
Clinical and Molecular Implications of Osteopontin in Heart Failure
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
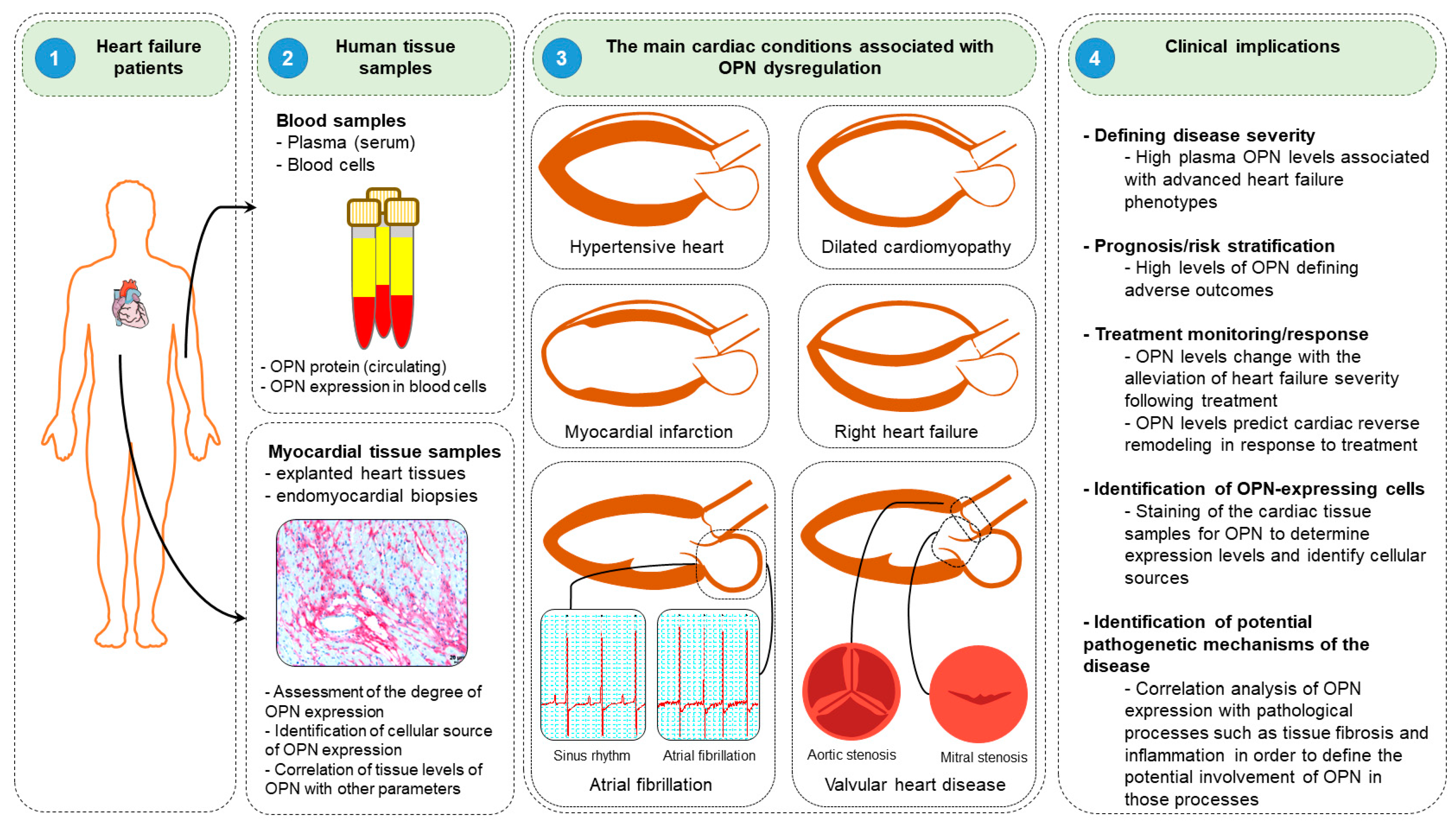{kind=link}
Abstract
:1. Introduction
2. Osteopontin Biology
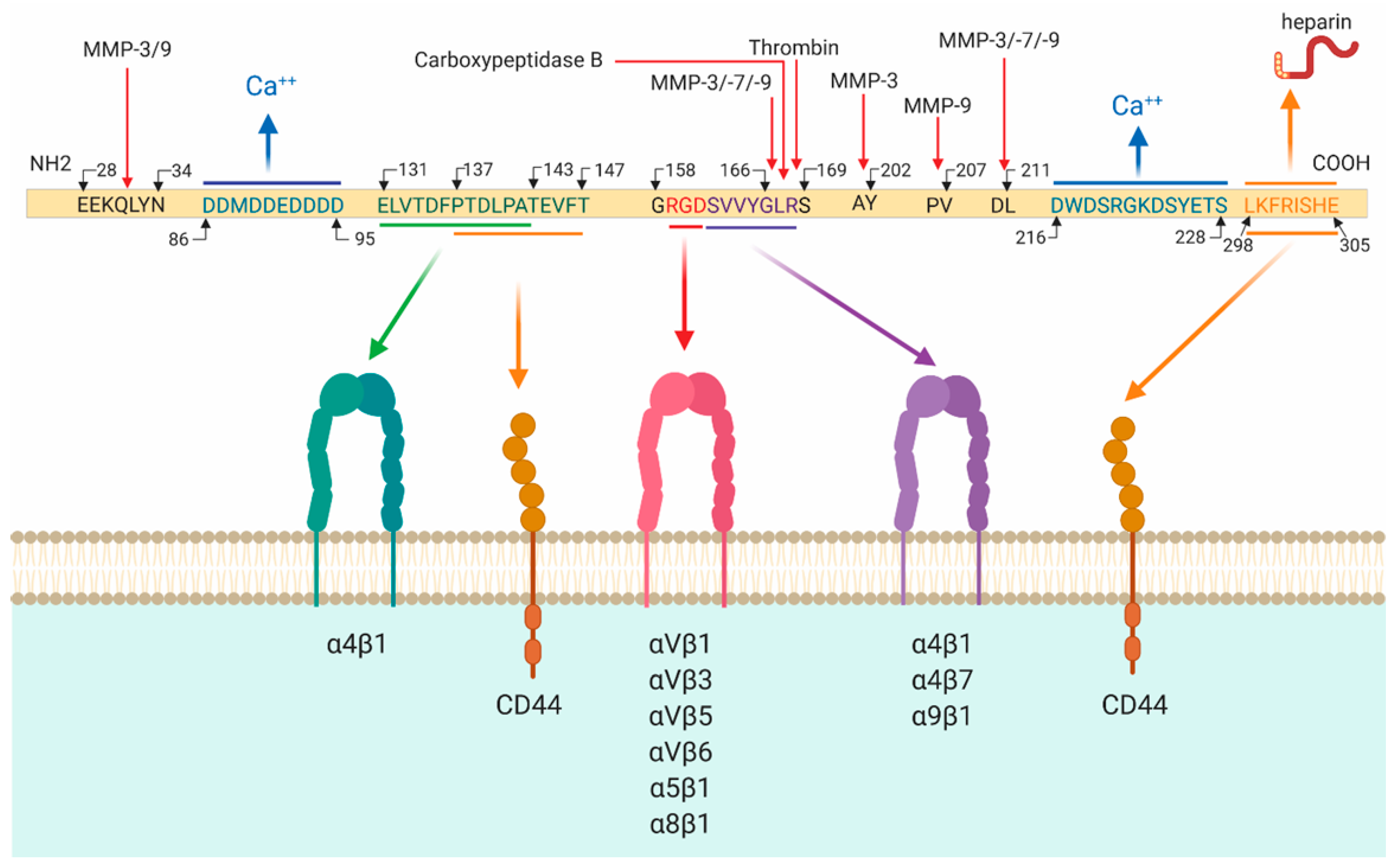
2.1. Osteopontin Cleavage
2.2. Osteopontin Receptors
3. Osteopontin in Heart Failure
3.1. Cell-Specific Regulation of Osteopontin
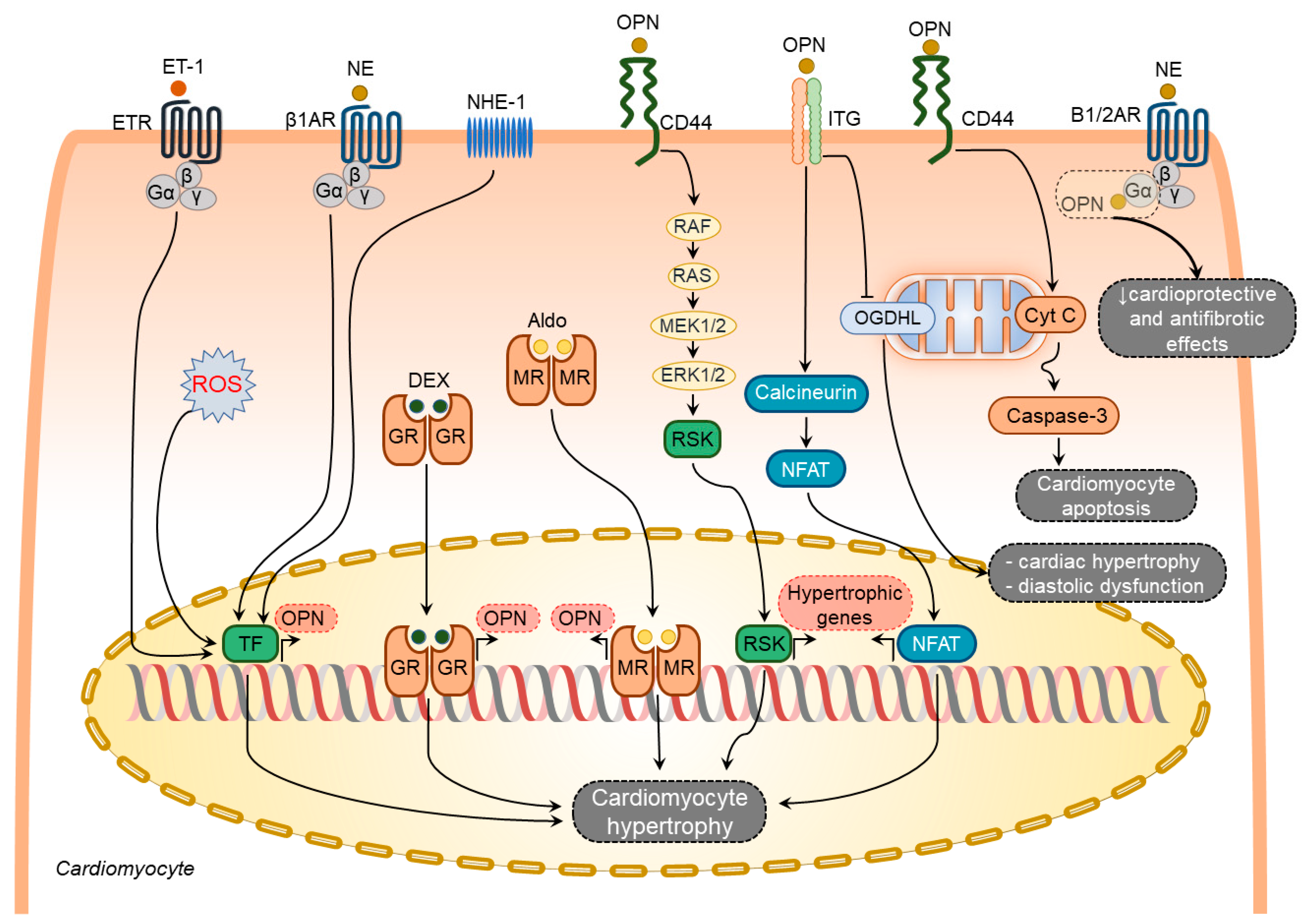
3.1.1. Osteopontin in Cardiomyocytes
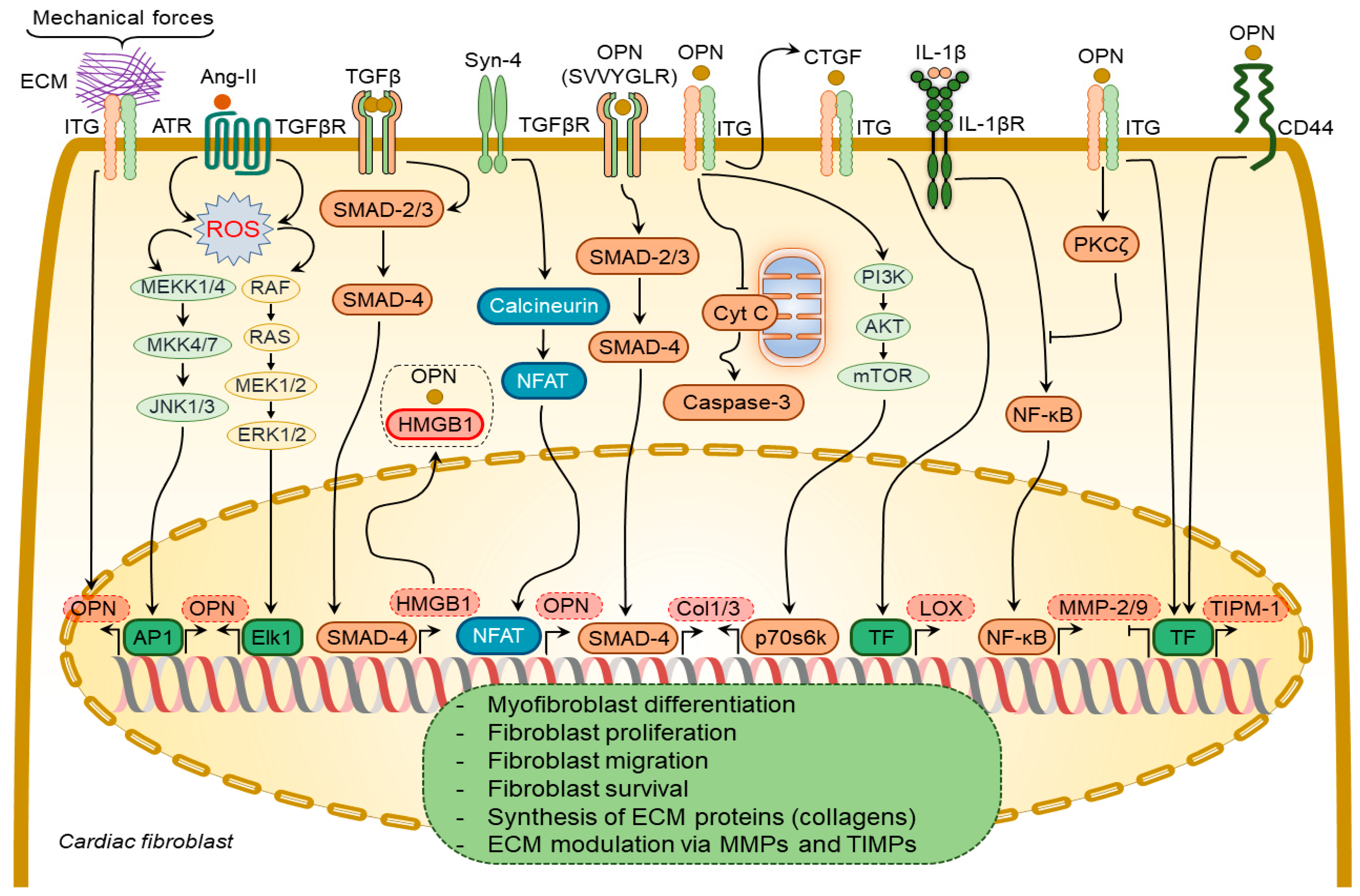
3.1.2. Osteopontin in Cardiac Fibroblasts
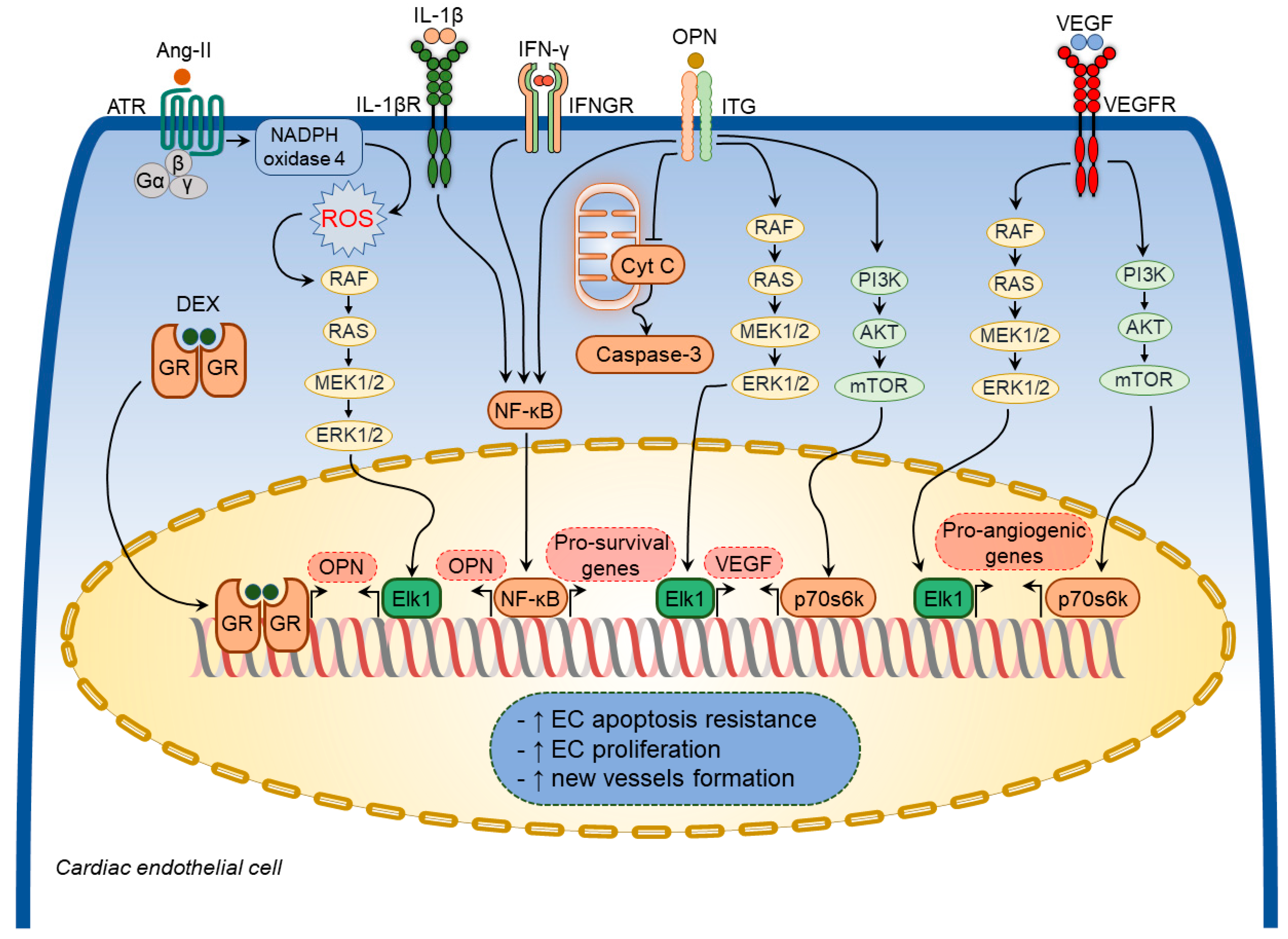
3.1.3. Osteopontin in Cardiac Endothelial Cells
3.1.4. Osteopontin in Cardiac Macrophages
3.2. Implications of Osteopontin in Specific Pathological Processes
3.2.1. Osteopontin in Cardiac Hypertrophy
3.2.2. Osteopontin in Cardiac Inflammation
3.2.3. Osteopontin in Cardiac Fibrosis
3.2.4. Osteopontin in Cardiac Capillarization
3.3. Clinical Implication of Osteopontin
3.3.1. Dilated Cardiomyopathy
3.3.2. Hypertensive Heart Failure
3.3.3. Myocardial Infarction
3.3.4. Atrial Fibrillation
3.3.5. Valvular Heart Disease
3.3.6. Right Ventricular Failure
4. Osteopontin as a Potential Therapeutic Target in Heart Failure
5. Summary
6. Current Challenges and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef] [Green Version]
- Vos, T.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abdulkader, R.S.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [Green Version]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef] [Green Version]
- Corris, P.A.; Seeger, W. Call It by the Correct Name—Pulmonary Hypertension Not Pulmonary Arterial Hypertension: Growing Recognition of the Global Health Impact for a Well-Recognized Condition and the Role of the Pulmonary Vascular Research Institute; American Physiological Society Bethesda: Rockville, MD, USA, 2020. [Google Scholar]
- Friedberg, M.K.; Redington, A.N. Right versus left ventricular failure: Differences, similarities, and interactions. Circulation 2014, 129, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Vonk-Noordegraaf, A.; Haddad, F.; Chin, K.M.; Forfia, P.R.; Kawut, S.M.; Lumens, J.; Naeije, R.; Newman, J.; Oudiz, R.J.; Provencher, S.; et al. Right heart adaptation to pulmonary arterial hypertension: Physiology and pathobiology. J. Am. Coll. Cardiol. 2013, 62 (Suppl. S25), D22–D33. [Google Scholar] [CrossRef]
- Egemnazarov, B.; Crnkovic, S.; Nagy, B.M.; Olschewski, H.; Kwapiszewska, G. Right ventricular fibrosis and dysfunction: Actual concepts and common misconceptions. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 68–69, 507–521. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [Green Version]
- Sydykov, A.; Mamazhakypov, A.; Petrovic, A.; Kosanovic, D.; Sarybaev, A.S.; Weissmann, N.; Ghofrani, H.A.; Schermuly, R.T. Inflammatory Mediators Drive Adverse Right Ventricular Remodeling and Dysfunction and Serve as Potential Biomarkers. Front. Physiol. 2018, 9, 609. [Google Scholar] [CrossRef]
- Carrillo-Salinas, F.J.; Ngwenyama, N.; Anastasiou, M.; Kaur, K.; Alcaide, P. Heart Inflammation: Immune Cell Roles and Roads to the Heart. Am. J. Pathol. 2019, 189, 1482–1494. [Google Scholar] [CrossRef] [Green Version]
- Frump, A.L.; Bonnet, S.; de Jesus Perez, V.A.; Lahm, T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L443–L460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, T.; Akazawa, H.; Naito, A.T.; Komuro, I. Angiogenesis and cardiac hypertrophy: Maintenance of cardiac function and causative roles in heart failure. Circ. Res. 2014, 114, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, G.; Mamazhakypov, A.; Schermuly, R.T.; Rajagopal, S. The Role of G Protein-Coupled Receptors in the Right Ventricle in Pulmonary Hypertension. Front. Cardiovasc. Med. 2018, 5, 179. [Google Scholar] [CrossRef]
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.J.; Archer, S.L. Emerging concepts in the molecular basis of pulmonary arterial hypertension: Part I: Metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015, 131, 1691–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Piao, L.; Marsboom, G.; Archer, S.L. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J. Mol. Med. 2010, 88, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Sabbah, H.N. Targeting the Mitochondria in Heart Failure: A Translational Perspective. JACC Basic Transl. Sci. 2020, 5, 88–106. [Google Scholar] [CrossRef]
- Shults, N.V.; Melnyk, O.; Suzuki, D.I.; Suzuki, Y.J. Redox Biology of Right-Sided Heart Failure. Antioxidants 2018, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Esfandiary, A.; Kutsche, H.S.; Schreckenberg, R.; Weber, M.; Pak, O.; Kojonazarov, B.; Sydykov, A.; Hirschhauser, C.; Wolf, A.; Haag, D.; et al. Protection against pressure overload-induced right heart failure by uncoupling protein 2 silencing. Cardiovasc. Res. 2019, 115, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- López, B.; Ravassa, S.; Moreno, M.U.; José, G.S.; Beaumont, J.; González, A.; Díez, J. Diffuse myocardial fibrosis: Mechanisms, diagnosis and therapeutic approaches. Nat. Rev. Cardiol. 2021, 18, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Matricellular proteins in cardiac adaptation and disease. Physiol. Rev. 2012, 92, 635–688. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Dalal, S.; Singh, K. Osteopontin: At the cross-roads of myocyte survival and myocardial function. Life Sci. 2014, 118, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Zahradka, P. Novel role for osteopontin in cardiac fibrosis. Circ. Res. 2008, 102, 270–272. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Jia, N.; Okamoto, H.; Kon, S.; Onozuka, H.; Akino, M.; Liu, L.; Morimoto, J.; Rittling, S.R.; Denhardt, D.; et al. Role of osteopontin in cardiac fibrosis and remodeling in angiotensin II-induced cardiac hypertrophy. Hypertension 2004, 43, 1195–1201. [Google Scholar] [CrossRef] [Green Version]
- Anwar, A.; Li, M.; Frid, M.G.; Kumar, B.; Gerasimovskaya, E.V.; Riddle, S.R.; McKeon, B.A.; Thukaram, R.; Meyrick, B.O.; Fini, M.A.; et al. Osteopontin is an endogenous modulator of the constitutively activated phenotype of pulmonary adventitial fibroblasts in hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L1–L11. [Google Scholar] [CrossRef]
- Singh, M.; Foster, C.R.; Dalal, S.; Singh, K. Role of osteopontin in heart failure associated with aging. Heart Fail. Rev. 2010, 15, 487–494. [Google Scholar] [CrossRef]
- Sørensen, E.S.; Petersen, T.E.; Højrup, P. Posttranslational modifications of bovine osteopontin: Identification of twenty-eight phosphorylation and three O-glycosylation sites. Protein Sci. 1995, 4, 2040–2049. [Google Scholar] [CrossRef]
- O’Regan, A. The Role of osteopontin in lung disease. Cytokine Growth Factor Rev. 2003, 14, 479–488. [Google Scholar] [CrossRef]
- Mazzali, M.; Kipari, T.; Ophascharoensuk, V.; Wesson, J.; Johnson, R.; Hughes, J. Osteopontin—A molecule for all seasons. Qjm 2002, 95, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef]
- Lee, G.S.; Salazar, H.F.; Joseph, G.; Lok, Z.S.Y.; Caroti, C.M.; Weiss, D.; Taylor, W.R.; Lyle, A.N. Osteopontin isoforms differentially promote arteriogenesis in response to ischemia via macrophage accumulation and survival. Lab. Investig. J. Tech. Methods Pathol. 2019, 99, 331–345. [Google Scholar] [CrossRef]
- Anborgh, P.H.; Mutrie, J.C.; Tuck, A.B.; Chambers, A.F. Pre- and post-translational regulation of osteopontin in cancer. J. Cell Commun. Signal. 2011, 5, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, M.L.; Zouein, F.A.; Tian, Y.; Padmanabhan Iyer, R.; de Castro Bras, L.E. Osteopontin is proteolytically processed by matrix metalloproteinase 9. Can. J. Physiol. Pharmacol. 2015, 93, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, N.; Sakai, F.; Kon, S.; Morimoto, J.; Kimura, C.; Yamazaki, H.; Okazaki, I.; Seki, N.; Fujii, T.; Uede, T. Essential role of the cryptic epitope SLAYGLR within osteopontin in a murine model of rheumatoid arthritis. J. Clin. Investig. 2003, 112, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Shao, Z.; Sharif, S.; Du, X.Y.; Myles, T.; Merchant, M.; Harsh, G.; Glantz, M.; Recht, L.; Morser, J.; et al. Thrombin-cleaved fragments of osteopontin are overexpressed in malignant glial tumors and provide a molecular niche with survival advantage. J. Biol. Chem. 2013, 288, 3097–3111. [Google Scholar] [CrossRef] [Green Version]
- Christensen, B.; Schack, L.; Klaning, E.; Sorensen, E.S. Osteopontin is cleaved at multiple sites close to its integrin-binding motifs in milk and is a novel substrate for plasmin and cathepsin D. J. Biol. Chem. 2010, 285, 7929–7937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokosaki, Y.; Matsuura, N.; Sasaki, T.; Murakami, I.; Schneider, H.; Higashiyama, S.; Saitoh, Y.; Yamakido, M.; Taooka, Y.; Sheppard, D. The integrin alpha(9)beta(1) binds to a novel recognition sequence (SVVYGLR) in the thrombin-cleaved amino-terminal fragment of osteopontin. J. Biol. Chem. 1999, 274, 36328–36334. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Kon, S.; Nakayama, Y.; Kurotaki, D.; Saito, Y.; Kanayama, M.; Kimura, C.; Diao, H.; Morimoto, J.; Matsui, Y.; et al. The differential amino acid requirement within osteopontin in alpha4 and alpha9 integrin-mediated cell binding and migration. Matrix Biol. J. Int. Soc. Matrix Biol. 2009, 28, 11–19. [Google Scholar] [CrossRef]
- Yokosaki, Y.; Tanaka, K.; Higashikawa, F.; Yamashita, K.; Eboshida, A. Distinct structural requirements for binding of the integrins alphavbeta6, alphavbeta3, alphavbeta5, alpha5beta1 and alpha9beta1 to osteopontin. Matrix Biol. J. Int. Soc. Matrix Biol. 2005, 24, 418–427. [Google Scholar] [CrossRef]
- Green, P.M.; Ludbrook, S.B.; Miller, D.D.; Horgan, C.M.; Barry, S.T. Structural elements of the osteopontin SVVYGLR motif important for the interaction with alpha(4) integrins. FEBS Lett. 2001, 503, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Rangaswami, H.; Bulbule, A.; Kundu, G.C. Osteopontin: Role in cell signaling and cancer progression. Trends Cell Biol. 2006, 16, 79–87. [Google Scholar] [CrossRef]
- Zheng, W.; Li, R.; Pan, H.; He, D.; Xu, R.; Guo, T.B.; Guo, Y.; Zhang, J.Z. Role of osteopontin in induction of monocyte chemoattractant protein 1 and macrophage inflammatory protein 1beta through the NF-kappaB and MAPK pathways in rheumatoid arthritis. Arthritis Rheum. 2009, 60, 1957–1965. [Google Scholar] [CrossRef]
- Barry, S.T.; Ludbrook, S.B.; Murrison, E.; Horgan, C.M. A regulated interaction between alpha5beta1 integrin and osteopontin. Biochem. Biophys. Res. Commun. 2000, 267, 764–769. [Google Scholar] [CrossRef]
- Smith, L.L.; Cheung, H.K.; Ling, L.E.; Chen, J.; Sheppard, D.; Pytela, R.; Giachelli, C.M. Osteopontin N-terminal domain contains a cryptic adhesive sequence recognized by alpha9beta1 integrin. J. Biol. Chem. 1996, 271, 28485–28491. [Google Scholar] [CrossRef] [Green Version]
- Scatena, M.; Liaw, L.; Giachelli, C.M. Osteopontin: A multifunctional molecule regulating chronic inflammation and vascular disease. Arter. Thromb. Vasc. Biol. 2007, 27, 2302–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayless, K.J.; Davis, G.E. Identification of dual α4β1 integrin binding sites within a 38 amino acid domain in the N-terminal thrombin fragment of human osteopontin. J. Biol. Chem. 2001, 276, 13483–13489. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.S.; Li, Y.; Zhang, Z.F.; You, J.; Wang, C.L. Osteopontin combined with CD44v6, a novel prognostic biomarker in non-small cell lung cancer undergoing curative resection. Ann. Thorac. Surg. 2013, 96, 1943–1951. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Bashir, M.M.; Werth, V.P. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: A unique molecular signature. J. Investig. Dermatol. 2012, 132, 1825–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, B.B.; Nemir, M.; Beninati, S.; Cordella-Miele, E.; Singh, K.; Chackalaparampil, I.; Shanmugam, V.; DeVouge, M.W.; Mukherjee, A.B. Interaction of osteopontin with fibronectin and other extracellular matrix molecules. Ann. N. Y. Acad. Sci. 1995, 760, 201–212. [Google Scholar] [CrossRef]
- Kaartinen, M.T.; Pirhonen, A.; Linnala-Kankkunen, A.; Maenpaa, P.H. Cross-linking of osteopontin by tissue transglutaminase increases its collagen binding properties. J. Biol. Chem. 1999, 274, 1729–1735. [Google Scholar] [CrossRef] [Green Version]
- Giachelli, C.M.; Schwartz, S.M.; Liaw, L. Molecular and cellular biology of osteopontin Potential role in cardiovascular disease. Trends Cardiovasc. Med. 1995, 5, 88–95. [Google Scholar] [CrossRef]
- Thayer, J.M.; Giachelli, C.M.; Mirkes, P.E.; Schwartz, S.M. Expression of osteopontin in the head process late in gastrulation in the rat. J. Exp. Zool. 1995, 272, 240–244. [Google Scholar] [CrossRef]
- Graf, K.; Do, Y.S.; Ashizawa, N.; Meehan, W.P.; Giachelli, C.M.; Marboe, C.C.; Fleck, E.; Hsueh, W.A. Myocardial osteopontin expression is associated with left ventricular hypertrophy. Circulation 1997, 96, 3063–3071. [Google Scholar] [CrossRef]
- Collins, A.R.; Schnee, J.; Wang, W.; Kim, S.; Fishbein, M.C.; Bruemmer, D.; Law, R.E.; Nicholas, S.; Ross, R.S.; Hsueh, W.A. Osteopontin modulates angiotensin II-induced fibrosis in the intact murine heart. J. Am. Coll. Cardiol. 2004, 43, 1698–1705. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Foster, C.R.; Dalal, S.; Singh, K. Osteopontin: Role in extracellular matrix deposition and myocardial remodeling post-MI. J. Mol. Cell. Cardiol. 2010, 48, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Denhardt, D.T.; Guo, X. Osteopontin: A protein with diverse functions. FASEB J. 1993, 7, 1475–1482. [Google Scholar] [CrossRef]
- Williams, E.B.; Halpert, I.; Wickline, S.; Davison, G.; Parks, W.C.; Rottman, J.N. Osteopontin expression is increased in the heritable cardiomyopathy of Syrian hamsters. Circulation 1995, 92, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Giachelli, C.M.; Schwartz, S.M.; Vracko, R. Macrophages express osteopontin during repair of myocardial necrosis. Am. J. Pathol. 1994, 145, 1450–1462. [Google Scholar] [PubMed]
- Singh, K.; Balligand, J.L.; Fischer, T.A.; Smith, T.W.; Kelly, R.A. Glucocorticoids increase osteopontin expression in cardiac myocytes and microvascular endothelial cells. Role in regulation of inducible nitric oxide synthase. J. Biol. Chem. 1995, 270, 28471–28478. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Singh, M.; Singh, K. ERK1/2 and JNKs, but not p38 kinase, are involved in reactive oxygen species-mediated induction of osteopontin gene expression by angiotensin II and interleukin-1beta in adult rat cardiac fibroblasts. J. Cell Physiol. 2004, 198, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Lund, S.A.; Giachelli, C.M.; Scatena, M. The role of osteopontin in inflammatory processes. J. Cell Commun. Signal. 2009, 3, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sam, F.; Xie, Z.; Ooi, H.; Kerstetter, D.L.; Colucci, W.S.; Singh, M.; Singh, K. Mice lacking osteopontin exhibit increased left ventricular dilation and reduced fibrosis after aldosterone infusion. Am. J. Hypertens. 2004, 17, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Singh, M.; Singh, K. Osteopontin modulates myocardial hypertrophy in response to chronic pressure overload in mice. Hypertension 2004, 44, 826–831. [Google Scholar] [CrossRef] [Green Version]
- Psarras, S.; Mavroidis, M.; Sanoudou, D.; Davos, C.H.; Xanthou, G.; Varela, A.E.; Panoutsakopoulou, V.; Capetanaki, Y. Regulation of adverse remodelling by osteopontin in a genetic heart failure model. Eur. Heart J. 2012, 33, 1954–1963. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, V.; Krishnamurthy, P.; Singh, K.; Singh, M. Lack of osteopontin improves cardiac function in streptozotocin-induced diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H673–H683. [Google Scholar] [CrossRef]
- Trueblood, N.A.; Xie, Z.; Communal, C.; Sam, F.; Ngoy, S.; Liaw, L.; Jenkins, A.W.; Wang, J.; Sawyer, D.B.; Bing, O.H.; et al. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ. Res. 2001, 88, 1080–1087. [Google Scholar] [CrossRef] [Green Version]
- Duerr, G.D.; Mesenholl, B.; Heinemann, J.C.; Zoerlein, M.; Huebener, P.; Schneider, P.; Feisst, A.; Ghanem, A.; Tiemann, K.; Dewald, D.; et al. Cardioprotective effects of osteopontin-1 during development of murine ischemic cardiomyopathy. BioMed Res. Int. 2014, 2014, 124063. [Google Scholar] [CrossRef] [PubMed]
- Pollard, C.M.; Desimine, V.L.; Wertz, S.L.; Perez, A.; Parker, B.M.; Maning, J.; McCrink, K.A.; Shehadeh, L.A.; Lymperopoulos, A. Deletion of Osteopontin Enhances beta(2)-Adrenergic Receptor-Dependent Anti-Fibrotic Signaling in Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Zhang, D.; Qiu, H.; Yi, X.; Zhang, Y.; Cao, Y.; Zhao, B.; Xia, Z.; Wang, C. Tiron ameliorates high glucose-induced cardiac myocyte apoptosis by PKCdelta-dependent inhibition of osteopontin. Clin. Exp. Pharmacol. Physiol. 2017, 44, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Pimental, D.R.; Lohan, S.; Vasertriger, A.; Pligavko, C.; Colucci, W.S.; Singh, K. Regulation of angiotensin II-stimulated osteopontin expression in cardiac microvascular endothelial cells: Role of p42/44 mitogen-activated protein kinase and reactive oxygen species. J. Cell Physiol. 2001, 188, 132–138. [Google Scholar] [CrossRef]
- Abdulrahman, N.; Jaspard-Vinassa, B.; Fliegel, L.; Jabeen, A.; Riaz, S.; Gadeau, A.P.; Mraiche, F. Na(+)/H(+) exchanger isoform 1-induced osteopontin expression facilitates cardiac hypertrophy through p90 ribosomal S6 kinase. Physiol. Genom. 2018, 50, 332–342. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, I.A.; Gadeau, A.P.; Fliegel, L.; Lopaschuk, G.; Mlih, M.; Abdulrahman, N.; Fillmore, N.; Mraiche, F. Na+/H+ exchanger isoform 1-induced osteopontin expression facilitates cardiomyocyte hypertrophy. PLoS ONE 2015, 10, e0123318. [Google Scholar] [CrossRef]
- Mlih, M.; Abdulrahman, N.; Gadeau, A.P.; Mohamed, I.A.; Jaballah, M.; Mraiche, F. Na(+)/H (+) exchanger isoform 1 induced osteopontin expression in cardiomyocytes involves NFAT3/Gata4. Mol. Cell. Biochem. 2015, 404, 211–220. [Google Scholar] [CrossRef]
- Dalal, S.; Zha, Q.; Daniels, C.R.; Steagall, R.J.; Joyner, W.L.; Gadeau, A.P.; Singh, M.; Singh, K. Osteopontin stimulates apoptosis in adult cardiac myocytes via the involvement of CD44 receptors, mitochondrial death pathway, and endoplasmic reticulum stress. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1182–H1191. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, K.; Irion, C.I.; Takeuchi, L.M.; Ding, W.; Lambert, G.; Eisenberg, T.; Sukkar, S.; Granzier, H.L.; Methawasin, M.; Lee, D.I.; et al. Osteopontin Promotes Left Ventricular Diastolic Dysfunction Through a Mitochondrial Pathway. J. Am. Coll. Cardiol. 2019, 73, 2705–2718. [Google Scholar] [CrossRef]
- Cabiati, M.; Svezia, B.; Matteucci, M.; Botta, L.; Pucci, A.; Rinaldi, M.; Caselli, C.; Lionetti, V.; Del Ry, S. Myocardial Expression Analysis of Osteopontin and Its Splice Variants in Patients Affected by End-Stage Idiopathic or Ischemic Dilated Cardiomyopathy. PLoS ONE 2016, 11, e0160110. [Google Scholar] [CrossRef] [Green Version]
- Sawaki, D.; Czibik, G.; Pini, M.; Ternacle, J.; Suffee, N.; Mercedes, R.; Marcelin, G.; Surenaud, M.; Marcos, E.; Gual, P.; et al. Visceral Adipose Tissue Drives Cardiac Aging Through Modulation of Fibroblast Senescence by Osteopontin Production. Circulation 2018, 138, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Lenga, Y.; Koh, A.; Perera, A.S.; McCulloch, C.A.; Sodek, J.; Zohar, R. Osteopontin expression is required for myofibroblast differentiation. Circ. Res. 2008, 102, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Herum, K.M.; Lunde, I.G.; Skrbic, B.; Louch, W.E.; Hasic, A.; Boye, S.; Unger, A.; Brorson, S.H.; Sjaastad, I.; Tonnessen, T.; et al. Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc. Res. 2015, 106, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Herum, K.M.; Romaine, A.; Wang, A.; Melleby, A.O.; Strand, M.E.; Pacheco, J.; Braathen, B.; Duner, P.; Tonnessen, T.; Lunde, I.G.; et al. Syndecan-4 Protects the Heart From the Profibrotic Effects of Thrombin-Cleaved Osteopontin. J. Am. Heart Assoc. 2020, 9, e013518. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, W.; Zhang, J.; Liang, T.; Fan, G.P.; Wang, Z.W.; Zhang, P.D.; Wang, X.; Zhang, J. Inhibition of osteopontin reduce the cardiac myofibrosis in dilated cardiomyopathy via focal adhesion kinase mediated signaling pathway. Am. J. Transl. Res. 2016, 8, 3645–3655. [Google Scholar] [PubMed]
- Ashizawa, N.; Graf, K.; Do, Y.S.; Nunohiro, T.; Giachelli, C.M.; Meehan, W.P.; Tuan, T.L.; Hsueh, W.A. Osteopontin is produced by rat cardiac fibroblasts and mediates A(II)-induced DNA synthesis and collagen gel contraction. J. Clin. Investig. 1996, 98, 2218–2227. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Singh, M.; Siwik, D.A.; Joyner, W.L.; Singh, K. Osteopontin inhibits interleukin-1beta-stimulated increases in matrix metalloproteinase activity in adult rat cardiac fibroblasts: Role of protein kinase C-zeta. J. Biol. Chem. 2003, 278, 48546–48552. [Google Scholar] [CrossRef] [Green Version]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M.; et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005, 2, e251. [Google Scholar] [CrossRef] [Green Version]
- Lopez, B.; Gonzalez, A.; Lindner, D.; Westermann, D.; Ravassa, S.; Beaumont, J.; Gallego, I.; Zudaire, A.; Brugnolaro, C.; Querejeta, R.; et al. Osteopontin-mediated myocardial fibrosis in heart failure: A role for lysyl oxidase? Cardiovasc. Res. 2013, 99, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Peng, L.; Fan, K.; Wang, H.; Wei, R.; Ji, G.; Cai, J.; Lu, B.; Li, B.; Zhang, D.; et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene 2009, 28, 3412–3422. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Lopez-Chua, C.A.; Zhang, J.; Fisher, L.W.; Sorensen, E.S.; Denhardt, D.T. Soluble osteopontin inhibits apoptosis of adherent endothelial cells deprived of growth factors. J. Cell. Biochem. 2002, 85, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Scatena, M.; Almeida, M.; Chaisson, M.L.; Fausto, N.; Nicosia, R.F.; Giachelli, C.M. NF-kappaB mediates alphavbeta3 integrin-induced endothelial cell survival. J. Cell Biol. 1998, 141, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Liaw, L.; Lindner, V.; Schwartz, S.M.; Chambers, A.F.; Giachelli, C.M. Osteopontin and beta 3 integrin are coordinately expressed in regenerating endothelium in vivo and stimulate Arg-Gly-Asp-dependent endothelial migration in vitro. Circ. Res. 1995, 77, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Johnson, J.N.; Singh, K.; Singh, M. Impairment of myocardial angiogenic response in the absence of osteopontin. Microcirculation 2007, 14, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Rittling, S.R. Osteopontin in macrophage function. Expert Rev. Mol. Med. 2011, 13, e15. [Google Scholar] [CrossRef] [PubMed]
- Krause, S.W.; Rehli, M.; Kreutz, M.; Schwarzfischer, L.; Paulauskis, J.D.; Andreesen, R. Differential screening identifies genetic markers of monocyte to macrophage maturation. J. Leukoc. Biol. 1996, 60, 540–545. [Google Scholar] [CrossRef]
- Komatsubara, I.; Murakami, T.; Kusachi, S.; Nakamura, K.; Hirohata, S.; Hayashi, J.; Takemoto, S.; Suezawa, C.; Ninomiya, Y.; Shiratori, Y. Spatially and temporally different expression of osteonectin and osteopontin in the infarct zone of experimentally induced myocardial infarction in rats. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2003, 12, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yoshida, N.; Yamamoto, T.; Isobe, S.; Moriyama, H.; Goto, S.; Kitakata, H.; et al. IL (Interleukin)-10-STAT3-Galectin-3 Axis Is Essential for Osteopontin-Producing Reparative Macrophage Polarization After Myocardial Infarction. Circulation 2018, 138, 2021–2035. [Google Scholar] [CrossRef]
- Shirakawa, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Anzai, A.; Moriyama, H.; Kitakata, H.; Hiraide, T.; Ko, S.; Goto, S.; et al. MerTK Expression and ERK Activation Are Essential for the Functional Maturation of Osteopontin-Producing Reparative Macrophages After Myocardial Infarction. J. Am. Heart Assoc. 2020, 9, e017071. [Google Scholar] [CrossRef]
- Frey, N.; Katus, H.A.; Olson, E.N.; Hill, J.A. Hypertrophy of the heart: A new therapeutic target? Circulation 2004, 109, 1580–1589. [Google Scholar] [CrossRef] [Green Version]
- Ndisang, J.F.; Chibbar, R.; Lane, N. Heme oxygenase suppresses markers of heart failure and ameliorates cardiomyopathy in L-NAME-induced hypertension. Eur. J. Pharmacol. 2014, 734, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Sirokman, G.; Communal, C.; Robinson, K.G.; Conrad, C.H.; Brooks, W.W.; Bing, O.H.; Colucci, W.S. Myocardial osteopontin expression coincides with the development of heart failure. Hypertension 1999, 33, 663–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Weisheit, C.; Zhang, Y.; Faron, A.; Köpke, O.; Weisheit, G.; Steinsträsser, A.; Frede, S.; Meyer, R.; Boehm, O.; Hoeft, A.; et al. Ly6C(low) and not Ly6C(high) macrophages accumulate first in the heart in a model of murine pressure-overload. PLoS ONE 2014, 9, e112710. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.H.; Zhou, X.; Ji, W.J.; Zeng, S.; Dong, Y.; Tian, L.; Bi, Y.; Guo, Z.Z.; Gao, F.; Chen, H.; et al. Overexpression of VEGF-C attenuates chronic high salt intake-induced left ventricular maladaptive remodeling in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H598–H609. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, J.M.; Schauerte, C.; Hubner, A.; Kolling, M.; Martino, F.; Scherf, K.; Batkai, S.; Zimmer, K.; Foinquinos, A.; Kaucsar, T.; et al. Osteopontin is indispensible for AP1-mediated angiotensin II-related miR-21 transcription during cardiac fibrosis. Eur. Heart J. 2015, 36, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Soejima, H.; Irie, A.; Fukunaga, T.; Oe, Y.; Kojima, S.; Kaikita, K.; Kawano, H.; Sugiyama, S.; Yoshimura, M.; Kishikawa, H.; et al. Osteopontin expression of circulating T cells and plasma osteopontin levels are increased in relation to severity of heart failure. Circ. J. Off. J. Jpn. Circ. Soc. 2007, 71, 1879–1884. [Google Scholar] [CrossRef] [Green Version]
- Soejima, H.; Irie, A.; Fukunaga, T.; Sugamura, K.; Kojima, S.; Sakamoto, T.; Yoshimura, M.; Kishikawa, H.; Nishimura, Y.; Ogawa, H. Elevated plasma osteopontin levels were associated with osteopontin expression of CD4+ T cells in patients with unstable angina. Circ. J. Off. J. Jpn. Circ. Soc. 2006, 70, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Van Linthout, S.; Miteva, K.; Tschöpe, C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc. Res. 2014, 102, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Renault, M.A.; Robbesyn, F.; Reant, P.; Douin, V.; Daret, D.; Allieres, C.; Belloc, I.; Couffinhal, T.; Arnal, J.F.; Klingel, K.; et al. Osteopontin expression in cardiomyocytes induces dilated cardiomyopathy. Circ. Heart Fail. 2010, 3, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, M.; Nakamura, M.; Akatsu, T.; Shimoda, Y.; Segawa, I.; Hiramori, K. Myocardial osteopontin expression is associated with collagen fibrillogenesis in human dilated cardiomyopathy. Eur. J. Heart Fail. 2005, 7, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, S.; Rickelt, S.; Soriano, M.E.; Schips, T.G.; Maier, H.J.; Davos, C.H.; Varela, A.; Kaklamanis, L.; Mann, D.L.; Capetanaki, Y. Tumor necrosis factor-alpha confers cardioprotection through ectopic expression of keratins K8 and K18. Nat. Med. 2015, 21, 1076–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Matsui, T.; Abel, E.D.; Dedhar, S.; Gerszten, R.E.; Seidman, C.E.; Seidman, J.G.; Rosenzweig, A. Deep sequence analysis of gene expression identifies osteopontin as a downstream effector of integrin-linked kinase (ILK) in cardiac-specific ILK knockout mice. Circ. Heart Fail. 2014, 7, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Caballero, E.P.; Santamaria, M.H.; Corral, R.S. Endogenous osteopontin induces myocardial CCL5 and MMP-2 activation that contributes to inflammation and cardiac remodeling in a mouse model of chronic Chagas heart disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 11–23. [Google Scholar] [CrossRef]
- Szalay, G.; Sauter, M.; Haberland, M.; Zuegel, U.; Steinmeyer, A.; Kandolf, R.; Klingel, K. Osteopontin: A fibrosis-related marker molecule in cardiac remodeling of enterovirus myocarditis in the susceptible host. Circ. Res. 2009, 104, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Berman, J.S.; Serlin, D.; Li, X.; Whitley, G.; Hayes, J.; Rishikof, D.C.; Ricupero, D.A.; Liaw, L.; Goetschkes, M.; O’Regan, A.W. Altered bleomycin-induced lung fibrosis in osteopontin-deficient mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L1311–L1318. [Google Scholar] [CrossRef] [Green Version]
- Leung, T.M.; Wang, X.; Kitamura, N.; Fiel, M.I.; Nieto, N. Osteopontin delays resolution of liver fibrosis. Lab. Investig. A J. Tech. Methods Pathol. 2013, 93, 1082–1089. [Google Scholar] [CrossRef]
- Sahai, A.; Malladi, P.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Upregulation of osteopontin expression is involved in the development of nonalcoholic steatohepatitis in a dietary murine model. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G264–G273. [Google Scholar] [CrossRef] [Green Version]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Peterson, J.T.; Subramanian, V.; Singh, M.; Singh, K. Inhibition of matrix metalloproteinases improves left ventricular function in mice lacking osteopontin after myocardial infarction. Mol. Cell. Biochem. 2009, 322, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahiya, S.; Givvimani, S.; Bhatnagar, S.; Qipshidze, N.; Tyagi, S.C.; Kumar, A. Osteopontin-stimulated expression of matrix metalloproteinase-9 causes cardiomyopathy in the mdx model of Duchenne muscular dystrophy. J. Immunol. 2011, 187, 2723–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagoshi, S. Osteopontin: Versatile modulator of liver diseases. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2014, 44, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Schneider, D.J.; Mayes, M.D.; Assassi, S.; Arnett, F.C.; Tan, F.K.; Blackburn, M.R.; Agarwal, S.K. Osteopontin in systemic sclerosis and its role in dermal fibrosis. J. Investig. Dermatol. 2012, 132, 1605–1614. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yousefi, K.; Ding, W.; Singh, J.; Shehadeh, L.A. Osteopontin RNA aptamer can prevent and reverse pressure overload-induced heart failure. Cardiovasc. Res. 2017, 113, 633–643. [Google Scholar] [CrossRef]
- Fan, D.; Takawale, A.; Basu, R.; Patel, V.; Lee, J.; Kandalam, V.; Wang, X.; Oudit, G.Y.; Kassiri, Z. Differential role of TIMP2 and TIMP3 in cardiac hypertrophy, fibrosis, and diastolic dysfunction. Cardiovasc. Res. 2014, 103, 268–280. [Google Scholar] [CrossRef] [Green Version]
- Engebretsen, K.V.; Skardal, K.; Bjornstad, S.; Marstein, H.S.; Skrbic, B.; Sjaastad, I.; Christensen, G.; Bjornstad, J.L.; Tonnessen, T. Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J. Mol. Cell. Cardiol. 2014, 76, 148–157. [Google Scholar] [CrossRef]
- Ren, G.; Michael, L.H.; Entman, M.L.; Frangogiannis, N.G. Morphological characteristics of the microvasculature in healing myocardial infarcts. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2002, 50, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Gogiraju, R.; Bochenek, M.L.; Schafer, K. Angiogenic Endothelial Cell Signaling in Cardiac Hypertrophy and Heart Failure. Front. Cardiovasc. Med. 2019, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Raja, R.; Kale, S.; Thorat, D.; Soundararajan, G.; Lohite, K.; Mane, A.; Karnik, S.; Kundu, G.C. Hypoxia-driven osteopontin contributes to breast tumor growth through modulation of HIF1alpha-mediated VEGF-dependent angiogenesis. Oncogene 2014, 33, 2053–2064. [Google Scholar] [CrossRef] [Green Version]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Stawowy, P.; Blaschke, F.; Pfautsch, P.; Goetze, S.; Lippek, F.; Wollert-Wulf, B.; Fleck, E.; Graf, K. Increased myocardial expression of osteopontin in patients with advanced heart failure. Eur. J. Heart Fail. 2002, 4, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, H. Osteopontin and cardiovascular system. Mol. Cell. Biochem. 2007, 300, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schipper, M.E.; Scheenstra, M.R.; van Kuik, J.; van Wichen, D.F.; van der Weide, P.; Dullens, H.F.; Lahpor, J.; de Jonge, N.; De Weger, R.A. Osteopontin: A potential biomarker for heart failure and reverse remodeling after left ventricular assist device support. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2011, 30, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Podzimkova, J.; Palecek, T.; Kuchynka, P.; Marek, J.; Danek, B.A.; Jachymova, M.; Safarikova, M.; Kalousova, M.; Zima, T.; Linhart, A. Plasma osteopontin levels, but not its myocardial expression, reflect heart failure severity in recently diagnosed dilated cardiomyopathy. Herz 2020, 45 (Suppl. S1), 105–110. [Google Scholar] [CrossRef]
- Podzimkova, J.; Palecek, T.; Kuchynka, P.; Marek, J.; Danek, B.A.; Jachymova, M.; Kalousova, M.; Zima, T.; Linhart, A. Plasma osteopontin levels in patients with dilated and hypertrophic cardiomyopathy. Herz 2019, 44, 347–353. [Google Scholar] [CrossRef]
- Rosenberg, M.; Zugck, C.; Nelles, M.; Juenger, C.; Frank, D.; Remppis, A.; Giannitsis, E.; Katus, H.A.; Frey, N. Osteopontin, a new prognostic biomarker in patients with chronic heart failure. Circ. Heart Fail. 2008, 1, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Rubis, P.; Wisniowska-Smialek, S.; Dziewiecka, E.; Rudnicka-Sosin, L.; Kozanecki, A.; Podolec, P. Prognostic value of fibrosis-related markers in dilated cardiomyopathy: A link between osteopontin and cardiovascular events. Adv. Med. Sci. 2018, 63, 160–166. [Google Scholar] [CrossRef]
- Yu, Q.; Vazquez, R.; Khojeini, E.V.; Patel, C.; Venkataramani, R.; Larson, D.F. IL-18 induction of osteopontin mediates cardiac fibrosis and diastolic dysfunction in mice. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H76–H85. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, Y.; Gao, P.J. Osteopontin associated with left ventricular hypertrophy and diastolic dysfunction in essential hypertension. J. Hum. Hypertens. 2020, 34, 388–396. [Google Scholar] [CrossRef]
- Hou, X.; Hu, Z.; Huang, X.; Chen, Y.; He, X.; Xu, H.; Wang, N. Serum osteopontin, but not OPN gene polymorphism, is associated with LVH in essential hypertensive patients. J. Mol. Med. 2014, 92, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.; von Ingersleben, N.; Lambers, M.; Rosenberg, M.; Freitag-Wolf, S.; Dempfle, A.; Lutter, G.; Frank, J.; Bramlage, P.; Frey, N.; et al. Osteopontin predicts clinical outcome in patients after treatment of severe aortic stenosis with transcatheter aortic valve implantation (TAVI). Open Heart 2017, 4, e000633. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Alpert, L.B.; Rellecke, P.; Petrov, G.; Albert, A.; Sixt, S.U.; Lichtenberg, A.; Akhyari, P. Osteopontin as novel biomarker for reversibility of pressure overload induced left ventricular hypertrophy. Biomark. Med. 2020, 14, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D.; Executive Group on behalf of the Joint European Society of Cardiology/American College of Cardiology/American Heart Association /World Heart Federation Task Force for the Universal Definition of Myocardial Infarction. Fourth Universal Definition of Myocardial Infarction (2018). Glob. Heart 2018, 13, 305–338. [Google Scholar] [CrossRef]
- Tamura, A.; Shingai, M.; Aso, N.; Hazuku, T.; Nasu, M. Osteopontin is released from the heart into the coronary circulation in patients with a previous anterior wall myocardial infarction. Circ. J. Off. J. Jpn. Circ. Soc. 2003, 67, 742–744. [Google Scholar] [CrossRef] [Green Version]
- Belal, S.; Farouk, W.; Khaled, M.; Abdelbary, A. Role of Osteopontin in Predicting Short-Term Major Adverse Cardiac Events and Infarction Size in STEMI. Egypt. J. Crit. Care Med. 2021, 8, 34–37. [Google Scholar]
- Bjerre, M.; Pedersen, S.H.; Mogelvang, R.; Lindberg, S.; Jensen, J.S.; Galatius, S.; Flyvbjerg, A. High osteopontin levels predict long-term outcome after STEMI and primary percutaneous coronary intervention. Eur. J. Prev. Cardiol. 2013, 20, 922–929. [Google Scholar] [CrossRef]
- Santhanakrishnan, R.; Wang, N.; Larson, M.G.; Magnani, J.W.; McManus, D.D.; Lubitz, S.A.; Ellinor, P.T.; Cheng, S.; Vasan, R.S.; Lee, D.S.; et al. Atrial Fibrillation Begets Heart Failure and Vice Versa: Temporal Associations and Differences in Preserved Versus Reduced Ejection Fraction. Circulation 2016, 133, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Bergau, L.; Bengel, P.; Sciacca, V.; Fink, T.; Sohns, C.; Sommer, P. Atrial Fibrillation and Heart Failure. J. Clin. Med. 2022, 11, 2510. [Google Scholar] [CrossRef]
- Anter, E.; Jessup, M.; Callans, D.J. Atrial fibrillation and heart failure: Treatment considerations for a dual epidemic. Circulation 2009, 119, 2516–2525. [Google Scholar] [CrossRef] [Green Version]
- Cunha, P.S.; Laranjo, S.; Heijman, J.; Oliveira, M.M. The Atrium in Atrial Fibrillation—A Clinical Review on How to Manage Atrial Fibrotic Substrates. Front. Cardiovasc. Med. 2022, 9, 879984. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.H.; Schotten, U.; Mahajan, R.; Antic, N.A.; Hatem, S.N.; Pathak, R.K.; Hendriks, J.M.; Kalman, J.M.; Sanders, P. Novel mechanisms in the pathogenesis of atrial fibrillation: Practical applications. Eur. Heart J. 2016, 37, 1573–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Liu, T.; Shen, C.; He, B.; Feng, M.; Liu, J.; Zhuo, W.; Fu, G.; Wang, B.; Xu, Y.; et al. Anti-fibrotic mechanism of SPP1 knockdown in atrial fibrosis associates with inhibited mitochondrial DNA damage and TGF-β/SREBP2/PCSK9 signaling. Cell Death Discov. 2022, 8, 246. [Google Scholar] [CrossRef]
- Künzel, S.R.; Hoffmann, M.; Weber, S.; Künzel, K.; Kämmerer, S.; Günscht, M.; Klapproth, E.; Rausch, J.S.E.; Sadek, M.S.; Kolanowski, T.; et al. Diminished PLK2 Induces Cardiac Fibrosis and Promotes Atrial Fibrillation. Circ. Res. 2021, 129, 804–820. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Wu, S.; Zhu, D.; Qin, M.; Liu, X. Osteopontin induces atrial fibrosis by activating Akt/GSK-3β/β-catenin pathway and suppressing autophagy. Life Sci. 2020, 245, 117328. [Google Scholar] [CrossRef] [PubMed]
- Molvin, J.; Jujic, A.; Melander, O.; Pareek, M.; Råstam, L.; Lindblad, U.; Daka, B.; Leosdottir, M.; Nilsson, P.; Olsen, M.; et al. Exploration of pathophysiological pathways for incident atrial fibrillation using a multiplex proteomic chip. Open Heart 2020, 7, e001190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Güneş, H.M.; Babur Güler, G.; Güler, E.; Demir, G.G.; Kızılırmak Yılmaz, F.; Omaygenç, M.O.; İstanbullu Tosun, A.; Akgün, T.; Boztosun, B.; Kılıçarslan, F. Relationship between serum osteopontin level and atrial fibrillation recurrence in patients undergoing cryoballoon catheter ablation. Turk Kardiyol. Dern. Ars. Turk Kardiyol. Dern. Yayin. Organidir 2017, 45, 26–32. [Google Scholar] [CrossRef]
- Hijazi, Z.; Wallentin, L.; Lindbäck, J.; Alexander, J.H.; Connolly, S.J.; Eikelboom, J.W.; Ezekowitz, M.D.; Granger, C.B.; Lopes, R.D.; Pol, T.; et al. Screening of Multiple Biomarkers Associated With Ischemic Stroke in Atrial Fibrillation. J. Am. Heart Assoc. 2020, 9, e018984. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, A.; Lindbäck, J.; Hijazi, Z.; Åberg, M.; Alexander, J.H.; Eikelboom, J.W.; Lopes, R.D.; Pol, T.; Oldgren, J.; Granger, C.B.; et al. Multiplex protein screening of biomarkers associated with major bleeding in patients with atrial fibrillation treated with oral anticoagulation. J. Thromb. Haemost. JTH 2021, 19, 2726–2737. [Google Scholar] [CrossRef] [PubMed]
- Adamo, M.; Alos, B.; Metra, M.; Lefèvre, T.; Swaans, M.J.; Gheorghe, L.; Tschöpe, C.; Krackhardt, F.; Alfieri, O.; Bouleti, C. Patient with heart failure: Importance to treat valvular diseases. Eur. Heart J. Suppl. J. Eur. Soc. Cardiol. 2020, 22, p38–p41. [Google Scholar] [CrossRef]
- Lok, Z.S.Y.; Lyle, A.N. Osteopontin in Vascular Disease. Arter. Thromb. Vasc. Biol. 2019, 39, 613–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atalar, E.; Ozturk, E.; Ozer, N.; Haznedaroglu, I.C.; Kepez, A.; Coskun, S.; Aksoyek, S.; Ovunc, K.; Kes, S.; Kirazli, S.; et al. Plasma soluble osteopontin concentrations are increased in patients with rheumatic mitral stenosis and associated with the severity of mitral valve calcium. Am. J. Cardiol. 2006, 98, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Sponder, M.; Reuter, C.; Fritzer-Szekeres, M.; Litschauer, B.; Binder, T.; Strametz-Juranek, J. Osteopontin is elevated in patients with mitral annulus calcification independent from classic cardiovascular risk factors. BMC Cardiovasc. Disord. 2016, 16, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, G.; Sainger, R.; Beckmann, E.; Keller, G.; Yu, P.J.; Monti, M.C.; Galloway, A.C.; Weiss, R.L.; Vernick, W.; Grau, J.B. Validation of plasma biomarkers in degenerative calcific aortic stenosis. J. Surg. Res. 2010, 163, 12–17. [Google Scholar] [CrossRef]
- Yu, P.J.; Skolnick, A.; Ferrari, G.; Heretis, K.; Mignatti, P.; Pintucci, G.; Rosenzweig, B.; Diaz-Cartelle, J.; Kronzon, I.; Perk, G.; et al. Correlation between plasma osteopontin levels and aortic valve calcification: Potential insights into the pathogenesis of aortic valve calcification and stenosis. J. Thorac. Cardiovasc. Surg. 2009, 138, 196–199. [Google Scholar] [CrossRef] [Green Version]
- Grau, J.B.; Poggio, P.; Sainger, R.; Vernick, W.J.; Seefried, W.F.; Branchetti, E.; Field, B.C.; Bavaria, J.E.; Acker, M.A.; Ferrari, G. Analysis of osteopontin levels for the identification of asymptomatic patients with calcific aortic valve disease. Ann. Thorac. Surg. 2012, 93, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Canver, C.C.; Gregory, R.D.; Cooler, S.D.; Voytovich, M.C. Association of osteopontin with calcification in human mitral valves. J. Cardiovasc. Surg. 2000, 41, 171–174. [Google Scholar]
- Kennedy, J.H.; Henrion, D.; Wassef, M.; Shanahan, C.M.; Bloch, G.; Tedgui, A. Osteopontin expression and calcium content in human aortic valves. J. Thorac. Cardiovasc. Surg. 2000, 120, 427. [Google Scholar] [CrossRef] [Green Version]
- Breyne, J.; Juthier, F.; Corseaux, D.; Marechaux, S.; Zawadzki, C.; Jeanpierre, E.; Ung, A.; Ennezat, P.V.; Susen, S.; Van Belle, E.; et al. Atherosclerotic-like process in aortic stenosis: Activation of the tissue factor-thrombin pathway and potential role through osteopontin alteration. Atherosclerosis 2010, 213, 369–376. [Google Scholar] [CrossRef]
- Kazimli, A.V.; Ryzhkov, A.V.; Goncharova, N.S.; Naymushin, A.V.; Moiseeva, O.M. Myeloperoxidase, osteopontin and asymmetrical dimethylarginine as biomarkers of pulmonary hypertension severity. Eur. Heart J. 2013, 34 (Suppl. S1), P332. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, J.M.; Nickel, N.; Kramer, R.; Golpon, H.; Westerkamp, V.; Olsson, K.M.; Haller, H.; Hoeper, M.M. Osteopontin in patients with idiopathic pulmonary hypertension. Chest 2011, 139, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.; Meyer, F.J.; Gruenig, E.; Schuster, T.; Lutz, M.; Lossnitzer, D.; Wipplinger, R.; Katus, H.A.; Frey, N. Osteopontin (OPN) improves risk stratification in pulmonary hypertension (PH). Int. J. Cardiol. 2012, 155, 504–505. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.; Wharton, J.; Howard, L.; Gibbs, J.; Wilkins, M. S96 Novel biomarkers in idiopathic pulmonary arterial hypertension. Thorax 2010, 65 (Suppl. S1), A44. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, M.; Meyer, F.J.; Gruenig, E.; Lutz, M.; Lossnitzer, D.; Wipplinger, R.; Katus, H.A.; Frey, N. Osteopontin predicts adverse right ventricular remodelling and dysfunction in pulmonary hypertension. Eur. J. Clin. Investig. 2012, 42, 933–942. [Google Scholar] [CrossRef]
- Keranov, S.; Dörr, O.; Jafari, L.; Liebetrau, C.; Keller, T.; Troidl, C.; Riehm, J.; Rutsatz, W.; Bauer, P.; Kriechbaum, S.; et al. Osteopontin and galectin-3 as biomarkers of maladaptive right ventricular remodeling in pulmonary hypertension. Biomark. Med. 2021, 15, 1021–1034. [Google Scholar] [CrossRef]
- Nadadur, R.D.; Umar, S.; Wong, G.; Eghbali, M.; Iorga, A.; Matori, H.; Partow-Navid, R.; Eghbali, M. Reverse right ventricular structural and extracellular matrix remodeling by estrogen in severe pulmonary hypertension. J. Appl. Physiol. 2012, 113, 149–158. [Google Scholar] [CrossRef]
- Imoto, K.; Okada, M.; Yamawaki, H. Expression profile of matricellular proteins in hypertrophied right ventricle of monocrotaline-induced pulmonary hypertensive rats. J. Vet. Med. Sci. 2017, 79, 1096–1102. [Google Scholar] [CrossRef] [Green Version]
- Bogaard, H.J.; Natarajan, R.; Henderson, S.C.; Long, C.S.; Kraskauskas, D.; Smithson, L.; Ockaili, R.; McCord, J.M.; Voelkel, N.F. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009, 120, 1951–1960. [Google Scholar] [CrossRef] [Green Version]
- Klusonova, P.; Rehakova, L.; Borchert, G.; Vagnerova, K.; Neckar, J.; Ergang, P.; Miksik, I.; Kolar, F.; Pacha, J. Chronic intermittent hypoxia induces 11beta-hydroxysteroid dehydrogenase in rat heart. Endocrinology 2009, 150, 4270–4277. [Google Scholar] [CrossRef]
- Behringer, A.; Trappiel, M.; Berghausen, E.M.; Ten Freyhaus, H.; Wellnhofer, E.; Odenthal, M.; Blaschke, F.; Er, F.; Gassanov, N.; Rosenkranz, S.; et al. Pioglitazone alleviates cardiac and vascular remodelling and improves survival in monocrotaline induced pulmonary arterial hypertension. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2016, 389, 369–379. [Google Scholar] [CrossRef]
- Byun, J. Recent Progress and Opportunities for Nucleic Acid Aptamers. Life 2021, 11, 193. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Chen, J.; Wu, Z.; Huang, H.; Wang, L.; Liang, Y.; Zeng, P.; Yang, J.; Uede, T.; Diao, H. Thrombin cleavage of osteopontin controls activation of hepatic stellate cells and is essential for liver fibrogenesis. J. Cell Physiol. 2019, 234, 8988–8997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, Y.; Nokihara, K.; Okazaki, M.; Fujitani, W.; Matsumoto, T.; Matsuo, M.; Umakoshi, Y.; Takahashi, J.; Matsuura, N. Angiogenic activity of osteopontin-derived peptide SVVYGLR. Biochem. Biophys. Res. Commun. 2003, 310, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Egusa, H.; Kaneda, Y.; Hirata, I.; Kawaguchi, N.; Hirao, T.; Matsumoto, T.; Yao, M.; Daito, K.; Suzuki, M.; et al. Synthetic osteopontin-derived peptide SVVYGLR can induce neovascularization in artificial bone marrow scaffold biomaterials. Dent. Mater. J. 2007, 26, 487–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, Y.; Uchinaka, A.; Horii, Y.; Mori, S.; Hamada, Y.; Miyagawa, S.; Saito, A.; Sawa, Y.; Matsuura, N.; Kawaguchi, N. Improvement of cardiac function after implanting the osteopontin-derived peptide SVVYGLR in a hamster model of dilated cardiomyopathy. Interact. Cardiovasc. Thorac. Surg. 2015, 21, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Sbarouni, E.; Georgiadou, P.; Mihas, C.; Chaidaroglou, A.; Degiannis, D.; Voudris, V. Significant peri-operative reduction in plasma osteopontin levels after coronary artery by-pass grafting. Clin. Biochem. 2012, 45, 1513–1515. [Google Scholar] [CrossRef]
- Inoue, M.; Shinohara, M.L. Intracellular osteopontin (iOPN) and immunity. Immunol. Res. 2011, 49, 160–172. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamazhakypov, A.; Sartmyrzaeva, M.; Sarybaev, A.S.; Schermuly, R.; Sydykov, A. Clinical and Molecular Implications of Osteopontin in Heart Failure. Curr. Issues Mol. Biol. 2022, 44, 3573-3597. https://doi.org/10.3390/cimb44080245
Mamazhakypov A, Sartmyrzaeva M, Sarybaev AS, Schermuly R, Sydykov A. Clinical and Molecular Implications of Osteopontin in Heart Failure. Current Issues in Molecular Biology. 2022; 44(8):3573-3597. https://doi.org/10.3390/cimb44080245
Chicago/Turabian StyleMamazhakypov, Argen, Meerim Sartmyrzaeva, Akpay Sh. Sarybaev, Ralph Schermuly, and Akylbek Sydykov. 2022. "Clinical and Molecular Implications of Osteopontin in Heart Failure" Current Issues in Molecular Biology 44, no. 8: 3573-3597. https://doi.org/10.3390/cimb44080245