
Multi-Omics Integration and Network Analysis Reveal Potential Hub Genes and Genetic Mechanisms Regulating Bovine Mastitis


 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
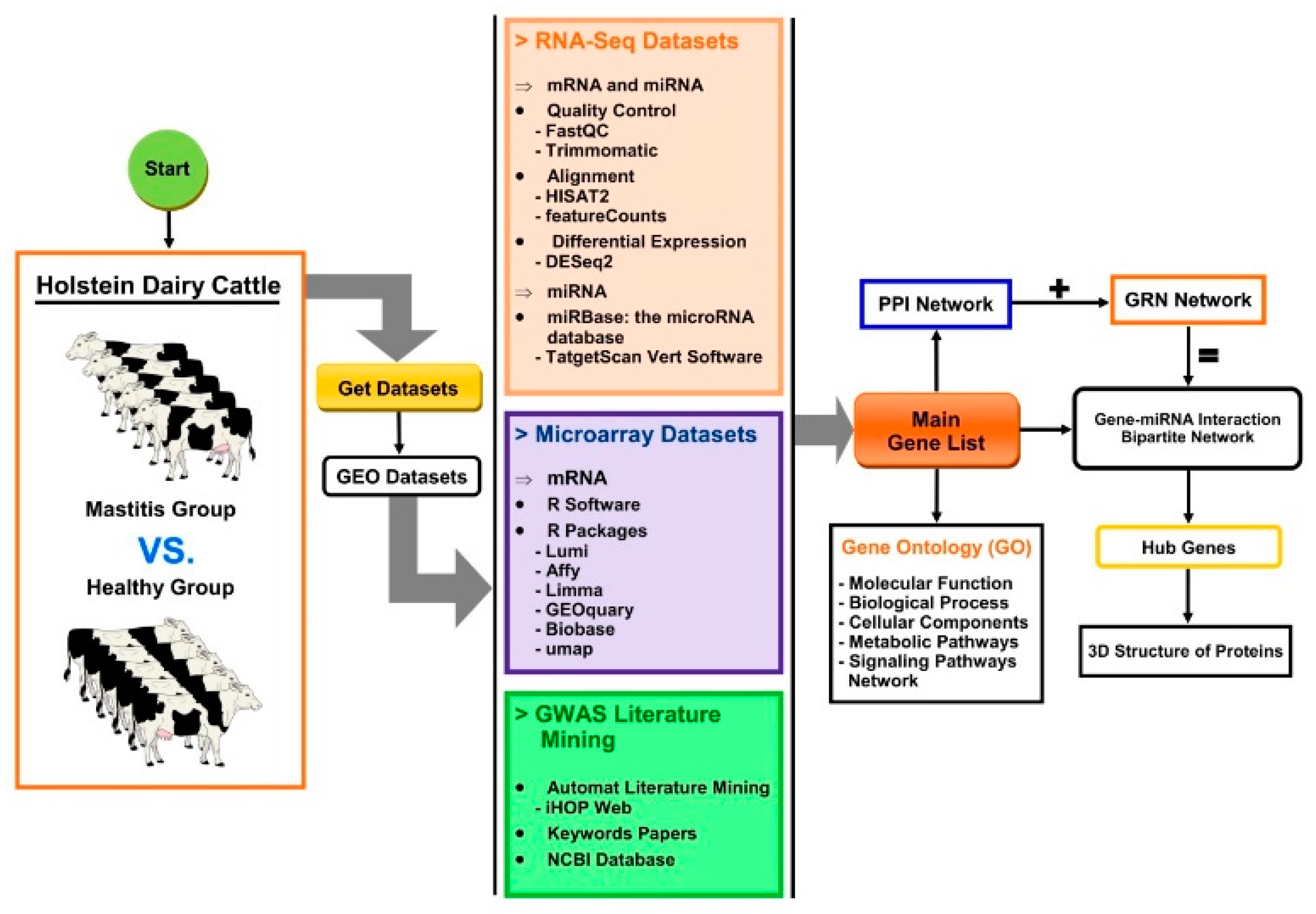
2. Materials and Methods
2.1. Data Collection
2.2. Differential Gene Expression Analysis
2.3. Literature Mining to Discover Candidate Genes for Mastitis
2.4. Determination of Main Gene List
2.5. Functional Enrichment and KEGG Pathway Analysis
2.6. Identification of miRNAs and Target Gene Prediction
2.7. Reconstruction of Omics Multi-Layers Networks
2.8. Modeling of Three-Dimensional (3D) Structure of Hub Proteins
3. Results
3.1. Transcriptome Analysis for Identifying Differentially Expressed Genes (DEGs)
3.2. Identification of miRNAs
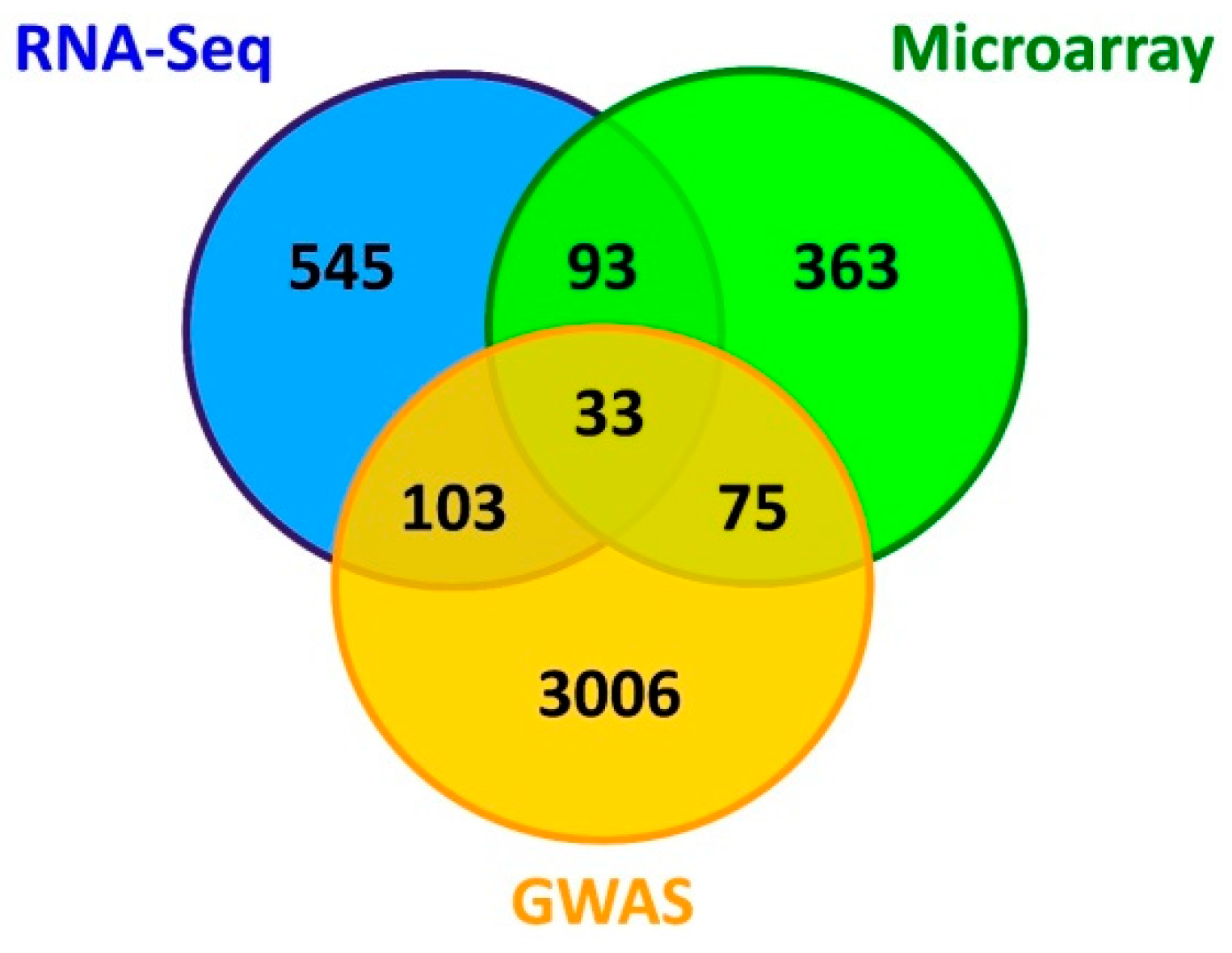
3.3. Literature Mining and Identification of Main Gene List
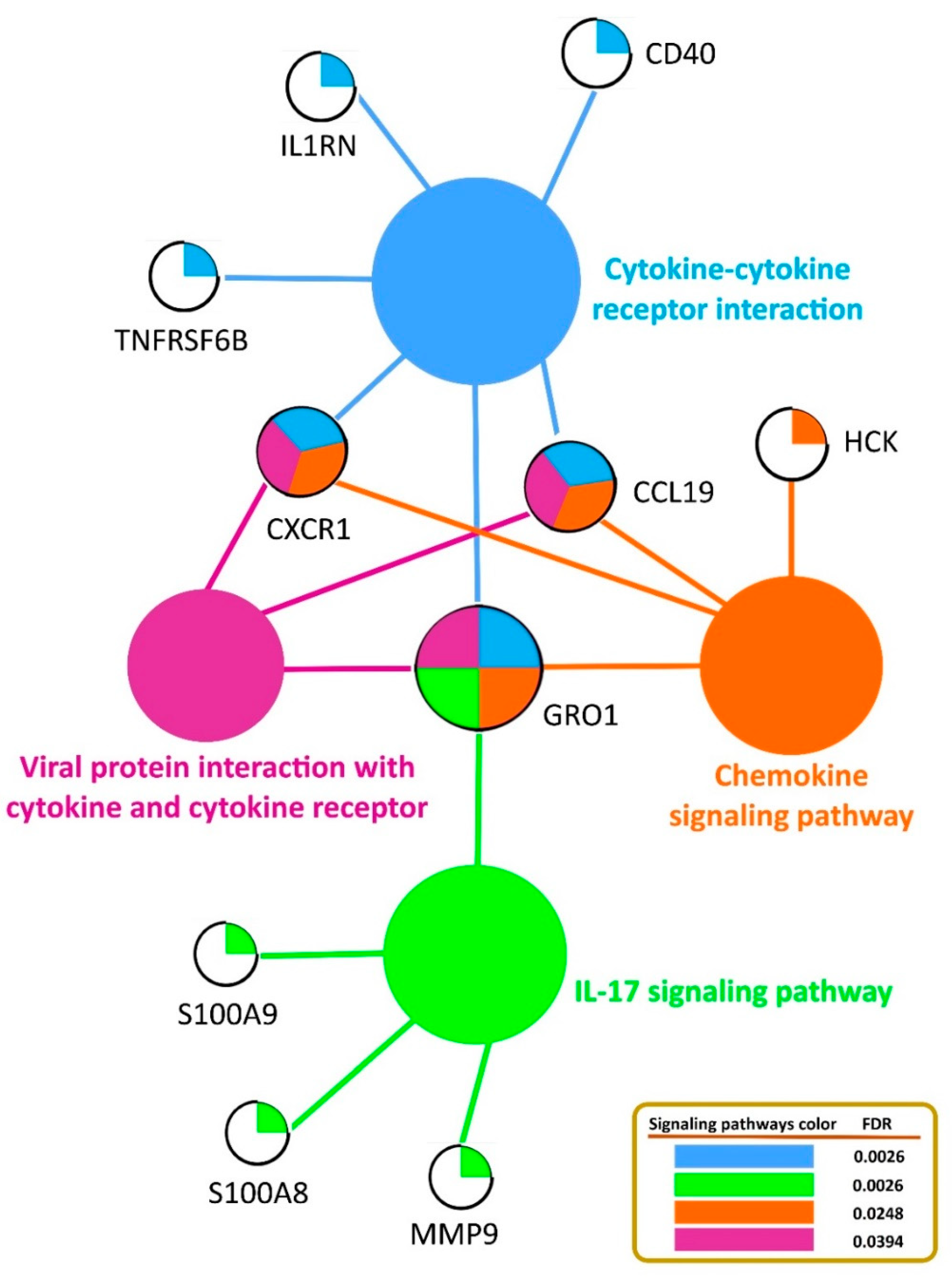
3.4. Functional Annotation and Pathway Enrichment Analysis
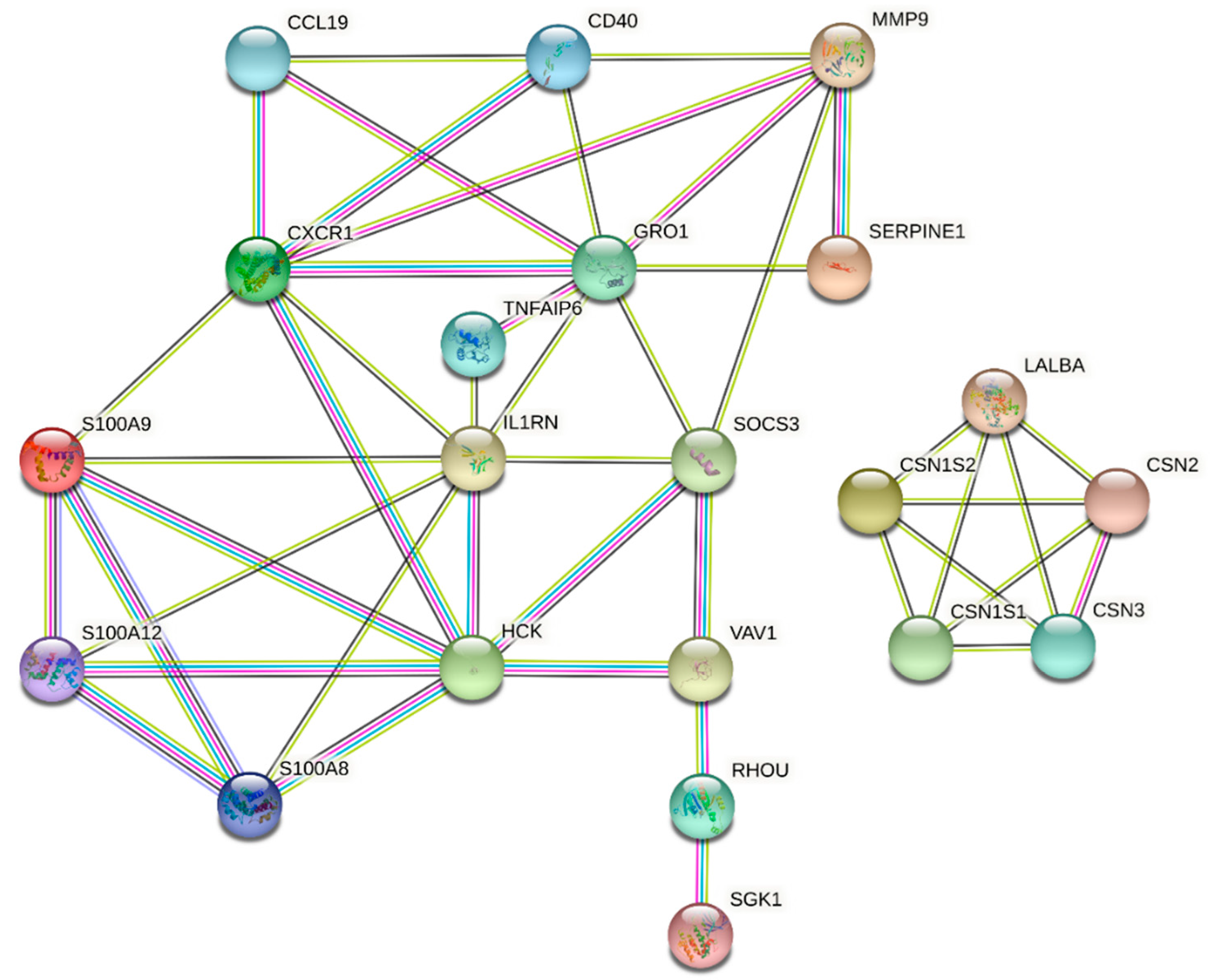
3.5. PPI Network and Identification of Hub Genes
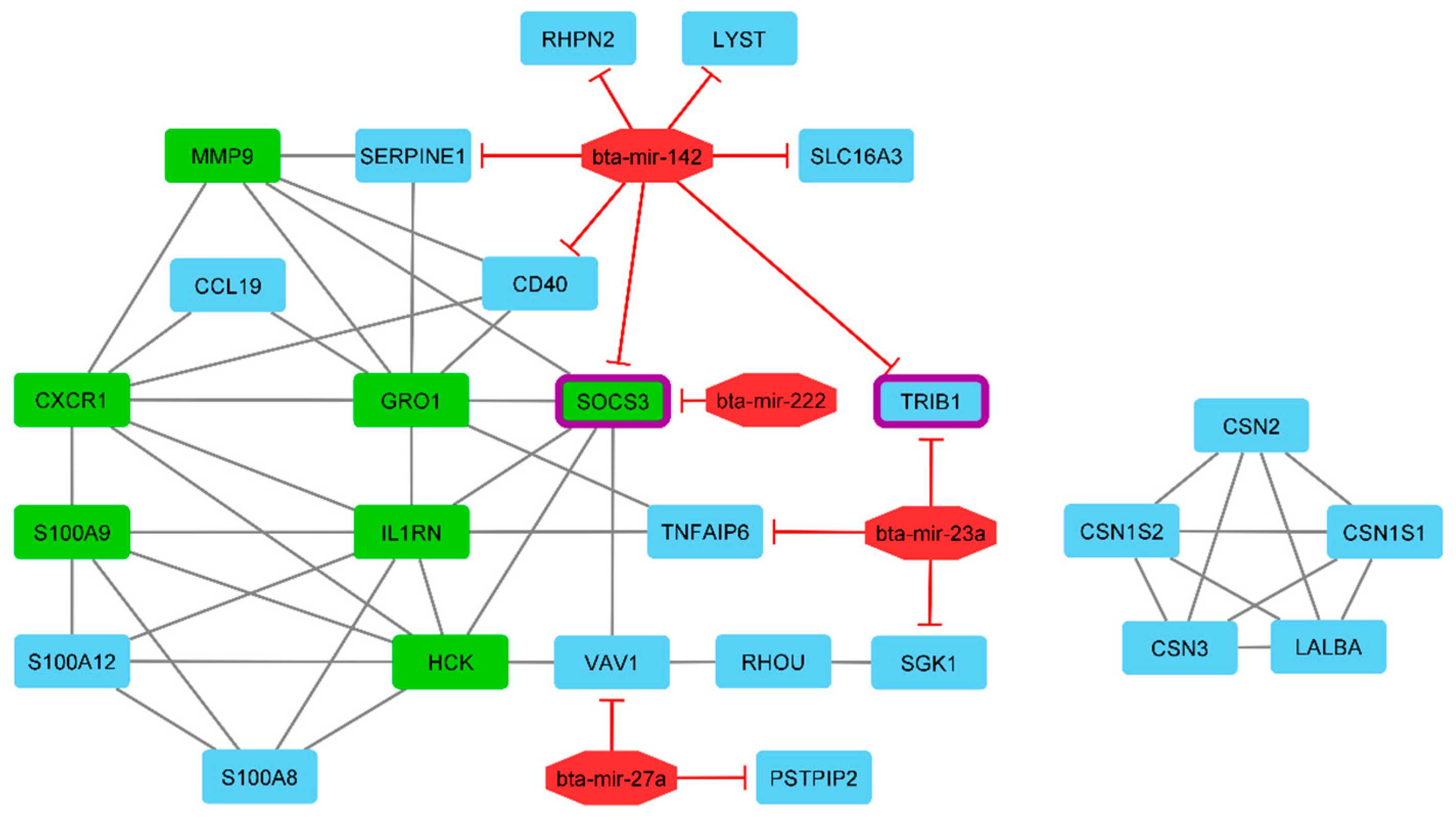
3.6. Prediction of miRNA-Target Genes and Gene Regulatory Network Reconstruction
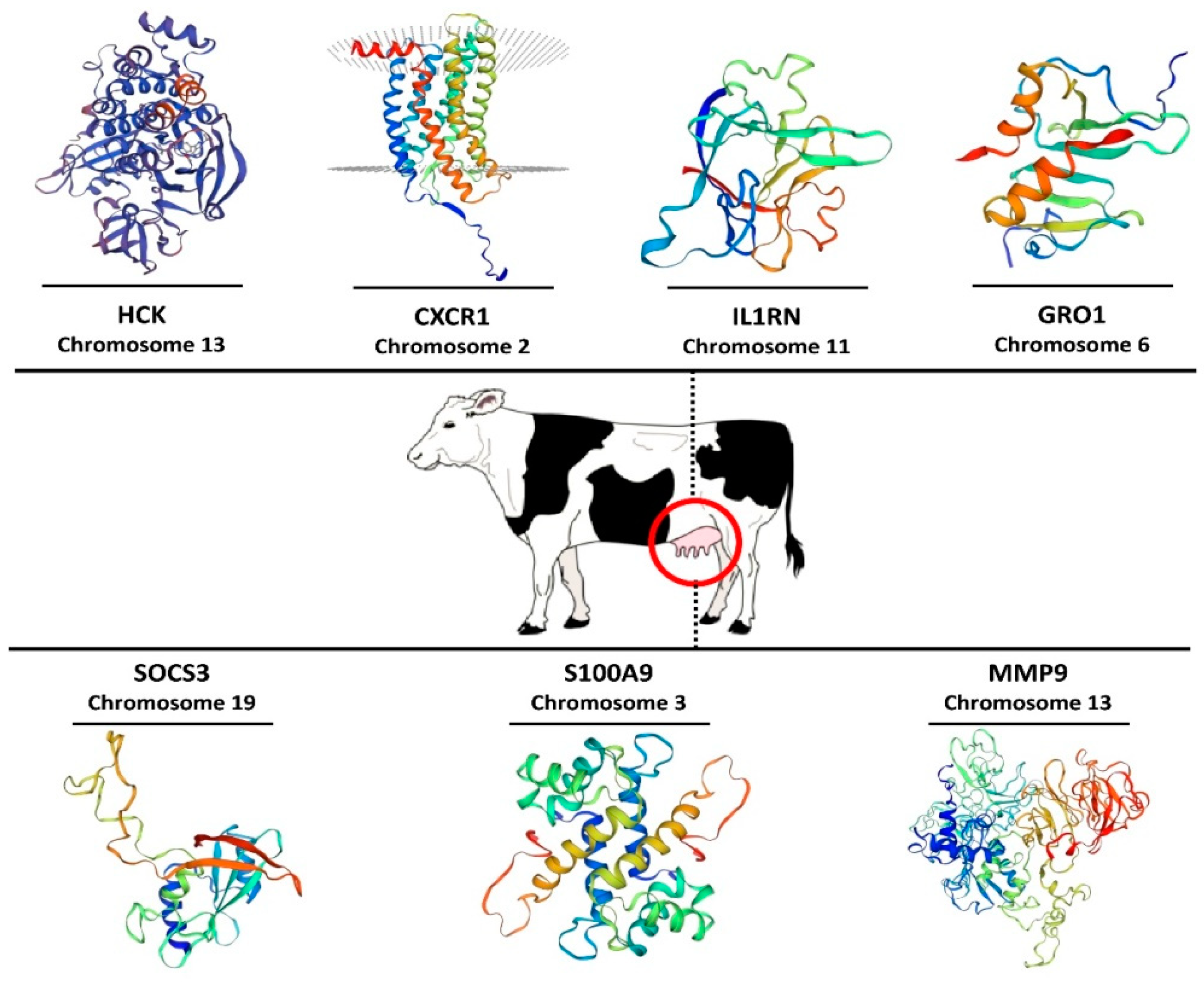
3.7. Three-Dimensional Modeling of Hub Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.; Sonstegard, T.S. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Fondi, M.; Liò, P. Multi-omics and metabolic modelling pipelines: Challenges and tools for systems microbiology. Microbiol. Res. 2015, 171, 52–64. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Ghafouri, F.; Bahrami, A.; Sadeghi, M.; Miraei-Ashtiani, S.R.; Bakherad, M.; Barkema, H.W.; Larose, S. Omics multi-layers networks provide novel mechanistic and functional insights into fat storage and lipid metabolism in poultry. Front. Genet. 2021, 12, 646297. [Google Scholar] [CrossRef]
- Fang, L.; Sahana, G.; Su, G.; Yu, Y.; Zhang, S.; Lund, M.S.; Sørensen, P. Integrating sequence-based GWAS and RNA-Seq provides novel insights into the genetic basis of mastitis and milk production in dairy cattle. Sci. Rep. 2017, 7, 45560. [Google Scholar] [CrossRef]
- Gòdia, M.; Reverter, A.; González-Prendes, R.; Ramayo-Caldas, Y.; Castelló, A.; Rodríguez-Gil, J.-E.; Sánchez, A.; Clop, A. A systems biology framework integrating GWAS and RNA-seq to shed light on the molecular basis of sperm quality in swine. Genet. Sel. Evol. 2020, 52, 72. [Google Scholar] [CrossRef]
- Ramayo-Caldas, Y.; Mármol-Sánchez, E.; Ballester, M.; Sánchez, J.P.; González-Prendes, R.; Amills, M.; Quintanilla, R. Integrating genome-wide co-association and gene expression to identify putative regulators and predictors of feed efficiency in pigs. Genet. Sel. Evol. 2019, 51, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kromer, J.O.; Sorgenfrei, O.; Klopprogge, K.; Heinzle, E.; Wittmann, C. In-depth profiling of lysine-producing Corynebacterium glutamicum by combined analysis of the transcriptome, metabolome, and fluxome. J. Bacteriol. 2004, 186, 1769–1784. [Google Scholar] [CrossRef] [Green Version]
- Sana, T.R.; Fischer, S.; Wohlgemuth, G.; Katrekar, A.; Jung, K.-H.; Ronald, P.C.; Fiehn, O. Metabolomic and transcriptomic analysis of the rice response to the bacterial blight pathogen Xanthomonas oryzae pv. oryzae. Metabolomics 2010, 6, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Tschaplinski, T.J.; Engle, N.L.; Carroll, S.L.; Martin, S.L.; Davison, B.H.; Palumbo, A.V.; Rodriguez, M.; Brown, S.D. Transcriptomic and metabolomic profiling of Zymomonas mobilis during aerobic and anaerobic fermentations. BMC Genom. 2009, 10, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Chen, L.; Te, R.; Qiao, J.; Wang, J.; Zhang, W. Complementary iTRAQ proteomics and RNA-seq transcriptomics reveal multiple levels of regulation in response to nitrogen starvation in Synechocystis sp. PCC 6803. Mol. BioSyst. 2013, 9, 2565–2574. [Google Scholar] [CrossRef]
- Kühl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.-G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 2017, 6, e30952. [Google Scholar] [CrossRef]
- Fu, F.; Cheng, V.W.; Wu, Y.; Tang, Y.; Weiner, J.H.; Li, L. Comparative proteomic and metabolomic analysis of Staphylococcus warneri SG1 cultured in the presence and absence of butanol. J. Proteome Res. 2013, 12, 4478–4489. [Google Scholar] [CrossRef]
- Ma, Q.; Zhou, J.; Zhang, W.; Meng, X.; Sun, J.; Yuan, Y.-J. Integrated proteomic and metabolomic analysis of an artificial microbial community for two-step production of vitamin C. PLoS ONE 2011, 6, e26108. [Google Scholar] [CrossRef] [Green Version]
- Asselstine, V.; Miglior, F.; Suarez-Vega, A.; Fonseca, P.; Mallard, B.; Karrow, N.; Islas-Trejo, A.; Medrano, J.; Cánovas, A. Genetic mechanisms regulating the host response during mastitis. J. Dairy Sci. 2019, 102, 9043–9059. [Google Scholar] [CrossRef]
- Cai, Z.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Prioritizing candidate genes post-GWAS using multiple sources of data for mastitis resistance in dairy cattle. BMC Genom. 2018, 19, 656. [Google Scholar] [CrossRef]
- Kumar, N.; Manimaran, A.; Kumaresan, A.; Jeyakumar, S.; Sreela, L.; Mooventhan, P.; Sivaram, M. Mastitis effects on reproductive performance in dairy cattle: A review. Trop. Anim. Health Prod. 2017, 49, 663–673. [Google Scholar] [CrossRef]
- Yang, F.; Chen, F.; Li, L.; Yan, L.; Badri, T.; Lv, C.; Yu, D.; Zhang, M.; Jang, X.; Li, J. Three novel players: PTK2B, SYK, and TNFRSF21 were identified to be involved in the regulation of bovine mastitis susceptibility via GWAS and post-transcriptional analysis. Front. Immunol. 2019, 10, 1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhtiarizadeh, M.R.; Mirzaei, S.; Norouzi, M.; Sheybani, N.; Vafaei Sadi, M.S. Identification of Gene Modules and Hub Genes Involved in Mastitis Development Using a Systems Biology Approach. Front. Genet. 2020, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Oviedo-Boyso, J.; Valdez-Alarcón, J.J.; Cajero-Juárez, M.; Ochoa-Zarzosa, A.; López-Meza, J.E.; Bravo-Patino, A.; Baizabal-Aguirre, V.M. Innate immune response of bovine mammary gland to pathogenic bacteria responsible for mastitis. J. Infect. 2007, 54, 399–409. [Google Scholar] [CrossRef]
- Heringstad, B.; Klemetsdal, G.; Ruane, J. Selection for mastitis resistance in dairy cattle: A review with focus on the situation in the Nordic countries. Livest. Prod. Sci. 2000, 64, 95–106. [Google Scholar] [CrossRef]
- Halasa, T.; Huijps, K.; Østerås, O.; Hogeveen, H. Economic effects of bovine mastitis and mastitis management: A review. Veter. Q. 2007, 29, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Heringstad, B.; Chang, Y.; Gianola, D.; Klemetsdal, G. Genetic association between susceptibility to clinical mastitis and protein yield in Norwegian dairy cattle. J. Dairy Sci. 2005, 88, 1509–1514. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Richoux, R.; Boutinaud, M.; Martin, P.; Gagnaire, V. Role of somatic cells on dairy processes and products: A review. Dairy Sci. Technol. 2014, 94, 517–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meredith, B.; Lynn, D.; Berry, D.; Kearney, F.; Bradley, D.; Finlay, E.; Fahey, A. A genome-wide association study for somatic cell score using the Illumina high-density bovine beadchip identifies several novel QTL potentially related to mastitis susceptibility. Front. Genet. 2013, 4, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodeland, M.; Kent, M.; Olsen, H.; Opsal, M.; Svendsen, M.; Sehested, E.; Hayes, B.; Lien, S. Quantitative trait loci for clinical mastitis on chromosomes 2, 6, 14 and 20 in Norwegian Red cattle. Anim. Genet. 2011, 42, 457–465. [Google Scholar] [CrossRef]
- Wagner, P.; Yin, T.; Brügemann, K.; Engel, P.; Weimann, C.; Schlez, K.; König, S. Genome-Wide Associations for Microscopic Differential Somatic Cell Count and Specific Mastitis Pathogens in Holstein Cows in Compost-Bedded Pack and Cubicle Farming Systems. Animals 2021, 11, 1839. [Google Scholar] [CrossRef]
- Wang, X.; Ma, P.; Liu, J.; Zhang, Q.; Zhang, Y.; Ding, X.; Jiang, L.; Wang, Y.; Zhang, Y.; Sun, D. Genome-wide association study in Chinese Holstein cows reveal two candidate genes for somatic cell score as an indicator for mastitis susceptibility. BMC Genet. 2015, 16, 111. [Google Scholar] [CrossRef]
- Welderufael, B.; Løvendahl, P.; De Koning, D.-J.; Janss, L.L.; Fikse, W. Genome-wide association study for susceptibility to and recoverability from mastitis in Danish Holstein cows. Front. Genet. 2018, 9, 141. [Google Scholar] [CrossRef] [Green Version]
- Mitterhuemer, S.; Petzl, W.; Krebs, S.; Mehne, D.; Klanner, A.; Wolf, E.; Zerbe, H.; Blum, H. Escherichia coli infection induces distinct local and systemic transcriptome responses in the mammary gland. BMC Genom. 2010, 11, 138. [Google Scholar] [CrossRef] [Green Version]
- Zoldan, K.; Schneider, J.; Moellmer, T.; Fueldner, C.; Knauer, J.; Fuerll, M.; Starke, A.; Specht, M.; Reiche, K.; Hackermueller, J. Discovery and validation of immunological biomarkers in milk for health monitoring of dairy cows-results from a multiomics approach. J. Adv. Dairy Res. 2017, 5, 2. [Google Scholar]
- Moyes, K.; Sørensen, P.; Bionaz, M. The impact of intramammary Escherichia coli challenge on liver and mammary transcriptome and cross-talk in dairy cows during early lactation using RNAseq. PLoS ONE 2016, 11, e0157480. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Kibbe, W.A.; Lin, S.M. lumi: A pipeline for processing Illumina microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.; Irizarry, R. Affy-Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Retrieved May 2010, 17, 2018. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, J.M.; Hoffmann, R.; Valencia, A. iHOP web services. Nucleic Acids Res. 2007, 35, W21–W26. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar]
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2012, 41, D377–D386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g: Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.-H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. Swiss-Model: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backman, M.; Flenkenthaler, F.; Blutke, A.; Dahlhoff, M.; Ländström, E.; Renner, S.; Philippou-Massier, J.; Krebs, S.; Rathkolb, B.; Prehn, C. Multi-omics insights into functional alterations of the liver in insulin-deficient diabetes mellitus. Mol. Metab. 2019, 26, 30–44. [Google Scholar] [CrossRef]
- Dao, M.C.; Sokolovska, N.; Brazeilles, R.; Affeldt, S.; Pelloux, V.; Prifti, E.; Chilloux, J.; Verger, E.O.; Kayser, B.D.; Aron-Wisnewsky, J. A data integration multi-omics approach to study calorie restriction-induced changes in insulin sensitivity. Front. Physiol. 2019, 9, 1958. [Google Scholar] [CrossRef]
- Lee, B.; Zhang, S.; Poleksic, A.; Xie, L. Heterogeneous multi-layered network model for omics data integration and analysis. Front. Genet. 2020, 10, 1381. [Google Scholar] [CrossRef]
- Farrell Jr, H.; Jimenez-Flores, R.; Bleck, G.; Brown, E.; Butler, J.; Creamer, L.; Hicks, C.; Hollar, C.; Ng-Kwai-Hang, K.; Swaisgood, H. Nomenclature of the proteins of cows’ milk—Sixth revision. J. Dairy Sci. 2004, 87, 1641–1674. [Google Scholar] [CrossRef] [Green Version]
- Zidi, A.; Casas, E.; Amills, M.; Jordana, J.; Carrizosa, J.; Urrutia, B.; Serradilla, J.M. Genetic variation at the caprine lactalbumin, alpha (LALBA) gene and its association with milk lactose concentration. Anim. Genet. 2014, 45, 612–613. [Google Scholar] [CrossRef]
- Seo, M.; Lee, H.-J.; Kim, K.; Caetano-Anolles, K.; Jeong, J.Y.; Park, S.; Oh, Y.K.; Cho, S.; Kim, H. Characterizing milk production related genes in Holstein using RNA-seq. Asian-Australas. J. Anim. Sci. 2016, 29, 343. [Google Scholar] [CrossRef] [Green Version]
- García-Gámez, E.; Gutiérrez-Gil, B.; Sahana, G.; Sánchez, J.-P.; Bayón, Y.; Arranz, J.-J. GWA analysis for milk production traits in dairy sheep and genetic support for a QTN influencing milk protein percentage in the LALBA gene. PLoS ONE 2012, 7, e47782. [Google Scholar] [CrossRef]
- Shi, H.; Zhu, J.; Luo, J.; Cao, W.; Shi, H.; Yao, D.; Li, J.; Sun, Y.; Xu, H.; Yu, K. Genes regulating lipid and protein metabolism are highly expressed in mammary gland of lactating dairy goats. Funct. Integr. Genom. 2015, 15, 309–321. [Google Scholar] [CrossRef]
- Bionaz, M.; Loor, J.J. Gene networks driving bovine milk fat synthesis during the lactation cycle. BMC Genom. 2008, 9, 366. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Cheng, Z.; Zhang, S.; Werling, D.; Wathes, D.C. Combining genome wide association studies and differential gene expression data analyses identifies candidate genes affecting mastitis caused by two different pathogens in the dairy cow. Open J. Anim. Sci. 2015, 5, 358–393. [Google Scholar] [CrossRef] [Green Version]
- Sharifi, S.; Pakdel, A.; Ebrahimi, M.; Reecy, J.M.; Fazeli Farsani, S.; Ebrahimie, E. Integration of machine learning and meta-analysis identifies the transcriptomic bio-signature of mastitis disease in cattle. PLoS ONE 2018, 13, e0191227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnzon, C.-F.; Artursson, K.; Söderlund, R.; Guss, B.; Rönnberg, E.; Pejler, G. Mastitis pathogens with high virulence in a mouse model produce a distinct cytokine profile in vivo. Front. Immunol. 2016, 7, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aderem, A.; Ulevitch, R.J. Toll-like receptors in the induction of the innate immune response. Nature 2000, 406, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Taraktsoglou, M.; Szalabska, U.; Magee, D.A.; Browne, J.A.; Sweeney, T.; Gormley, E.; MacHugh, D.E. Transcriptional profiling of immune genes in bovine monocyte-derived macrophages exposed to bacterial antigens. Vet. Immunol. Immunopathol. 2011, 140, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Proinflammatory cytokines. Chest 2000, 118, 503–508. [Google Scholar] [CrossRef]
- Lahouassa, H.; Rainard, P.; Caraty, A.; Riollet, C. Identification and characterization of a new interleukin-8 receptor in bovine species. Mol. Immunol. 2008, 45, 1153–1164. [Google Scholar] [CrossRef]
- Mao, Y.; Zhu, X.; Li, R.; Chen, D.; Xin, S.; Zhu, Y.; Liao, X.; Wang, X.; Zhang, H.; Yang, Z. Methylation analysis of CXCR1 in mammary gland tissue of cows with mastitis induced by Staphylococcus aureus. Genet. Mol. Res. 2015, 14, 12606–12615. [Google Scholar] [CrossRef]
- Pokorska, J.; Dusza, M.; Kułaj, D.; Żukowski, K.; Makulska, J. Single nucleotide polymorphisms in the CXCR1 gene and its association with clinical mastitis incidence in Polish Holstein-Friesian cows. Genet. Mol. Res. 2016, 15, gmr15017573. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, H.; Ju, Z.; Zhang, Y.; Huang, J.; Qi, C.; Hou, M.; An, L.; Zhong, J.; Wang, C. Association of novel single nucleotide polymorphisms of the CXCR1 gene with the milk performance traits of Chinese native cattle. Genet. Mol. Res. 2013, 12, 2725–2739. [Google Scholar] [CrossRef]
- Strillacci, M.G.; Frigo, E.; Schiavini, F.; Samoré, A.B.; Canavesi, F.; Vevey, M.; Cozzi, M.C.; Soller, M.; Lipkin, E.; Bagnato, A. Genome-wide association study for somatic cell score in Valdostana Red Pied cattle breed using pooled DNA. BMC Genet. 2014, 15, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Breems, N.Y.; Nguyen, L.K.; Kulasiri, D. Integrated signaling pathway and gene expression regulatory model to dissect dynamics of Escherichia coli challenged mammary epithelial cells. Biosystems 2014, 126, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Z.; Jiang, Q.; Liu, G.; Wang, X.; Luo, G.; Zhang, Y.; Zhang, J.; Zhong, J.; Huang, J. Solexa sequencing and custom micro RNA chip reveal repertoire of micro RNA s in mammary gland of bovine suffering from natural infectious mastitis. Anim. Genet. 2018, 49, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Lawless, N.; Foroushani, A.B.; McCabe, M.S.; O’Farrelly, C.; Lynn, D.J. Next generation sequencing reveals the expression of a unique miRNA profile in response to a gram-positive bacterial infection. PLoS ONE 2013, 8, e57543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Huang, J.; Zhang, X.; Ju, Z.; Qi, C.; Zhang, Y.; Li, Q.; Wang, C.; Miao, W.; Zhong, J. One SNP in the 3′-UTR of HMGB1 gene affects the binding of target bta-miR-223 and is involved in mastitis in dairy cattle. Immunogenetics 2012, 64, 817–824. [Google Scholar] [CrossRef]
- Fiser, A. Template-based protein structure modeling. Comput. Biol. 2010, 373, 73–94. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Data Type | GEO a Accession | Platforms | Samples (M:H) b | Citation |
|---|---|---|---|---|---|
| 1 | RNA-Seq | GSE131607 | GPL15749 (Illumina HiSeq 2000) | 12 (6:6) | Asselstine et al. [16] |
| 2 | RNA-Seq | GSE75379 | GPL15749 (Illumina HiSeq 2000) | 18 (6:12) | Moyes et al. [33] |
| 3 | Microarray | GSE93082 | GPL2112 ((Bovine) Affymetrix Bovine Genome Array) | 12 (6:6) | Zoldan et al. [32] |
| 4 | Microarray | GSE15020 | GPL2112 ((Bovine) Affymetrix Bovine Genome Array) | 10 (5:5) | Mitterhuemer et al. [31] |
| 5 | Microarray | GSE15022 | GPL2112 ((Bovine) Affymetrix Bovine Genome Array) | 10 (5:5) | Mitterhuemer et al. [31] |
| miRNA Name | miRNA Region | Fold Change | p-Value | FDR | ||
|---|---|---|---|---|---|---|
| BTA | miRNA Start | miRNA End | ||||
| bta-mir-339a | 25 | 41736134 | 41736211 | 2.0472 | 0.0032 | 0.0490 |
| bta-mir-24-2 | 7 | 11839032 | 11839103 | 2.5302 | 0.0001 | 0.0056 |
| bta-mir-222 | X | 98125920 | 98126030 | 3.2194 | 4.88 × 10−6 | 0.0002 |
| bta-mir-27a | 7 | 11838877 | 11838949 | 3.6647 | 1.13 × 10−8 | 1.16 × 10−6 |
| bta-mir-146a | 7 | 72071548 | 72071646 | 3.9677 | 2.26 × 10−8 | 2.20 × 10−6 |
| bta-mir-23a | 7 | 11838702 | 11838776 | 3.9826 | 8.97 × 10−9 | 9.60 × 10−7 |
| bta-mir-142 | 19 | 9301432 | 9301518 | 4.2675 | 2.69 × 10−13 | 6.83 × 10−11 |
| bta-mir-223 | X | 94562822 | 94562929 | 4.4983 | 3.05 × 10−11 | 5.58 × 10−9 |
| Gene Symbol | Gene Name | Gene Region | Fold Change | p-Value | FDR | ||
|---|---|---|---|---|---|---|---|
| Chr | Gene Start | Gene End | |||||
| CSN3 | casein kappa | 6 | 85645854 | 85658926 | −4.1767 | 8.29 × 10−10 | 1.55 × 10−6 |
| CSN1S2 | casein alpha-S2 | 6 | 85529905 | 85548556 | −3.9696 | 3.53 × 10−8 | 1.85 × 10−5 |
| CSN2 | casein beta | 6 | 85449164 | 85457744 | −3.7916 | 2.92 × 10−8 | 1.74 × 10−5 |
| RHPN2 | rhophilin Rho GTPase binding protein 2 | 18 | 43404074 | 43474596 | −3.4556 | 1.38 × 10−6 | 0.0002 |
| CSN1S1 | casein alpha s1 | 6 | 85411118 | 85429268 | −3.4530 | 2.94 × 10−7 | 7.41 × 10−5 |
| LALBA | lactalbumin alpha | 5 | 31183432 | 31213145 | −3.1099 | 1.23 × 10−5 | 0.0009 |
| ACSS2 | acyl-CoA synthetase short chain family member 2 | 13 | 64186743 | 64233568 | −2.6729 | 4.02 × 10−10 | 1.05 × 10−6 |
| RHOU | ras homolog family member U | 28 | 697339 | 706882 | −2.4960 | 0.0001 | 0.0047 |
| KRT7 | keratin 7 | 5 | 27674854 | 27689030 | −2.3804 | 0.0003 | 0.0110 |
| SGK1 | serum/glucocorticoid regulated kinase 1 | 9 | 72305979 | 72418535 | 2.0107 | 1.85 × 10−14 | 5.70 × 10−12 |
| TRIB1 | tribbles pseudokinase 1 | 14 | 14779050 | 14787206 | 2.0650 | 4.36 × 10−5 | 0.0024 |
| LYST | lysosomal trafficking regulator | 28 | 8379173 | 8523114 | 2.1066 | 0.0009 | 0.0212 |
| VAV1 | vav guanine nucleotide exchange factor 1 | 7 | 17664498 | 17728163 | 2.1138 | 3.31 × 10−5 | 0.0013 |
| GRO1 | chemokine (C-X-C motif) ligand 1 (melanoma growth stimulating activity, alpha) | 6 | 89072611 | 89075133 | 2.2062 | 0.0003 | 0.0105 |
| F5 | coagulation factor V | 16 | 37159073 | 37238306 | 2.2729 | 3.94 × 10−10 | 6.01 × 10−8 |
| SERPINE1 | serpin family E member 1 | 25 | 35596139 | 35617193 | 2.3498 | 0.0002 | 0.0065 |
| BASP1 | brain abundant membrane attached signal protein 1 | 20 | 55908762 | 55964145 | 2.4174 | 3.40 × 10−8 | 3.14 × 10−6 |
| CD40 | CD40 molecule | 13 | 74842191 | 74853116 | 2.4243 | 1.20 × 10−5 | 0.0005 |
| TNFRSF6B | TNF receptor superfamily member 6b | 13 | 54054302 | 54055810 | 2.4416 | 0.0001 | 0.0035 |
| SLC16A3 | solute carrier family 16 member 3 | 19 | 50634317 | 50642204 | 2.4821 | 8.56 × 10−6 | 0.0004 |
| CXCR1 | chemokine (C-X-C motif) receptor 1 | 2 | 106215131 | 106219158 | 2.5332 | 0.0002 | 0.0065 |
| SOCS3 | suppressor of cytokine signaling 3 | 19 | 53840159 | 53840858 | 2.5589 | 0.0001 | 0.0047 |
| CCDC88B | coiled-coil domain containing 88B | 29 | 42630756 | 42645750 | 2.6392 | 4.24 × 10−8 | 3.81 × 10−6 |
| TNFAIP6 | TNF alpha induced protein 6 | 2 | 44747145 | 44764214 | 2.7306 | 0.0001 | 0.0040 |
| S100A9 | S100 calcium binding protein A9 | 3 | 17115128 | 17117984 | 2.9459 | 5.39 × 10−6 | 0.0005 |
| PSTPIP2 | proline-serine-threonine phosphatase interacting protein 2 | 24 | 45737786 | 45832060 | 2.9685 | 3.43 × 10−10 | 1.05 × 10−6 |
| ALOX5AP | arachidonate 5-lipoxygenase activating protein | 12 | 30108987 | 30138259 | 3.2299 | 1.64 × 10−11 | 3.14 × 10−9 |
| CCL19 | C-C motif chemokine ligand 19 | 8 | 76054024 | 76055932 | 3.5366 | 4.73 × 10−7 | 3.31 × 10−5 |
| MMP9 | matrix metallopeptidase 9 | 13 | 74746976 | 74754303 | 3.5921 | 8.64 × 10−8 | 7.35 × 10−6 |
| HCK | HCK proto-onco, Src family tyrosine kinase | 13 | 61563070 | 61608503 | 4.2225 | 3.23 × 10−18 | 1.69 × 10−15 |
| S100A12 | S100 calcium binding protein A12 | 3 | 17102722 | 17104173 | 4.4133 | 4.22 × 10−11 | 7.47 × 10−9 |
| S100A8 | S100 calcium binding protein A8 | 3 | 17085577 | 17086827 | 4.7179 | 6.81 × 10−12 | 1.36 × 10−9 |
| IL1RN | interleukin 1 receptor antagonist | 11 | 46815591 | 46837831 | 4.9613 | 8.55 × 10−16 | 3.36 × 10−13 |
| Category | Term_ID | Term | Count | FDR | Genes |
|---|---|---|---|---|---|
| BP 1_DIRECT | GO:0050896 | Response to stimulus | 20 | 3.26 × 10−9 | CSN2, RHPN2, SGK1, CSN1S2, LALBA, S100A9, CSN1S1, SOCS3, S100A8, ALOX5AP, LYST, F5, RHOU, IL1RN, MMP9, CD40, CSN3, GRO1, S100A12, CXCR1 |
| BP_DIRECT | GO:0032570 | Response to progesterone | 5 | 5.21 × 10−8 | CSN2, CSN1S2, LALBA, CSN1S1, CSN3 |
| BP_DIRECT | GO:0032355 | Response to estradiol | 5 | 3.34 × 10−7 | CSN2, CSN1S2, LALBA, CSN1S1, CSN3 |
| BP_DIRECT | GO:0006952 | Defense response | 9 | 3.93 × 10−6 | CSN1S2, LALBA, S100A9, S100A8, LYST, IL1RN, CD40, GRO1, S100A12 |
| BP_DIRECT | GO:0006950 | Response to stress | 12 | 7.04 × 10−6 | CSN2, CSN1S2, LALBA, S100A9, S100A8, LYST, F5, IL1RN, MMP9, CD40, GRO1, S100A12 |
| BP_DIRECT | GO:0006955 | Immune response | 8 | 2.06 × 10−5 | S100A9, S100A8, LYST, IL1RN, CD40, GRO1, S100A12, CXCR1 |
| BP_DIRECT | GO:0051716 | Cellular response to stimulus | 14 | 2.06 × 10−5 | CSN2, RHPN2, SGK1, S100A9, CSN1S1, SOCS3, S100A8, ALOX5AP, RHOU, IL1RN, MMP9, CD40, GRO1, CXCR1 |
| BP_DIRECT | GO:0030593 | Neutrophil chemotaxis | 4 | 6.46 × 10−5 | S100A9, S100A8, GRO1, CXCR1 |
| BP_DIRECT | GO:0098542 | Defense response to other organisms | 7 | 6.46 × 10−5 | CSN1S2, LALBA, S100A9, S100A8, LYST, CD40, S100A12 |
| BP_DIRECT | GO:0006954 | Inflammatory response | 6 | 6.92 × 10−5 | S100A9, S100A8, IL1RN, CD40, GRO1, S100A12 |
| BP_DIRECT | GO:0033993 | Response to lipids | 6 | 9.95 × 10−5 | CSN2, CSN1S2, LALBA, CSN1S1, CSN3, GRO1 |
| BP_DIRECT | GO:0065007 | Biological regulation | 17 | 0.00029 | CSN2, RHPN2, SGK1, S100A9, CSN1S1, SOCS3, S100A8, ALOX5AP, SERPINE1, F5, RHOU, IL1RN, MMP9, CD40, CSN3, GRO1, CXCR1 |
| BP_DIRECT | GO:0052548 | Regulation of endopeptidase activity | 5 | 0.00063 | CSN2, S100A9, S100A8, SERPINE1, MMP9 |
| BP_DIRECT | GO:0045087 | Innate immune response | 5 | 0.0028 | S100A9, S100A8, LYST, CD40, S100A12 |
| BP_DIRECT | GO:0023051 | Regulation of signaling | 8 | 0.0041 | CSN2, S100A9, CSN1S1, SOCS3, S100A8, IL1RN, MMP9, CD40 |
| BP_DIRECT | GO:0042981 | Regulation of apoptotic process | 6 | 0.0041 | S100A9, CSN1S1, SOCS3, S100A8, MMP9, CD40 |
| BP_DIRECT | GO:0050727 | Regulation of inflammatory response | 4 | 0.0046 | CSN2, S100A9, SOCS3, S100A8 |
| BP_DIRECT | GO:0070488 | Neutrophil aggregation | 2 | 0.0046 | S100A9, S100A8 |
| BP_DIRECT | GO:0002523 | Leukocyte migration involved in inflammatory response | 2 | 0.01 | S100A9, S100A8 |
| BP_DIRECT | GO:0009987 | Cellular process | 18 | 0.0104 | CSN2, RHPN2, SGK1, LALBA, S100A9, CSN1S1, SOCS3, S100A8, ALOX5AP, SERPINE1, LYST, RHOU, IL1RN, MMP9, CD40, GRO1, S100A12, CXCR1 |
| BP_DIRECT | GO:0032268 | Regulation of cellular protein metabolic process | 7 | 0.0221 | CSN2, S100A9, SOCS3, S100A8, SERPINE1, MMP9, CD40 |
| BP_DIRECT | GO:0050793 | Regulation of developmental process | 6 | 0.0276 | CSN2, CSN1S1, SOCS3, RHOU, MMP9, CD40 |
| BP_DIRECT | GO:0071345 | Cellular response to cytokine stimulus | 4 | 0.0302 | SOCS3, CD40, GRO1, CXCR1 |
| MF 2_DIRECT | GO:0016209 | Antioxidant activity | 5 | 2.82 × 10−5 | CSN2, S100A9, CSN1S1, S100A8, ALOX5AP |
| MF_DIRECT | GO:0005488 | Binding | 19 | 0.00034 | CSN2, SGK1, CSN1S2, LALBA, S100A9, CSN1S1, SOCS3, S100A8, ALOX5AP, SERPINE1, F5, RHOU, IL1RN, MMP9, CD40, CSN3, GRO1, S100A12, CXCR1 |
| MF_DIRECT | GO:0005515 | Protein binding | 14 | 0.00034 | CSN2, CSN1S2, LALBA, S100A9, SOCS3, S100A8, SERPINE1, RHOU, IL1RN, MMP9, CD40, CSN3, GRO1, CXCR1 |
| MF_DIRECT | GO:0035375 | Zymogen binding | 3 | 0.00034 | CSN1S2, SERPINE1, CSN3 |
| MF_DIRECT | GO:0050544 | Arachidonic acid binding | 3 | 0.00034 | S100A9, S100A8, ALOX5AP |
| MF_DIRECT | GO:0035662 | Toll-like receptor 4 binding | 2 | 0.007 | S100A9, S100A8 |
| MF_DIRECT | GO:0050786 | RAGE receptor binding | 2 | 0.033 | S100A9, S100A8 |
| CC 3_DIRECT | GO:0005576 | Extracellular region | 13 | 1.74 × 10−7 | CSN2, CSN1S2, LALBA, S100A9, CSN1S1, S100A8, SERPINE1, F5, IL1RN, MMP9, CSN3, GRO1, S100A12 |
| CC_DIRECT | GO:0005796 | Golgi lumen | 4 | 1.38 × 10−6 | CSN2, CSN1S2, CSN1S1, CSN3 |
| CC_DIRECT | GO:0005615 | Extracellular space | 10 | 2.57 × 10−6 | CSN2, CSN1S2, LALBA, CSN1S1, S100A8, SERPINE1, IL1RN, MMP9, CSN3, GRO1 |
| CC_DIRECT | GO:0110165 | Cellular anatomical entity | 22 | 0.00069 | CSN2, RHPN2, SGK1, CSN1S2, LALBA, S100A9, CSN1S1, SOCS3, S100A8, ALOX5AP, SERPINE1, KRT7, LYST, F5, RHOU, IL1RN, MMP9, CD40, CSN3, GRO1, S100A12, CXCR1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naserkheil, M.; Ghafouri, F.; Zakizadeh, S.; Pirany, N.; Manzari, Z.; Ghorbani, S.; Banabazi, M.H.; Bakhtiarizadeh, M.R.; Huq, M.A.; Park, M.N.; et al. Multi-Omics Integration and Network Analysis Reveal Potential Hub Genes and Genetic Mechanisms Regulating Bovine Mastitis. Curr. Issues Mol. Biol. 2022, 44, 309-328. https://doi.org/10.3390/cimb44010023
Naserkheil M, Ghafouri F, Zakizadeh S, Pirany N, Manzari Z, Ghorbani S, Banabazi MH, Bakhtiarizadeh MR, Huq MA, Park MN, et al. Multi-Omics Integration and Network Analysis Reveal Potential Hub Genes and Genetic Mechanisms Regulating Bovine Mastitis. Current Issues in Molecular Biology. 2022; 44(1):309-328. https://doi.org/10.3390/cimb44010023
Chicago/Turabian StyleNaserkheil, Masoumeh, Farzad Ghafouri, Sonia Zakizadeh, Nasrollah Pirany, Zeinab Manzari, Sholeh Ghorbani, Mohammad Hossein Banabazi, Mohammad Reza Bakhtiarizadeh, Md. Amdadul Huq, Mi Na Park, and et al. 2022. "Multi-Omics Integration and Network Analysis Reveal Potential Hub Genes and Genetic Mechanisms Regulating Bovine Mastitis" Current Issues in Molecular Biology 44, no. 1: 309-328. https://doi.org/10.3390/cimb44010023