; Guihong, T.; Zhangliang, C. T-linker-specific ligation PCR (T-linker PCR): An advanced PCR technique for chromosome walking or for isolation of tagged DNA ends. Nucleic Acids Res. 2003, 31, e68. [Google Scholar] [CrossRef]
- Yik, M.H.Y.; Lo, Y.T.; Lin, X.; Sun, W.; Chan, T.F.; Shaw, P.C. Authentication of Hedyotis products by adaptor ligation-mediated PCR and metabarcoding. J. Pharm. Biomed. 2021, 196, 113920. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.G.; Whittier, R.F. Thermal asymmetric interlaced PCR: Automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 1995, 25, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jia, M.; Li, Z.; Liu, X.; Sun, T.; Pei, J.; Wei, C.; Lin, Z.; Li, H. Wristwatch PCR: A Versatile and Efficient Genome Walking Strategy. Front. Bioeng. Biotechnol. 2022, 10, 458. [Google Scholar] [CrossRef]
- Triglia, T.; Peterson, M.G.; Kemp, D.J. A procedure for in vitro amplification of DNA segments that lie outside the boundaries of known sequences. Nucleic Acids Res. 1988, 16, 8186. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.; Wang, Q.; Shi, X.; Wang, S.; Wu, H.; Nie, L.; Li, H. Stepwise partially overlapping primer-based PCR for genome walking. AMB Express 2018, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Braam, J. Restriction site extension PCR: A novel method for high-throughput characterization of tagged DNA fragments and genome walking. PLoS ONE 2010, 5, e10577. [Google Scholar] [CrossRef] [Green Version]
- Kotik, M. Novel genes retrieved from environmental DNA by polymerase chain reaction: Current genome-walking techniques for future metagenome applications. J. Biotechnol. 2009, 144, 75–82. [Google Scholar] [CrossRef]
- Jia, X.B.; Lin, X.J.; Chen, J.C. Linear and exponential TAIL-PCR: A method for efficient and quick amplification of flanking sequences adjacent to Tn5 transposon insertion sites. AMB Express 2017, 7, 195. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Chang, K.; Ding, G.; Wu, H.; Chen, Y.; Jia, M.; Liu, X.; Wang, S.; Jin, Y.; Pan, H.; et al. Genomic insights into a robust gamma-aminobutyric acid-producer Lactobacillus brevis CD0817. AMB Express 2019, 9, 72. [Google Scholar] [CrossRef]
- Jia, M.; Zhu, Y.; Wang, L.; Sun, T.; Pan, H.; Li, H. pH Auto-Sustain-Based Fermentation Supports Efficient Gamma-Aminobutyric Acid Production by Lactobacillus brevis CD0817. Fermentation 2022, 8, 208. [Google Scholar] [CrossRef]
- Li, H.; Sun, T.; Jia, M.; Wang, L.; Wei, C.; Pei, J.; Lin, Z.; Wang, S. Production of Gamma-Aminobutyric Acid by Levilactobacillus brevis CD0817 by Coupling Fermentation with Self-Buffered Whole-Cell Catalysis. Fermentation 2022, 8, 321. [Google Scholar] [CrossRef]
- Sun, T.; Jia, M.; Wang, L.; Li, Z.; Lin, Z.; Wei, C.; Pei, J.; Li, H. DAR-PCR: A new tool for efficient retrieval of unknown flanking genomic DNA. AMB Express 2022, 12, 131. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Wei, C.; Pei, J.; Li, H. Bridging PCR: An efficient and reliable scheme implemented for genome-walking. Curr. Issues Mol. Biol. 2023, 45, 501–511. [Google Scholar]
- Pei, J.; Sun, T.; Wang, L.; Pan, Z.; Guo, X.; Li, H. Fusion primer driven racket PCR: A novel tool for genome walking. Front. Genet. 2022, 13, 969840. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.D.; Rabinovitch, P.S.; Burmer, G.C. Targeted gene walking polymerase chain reaction. Nucleic Acids Res. 1991, 19, 3055–3060. [Google Scholar] [CrossRef] [Green Version]
- Parks, C.L.; Chang, L.S.; Shenk, T. A polymerase chain reaction mediated by a single primer: Cloning of genomic sequences adjacent to a serotonin receptor protein coding region. Nucleic Acids Res. 1991, 19, 7155–7160. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primary PCR | Secondary PCR | |
|---|---|---|---|
| oSSP | semi-oSSP | iSSP | |
| gadA | I: GGATGCTGCCTTCGGTGGGTTATTT II: GGTTTAGGGTGGATCGTATGGCGT III: ACAACAATGCTGATACGCTGCCAGA | I: ACTCCAACGGCATCCTGGGTTATTT II: TAAGGTCTTCACTGCGTATGGCGT III: TACTTTCCATAACACCGCTGCCAGA | ACGGTTGACTCCATTGCCATTAACT |
| gadR | I: TAGCCAACCGTAAACCTGCGTAAAA II: AACTATCACCCCACAACGTCATCTC III: GGATACTGGCTAAAATGAATTAACTCGGAT | I: AGCTGCGATACTCCACTGCGTAAAA II: GGTCCAGCATAGGGTACGTCATCTC III: CATGTAATAACCTCGCTTCATAACTCGGAT | ACCGTTCATAGGCGAAATTGTTTGT |
| hyg | I: AAGACCTGCCTGAAACCGAACTGC II: AGTTTAGCGAGAGCCTGACCTATTG III: GGAAGTGCTTGACATTGGGGAGT | I: TTAGAACTGACCCGACCGAACTGC II: CCGCCTAGCCACTGATGACCTATTG III: CTTCTCAGCCTGGATTGGGGAGT | CAAGGAATCGGTCAATACATACATGGC |
| Round of PCR | Thermal Parameters | Cycle Number |
|---|---|---|
| Primary | 94 °C, 2 min | |
| 94 °C, 30 s; 60 °C, 30 s; 72 °C, 3 min | 5 | |
| 94 °C, 30 s; 25 °C, 30 s; 72 °C, 3 min | 1 | |
| 94 °C, 30 s; 60 °C, 30 s; 72 °C, 3 min | 25 | |
| 72 °C, 10 min | ||
| Secondary | 94 °C, 2 min | |
| 94 °C, 30 s; 65 °C, 30 s; 72 °C, 3 min | 5 | |
| 94 °C, 30 s; 40 °C, 30 s; 72 °C, 3 min | 1 | |
| 94 °C, 30 s; 65 °C, 30 s; 72 °C, 3 min | 25 | |
| 72 °C, 10 min |
| Approach | Rationale and Process | ESGM | Walk Range | Reference |
|---|---|---|---|---|
| Inverse PCR | Genomic DNA is digested with an endonuclease then self-circularized. The resultant DNA undergoes PCR performed by two SSPs with opposite extension orientations. | No | 0.3–1.8 | [1,8] |
| DL-PCR | DNA is digested with an endonuclease then ligated to a random oligo. The resultant DNA undergoes 2–3 consecutive PCRs by nested pairing the oligo primer with SSPs. | No | 0.3–3.0 | [1] |
| TAIL-PCR | A short random oligo is used as a walking primer. One low-stringency cycle has to be performed in every three cycles to aid walking primer annealing. The amplification of a target DNA is over a non-target one due to the differential amplification efficiency. | Yes | 0.3–3.5 | [1,12] |
| POP-PCR | A set of walking primers with 3′-overlaps are individually paired with nested SSPs. A walking primer can partially anneal to a DNA template only in the one relaxed-stringency cycle of each PCR, generating a pool of ssDNAs. A target ssDNA can be converted into dsDNA by the SSP in the next high-stringency cycle, but a non-target one cannot. | Yes | 0.3–3.5 | [7] |
| 3SP-PCR | The rationale and process are shown in the section of “3.1 Principle of 3SP-PCR” of this study. | Yes | 0.4–8.0 | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, C.; Lin, Z.; Pei, J.; Pan, H.; Li, H. Semi-Site-Specific Primer PCR: A Simple but Reliable Genome-Walking Tool. Curr. Issues Mol. Biol. 2023, 45, 512-523. https://doi.org/10.3390/cimb45010034
Wei C, Lin Z, Pei J, Pan H, Li H. Semi-Site-Specific Primer PCR: A Simple but Reliable Genome-Walking Tool. Current Issues in Molecular Biology. 2023; 45(1):512-523. https://doi.org/10.3390/cimb45010034
Chicago/Turabian StyleWei, Cheng, Zhiyu Lin, Jinfeng Pei, Hao Pan, and Haixing Li. 2023. "Semi-Site-Specific Primer PCR: A Simple but Reliable Genome-Walking Tool" Current Issues in Molecular Biology 45, no. 1: 512-523. https://doi.org/10.3390/cimb45010034