

Dyslipidemia: A Narrative Review on Pharmacotherapy
 and
and
Abstract
:1. Introduction
2. Methods
3. HMG-CoA Reductase Inhibitors
3.1. Primary Prevention

{kind=link}
{kind=link}
| Study | Sample Size | Characteristics of Patients | Comparison Groups | Follow-Up | LDL-C Reduction | CV Effects |
|---|---|---|---|---|---|---|
| WOSCOPS (1995) [9] | 6595 | TC > 252 mg/dL | Pravastatin 40 mg vs. placebo | 4.9 years | 26% | Reduction in MI or coronary death (HR 0.69, 95% CI 0.57 to 0.83, NNT 111) |
| AFCAPS/TexCAPS (1998) [10] | 6605 | LDL-C 130–180 mg/dL | Lovastatin 20–40 mg vs. placebo | 5.3 years | 25% | Reduction in coronary events (HR 0.63, 95% CI 0.50 to 0.79, NNT 86) |
| ASCOT-LLA (2003) [12] | 10,305 | Hypertension and CV risk factors | Atorvastatin 10 mg vs. placebo | 3.3 years | 35% | Reduction in MI or coronary death (HR 0.64, 95% CI 0.50 to 0.83, NNT 83) |
| MEGA (2006) [13] | 7832 | TC 220–270 mg/dL | Pravastatin 10 mg vs. placebo | 5.3 years | 18% | Reduction in CAD (HR 0.67, 95% CI 0.49 to 0.91, NNT 119) |
| JUPITER (2008) [14] | 17,802 | LDL-C < 130 mg/dL + hsCRP ≥ 2 mg/L | Rosuvastatin 20 mg vs. placebo | 1.9 years | 50% | Reduction in CV death, MI, stroke, arterial revascularization, or UA hospitalization (HR 0.56, 95% CI 0.46 to 0.69, NNT 169) |
| HOPE-3 (2016) [15] | 12,705 | Intermediate CV risk (CV event rate 1%/year) | Rosuvastatin 10 mg vs. placebo | 5.6 years | 26.5% | Reduction in CV death, MI, or stroke (HR 0.76, 95% CI 0.64 to 91, NNT 91) |
3.2. Secondary Prevention
| Study | Sample Size | Characteristics of Patients | Comparison Groups | Follow-Up | LDL-C Reduction | CV Effects |
|---|---|---|---|---|---|---|
| 4S (1994) [16] | 4444 | Angina or previous MI | Simvastatin 20–40 mg vs. placebo | 5.4 years | 35% | Reduction in death (HR 0.70, 95% CI 0.58 to 0.85, NNT 30) |
| CARE (1996) [17] | 4159 | Previous MI TC < 240 mg/dL LDL-C 115–174 mg/dL | Pravastatin 40 mg vs. placebo | 5 years | 28% | Reduction in coronary death or MI (10.2% vs. 13.2%, NNT 34) |
| LIPID (1998) [18] | 9014 | Previous MI or UA TC 155–271 mg/dL | Pravastatin 40 mg vs. placebo | 6.1 years | 25% | Reduction in coronary death (6.4% vs. 8.3%, NNT 53) |
| FLORIDA (2000) [20] | 540 | MI | Fluvastatin 80 mg vs. placebo | 1 year | 21% | No significant differences in major coronary event |
| HPS * (2002) [19] | 20,536 | TC > 135 mg/dL + CAD or other arterial disease or DM or >65 years male w/HTN | Simvastatin 40 mg vs. placebo | 5 years | 35% | Reduction in all-cause mortality (12.9% vs. 14.7%, NNT 56) |
| PROVE-IT (2004) [22] | 4162 | ACS < 10 days | Atorvastatin 80 mg vs. pravastatin 40 mg | 24 months | 31% | Reduction in all-cause mortality, MI, UA hospitalization, revascularization in 30 days, or stroke (HR 0.84, 95% CI 0.74 to 0.95, NNT 53) |
| IDEAL (2005) [23] | 8888 | Previous MI | Atorvastatin 80 mg vs. simvastatin 20 mg | 4.8 years | 20% | No significant differences in major coronary event |
| TNT (2005) [24] | 10,001 | CAD | Atorvastatin 80 mg vs. atorvastatin 10 mg | 4.9 years | 24% | Reduction in CV death, MI, CA, or stroke (HR 0.78, 95% CI 0.69 to 0.89, NNT 45) |
| SEARCH (2010) [25] | 12,064 | Previous MI LDL-C > 135 mg/dL (statin use) or LDL-C > 193 mg/dL (no statin) | Simvastatin 20 mg vs. simvastatin 80 mg | 6.7 years | 14% | No significant differences in CV events |
3.3. Special Groups
| Study | Sample Size | Characteristics of Patients | Comparison Groups | Follow-Up | LDL-C Reduction | CV Effects |
|---|---|---|---|---|---|---|
| CARDS (2004) [26] | 2838 | DM (40–75 years) + LDL-C < 160 mg/dL + TGs < 600 mg/dL + additional risk factor | Atorvastatin 10 mg vs. placebo | 3.9 years | 40% | Reduction in ACS, revascularization, or stroke (HR 0.63, 95% CI 0.48 to 0.83, NNT 31) |
| ASPEN (2006) [27] | 2410 | Diabetes (40–75 years) + LDL < 160 mg/dL or < 140 mg/dL (prior MI or revascularization) | Atorvastatin 10 mg vs. placebo | 4 years | 29% | No significant differences in CV events |
| ALERT (2003) [30] | 2102 | Renal or combined renal and pancreas transplants > 6 months | Fluvastatin 40 mg vs. placebo | 5.1 years | 25% | No significant differences in CV events |
| 4D (2005) [31] | 1255 | Diabetes + CKD on dialysis | Atorvastatin 20 mg vs. placebo | 4 years | 42% | No significant differences in CV events |
| AURORA (2009) [32] | 2773 | CKD on dialysis | Rosuvastatin 10 mg vs. placebo | 3.8 years | 43% | No significant differences in CV events |
| SHARP (2011) [33] | 9270 | CKD | Simvastatin 20 mg + ezetimibe 10 mg vs. placebo | 4.9 years | 31% | Reduction in coronary death, MI, stroke, or revascularization (HR 0.83, 95% CI 0.74 to 0.94, NNT 53) |
| CORONA (2007) [34] | 5011 | LVEF < 40% + NYHA II–IV | Rosuvastatin 10 mg vs. placebo | 2.7 years | 45% | No significant differences in CV events |
| GISSI-HF (2008) [35] | 6975 | Heart failure NYHA II–IV | Rosuvastatin 10 mg vs. placebo | 3.9 years | 16% | No significant differences in CV events |
| PROSPER (2002) [36] | 5804 | Elderly (70–82 years) + high CV risk | Pravastatin 40 mg vs. placebo | 3.2 years | 34% | Reduction in coronary death, MI, or stroke (HR 0.85, 95% CI 0.74 to 0.97, NNT 48) |
| REPRIEVE (2023) [37] | 7769 | HIV | Pitavastatin 4 mg vs. placebo | 5.1 years | 30% | Reduction in MACEs (HR 0.65, 95% CI 0.48 to 0.90) |
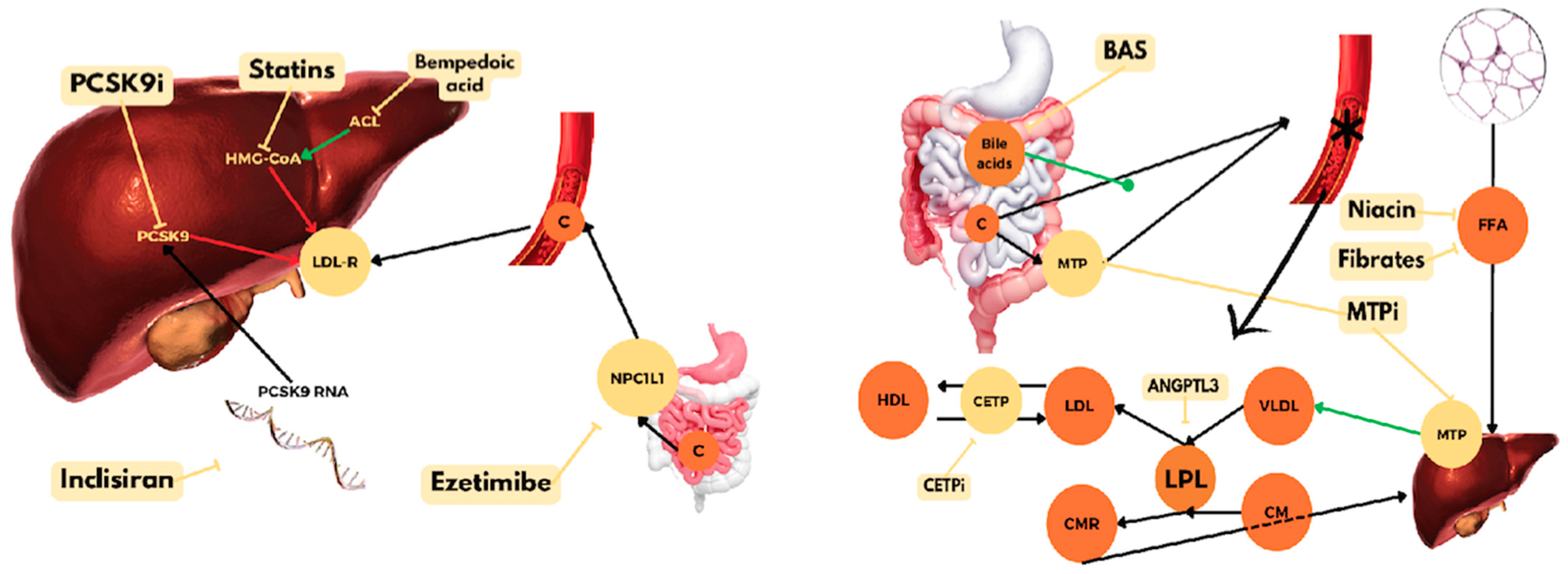
4. PCSK9 Inhibitors
4.1. Tafolecimab
4.2. Lerodalcibep
5. Ezetimibe
6. Bempedoic Acid
7. Lp(a)-Targeted Therapies
8. Bile Acid-Binding Resins
9. Nicotinic Acid
10. Fibric Acid Derivatives
11. Omega-3 Fatty Acids
| Study | Sample Size | Characteristics of Patients | Comparison Groups | Follow-Up | Lipids Effect | CV Effects |
|---|---|---|---|---|---|---|
| JELIS (2007) [102] | 18,645 | TC > 250 mg/dL | EPA (1.8 g/d) + statin vs. only statin | 4.6 years | Reduction LDL-C Reduction TC (no differences between groups) | Reduction in CV events (HR 0.81, 95% CI 0.69 to 0.95, NNT 143) |
| OMEGA (2010) [103] | 3818 | ACS | Omega-3 (1 g/d) vs. placebo | 1 year | NE | No significant differences in SCD |
| VITAL (2019) [104] | 25,871 | No known CV disease | Omega-3 (1 g/d) vs. placebo | 5.3 years | NE | No significant differences in CV events |
| REDUCE-IT (2019) [105] | 8179 | Age >45 years + CV disease or age > 50 years + diabetes and ≥1 risk factor + TGs 150–499 mg/dL + LDL-C 41–100 mg/dL | Icosapent ethyl (4 g/d) vs. placebo | 4.9 years | Reduction LDL-C 3.1% Reduction TGs 18.3% | Reduction in CV death, MI, stroke, coronary revascularization, or UA (HR 0.75, 95% CI 0.68 to 0.83, NNT 21) |
| STRENGTH (2020) [107] | 13,078 | High CV risk | Omega-3 CA (4 g/d) vs. placebo | 42 months | NE | No significant differences in CV events |
| OMEMI (2021) [108] | 1027 | Aged 70–82 years + recent MI (2–8 weeks) | n-3 PUFA (1.8 g/d) vs. placebo | 2 years | NE | No significant differences in CV events |
12. Cholesteryl Ester Transfer Protein (CETP) Inhibitors
13. Gene Therapy
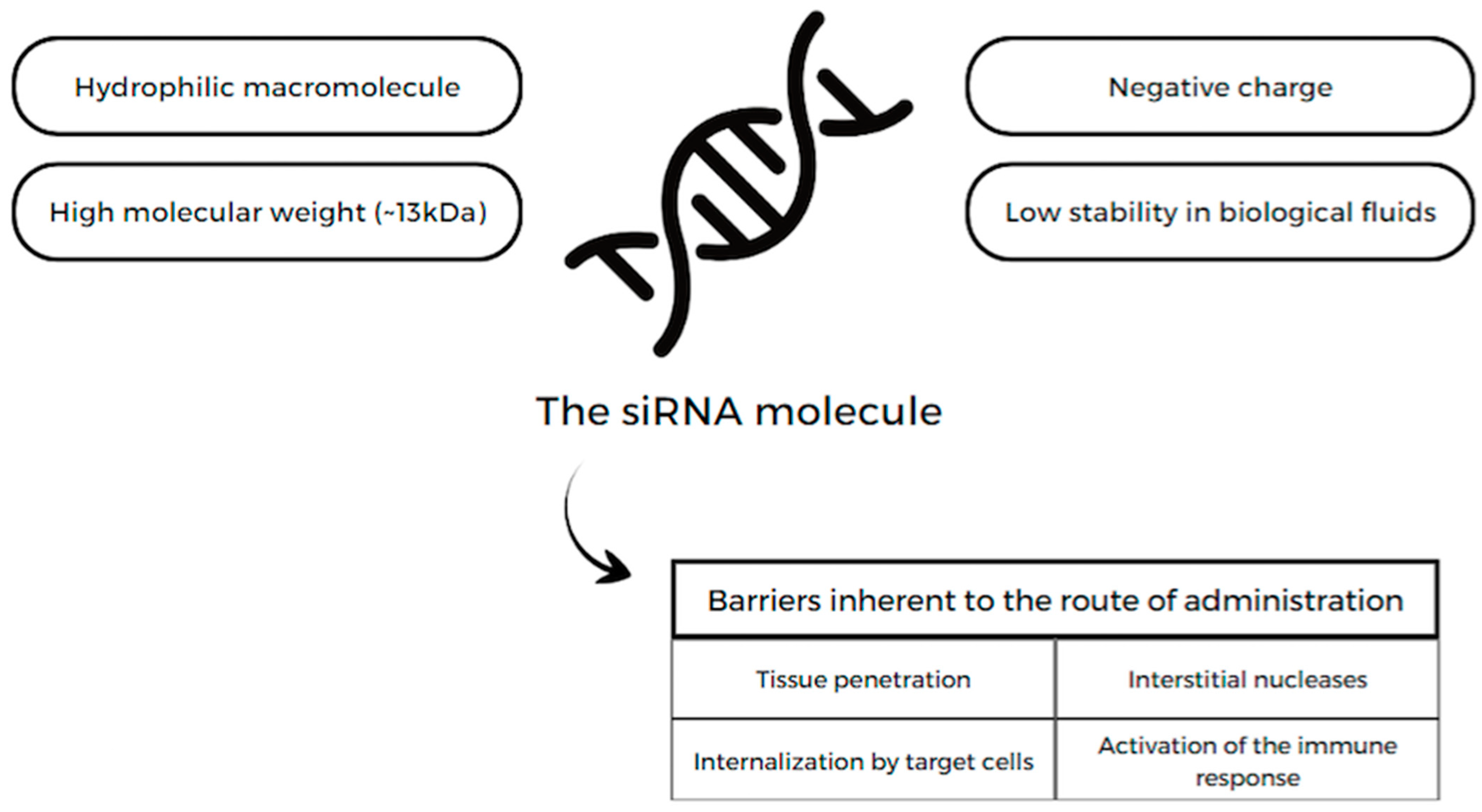
13.1. siRNA
13.1.1. Lepodisiran
13.1.2. Inclisiran
13.2. ANGPTL3 Inhibitors
13.3. CRISPR/Cas9
13.4. Antisense Oligonucleotides (ASO)
13.4.1. Mipomersen
13.4.2. Volanesorsen and Olezarsen
13.5. apoB and MTP Inhibitors
13.6. Inducible Degrader of LDLRs (IDOL)
14. Lomitapide
15. Vaccines against PCSK9
16. Plasmapheresis
17. Targeted Nanotherapy
18. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| ACL | ATP citrate lyase |
| ACS | acute coronary syndrome |
| ANGPTL3 | angiopoietin-like 3 |
| apo | apolipoprotein |
| ARR | absolute risk reduction |
| ASCVD | atherosclerotic cardiovascular disease |
| ASO | Antisense Oligonucleotides |
| BASs | Bile acid sequestrants |
| CA | cardiac arrest |
| CABG | coronary artery bypass graft |
| CAD | coronary artery disease |
| CETP | cholesteryl ester transfer protein |
| CKD | chronic kidney disease |
| CV | cardiovascular |
| DALI | direct adsorption of lipoproteins |
| DHA | docosahexaenoic acid |
| DM | diabetes mellitus |
| EPA | eicosapentaenoic acid |
| FDA | food and drug administration |
| FH | familial hypercholesterolemia |
| FPP | farnesyl diphosphate synthase |
| HDL-C | high-density lipoprotein–cholesterol |
| HeFH | heterozygous familial hypercholesterolemia |
| HF | heart failure |
| HIV | human immunodeficiency virus |
| HMG-CoA | hydroxymethylglutaryl-CoA |
| HoFH | Homozygous familial hypercholesterolemia |
| hsCRP | high sensitivity C-reactive protein |
| HTG-AP | hypertriglyceridemia-associated acute pancreatitis |
| IDOL | inducible degrader of LDL receptors |
| IFPTA | immunogenic fused PCSK9-tetanus |
| LDL-C | low-density lipoprotein–cholesterol |
| LDLRs | LDL receptors |
| Lp(a) | lipoprotein(a) |
| LPL | lipoprotein lipase |
| Lp-PLA2 | phospholipase A2 |
| LVEF | left ventricular ejection fraction |
| LXR | liver X receptor |
| MACEs | major adverse cardiovascular events |
| MI | Myocardial infarction |
| MTP | microsomal triglyceride transfer protein |
| NGAT | acyl-CoA:1,2-diacylglycerol acyltransferase |
| NNT | Number Needed to Treat |
| NPC1L1 | Niemann–Pick C1-like 1 |
| NYHA | New York Heart Association |
| OM3FAs | Omega-3 fatty acids |
| PAD | peripheral artery disease |
| PCI | percutaneous coronary intervention |
| PCSK9 | proprotein convertase subtilisin/kexin type 9 |
| PPARs | peroxisome proliferator-activated receptors |
| PUFA | polyunsaturated fatty acid |
| PVD | polyvascular disease |
| RCT | randomized clinical trial |
| RISC | RNA-induced silencing complex |
| RRR | relative risk reduction |
| SCD | sudden cardiac death |
| TGRL | triglyceride-rich lipoprotein |
| VLDL | very low-density lipoproteins |
References
- NCD Risk Factor Collaboration (NCD-RisC). Repositioning of the global epicenter of non-optimal cholesterol. Nature 2020, 582, 73–77. [Google Scholar] [CrossRef]
- Lazar, L.D.; Pletcher, M.J.; Coxson, P.G.; Bibbins-Domingo, K.; Goldman, L. Cost-effectiveness of statin therapy for primary prevention in a low-cost statin era. Circulation 2011, 124, 146–153. [Google Scholar] [CrossRef]
- McConnachie, A.; Walker, A.; Robertson, M.; Marchbank, L.; Peacock, J.; Packard, C.J.; Cobbe, S.M.; Ford, I. Long-term impact on healthcare resource utilization of statin treatment, and its cost effectiveness in the primary prevention of cardiovascular disease: A record linkage study. Eur. Heart J. 2014, 35, 290–298. [Google Scholar] [CrossRef]
- Heller, D.J.; Coxson, P.G.; Penko, J.; Pletcher, M.J.; Goldman, L.; Odden, M.C.; Kazi, D.S.; Bibbins-Domingo, K. Evaluating the Impact and Cost-Effectiveness of Statin Use Guidelines for Primary Prevention of Coronary Heart Disease and Stroke. Circulation 2017, 136, 1087–1098. [Google Scholar] [CrossRef]
- Wang, M.; Liu, J.; Bellows, B.K.; Qi, Y.; Sun, J.; Liu, J.; Moran, A.E.; Zhao, D. Impact of China’s Low Centralized Medicine Procurement Prices on the Cost -Effectiveness of Statins for the Primary Prevention of Atherosclerotic Cardiovascular Disease. Glob. Heart 2020, 15, 43. [Google Scholar] [CrossRef]
- Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry, 5th ed.; W. H. Freeman and Company: New York, NY, USA, 2008; pp. 343–389, 831–850. [Google Scholar]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, 3168–3209. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.; Cobbe, S.M.; Ford, I.; Isles, C.G.; Lorimer, A.R.; MacFarlane, P.W.; McKillop, J.H.; Packard, C.J. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N. Engl. J. Med. 1995, 333, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.R.; Clearfield, M.; Weis, S.; Whitney, E.; Shapiro, D.R.; Beere, P.A.; Langendorfer, A.; Stein, E.A.; Kruyer, W.; Gotto, A.M., Jr. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: Results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998, 279, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Bassler, D.; Briel, M.; Montori, V.M.; Lane, M.; Glasziou, P.; Zhou, Q.; Heels-Ansdell, D.; Walter, S.D.; Guyatt, G.H.; STOPIT-2 Study Group; et al. Stopping randomized trials early for benefit and estimation of treatment effects: Systematic review and meta-regression analysis. JAMA 2010, 303, 1180–1187. [Google Scholar] [CrossRef]
- Sever, P.S.; Dahlöf, B.; Poulter, N.R.; Wedel, H.; Beevers, G.; Caulfield, M.; Collins, R.; Kjeldsen, S.E.; Kristinsson, A.; McInnes, G.T.; et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): A multicentre randomised controlled trial. Lancet 2003, 361, 1149–1158. [Google Scholar]
- Nakamura, H.; Arakawa, K.; Itakura, H.; Kitabatake, A.; Goto, Y.; Toyota, T.; Nakaya, N.; Nishimoto, S.; Muranaka, M.; Yamamoto, A.; et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): A prospective randomised controlled trial. Lancet 2006, 368, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Bosch, J.; Dagenais, G.; Zhu, J.; Xavier, D.; Liu, L.; Pais, P.; López-Jaramillo, P.; Leiter, L.A.; Dans, A.; et al. Cholesterol Lowering in Intermediate-Risk Persons without Cardiovascular Disease. N. Engl. J. Med. 2016, 374, 2021–2031. [Google Scholar] [CrossRef] [PubMed]
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389.
- Sacks, F.M.; Pfeffer, M.A.; Moye, L.A.; Rouleau, J.L.; Rutherford, J.D.; Cole, T.G.; Brown, L.; Warnica, J.W.; Arnold, J.M.; Wun, C.C.; et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N. Engl. J. Med. 1996, 335, 1001–1009. [Google Scholar] [CrossRef]
- Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N. Engl. J. Med. 1998, 339, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 7–22. [Google Scholar] [CrossRef]
- Liem, A.; van Boven, A.J.; Withagen, A.P.; Robles de Medina, R.M.; Veeger, N.J.; Tijssen, J.G. Fluvastatin in Acute Myocardial Infarction: Effects on Early and Late Ischemia and Events: The FLORIDA Trial. Circulation 2000, 102, 2672. [Google Scholar] [CrossRef]
- Liem, A.H.; van Boven, A.J.; Veeger, N.J.; Withagen, A.J.; Robles de Medina, R.M.; Tijssen, J.G.; van Veldhuisen, D.J.; FLuvastatin On Risk Reduction after Acute Myocardial Infarction Study Group. Effect of fluvastatin on ischaemia following acute myocardial infarction: A randomized trial. Eur. Heart J. 2002, 23, 1931–1937. [Google Scholar] [CrossRef]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Rader, D.J.; Rouleau, J.L.; Belder, R.; Joyal, S.V.; Hill, K.A.; Pfeffer, M.A.; Skene, A.M.; et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N. Engl. J. Med. 2004, 350, 1495–1504. [Google Scholar] [CrossRef]
- Pedersen, T.R.; Faergeman, O.; Kastelein, J.J.; Olsson, A.G.; Tikkanen, M.J.; Holme, I.; Larsen, M.L.; Bendiksen, F.S.; Lindahl, C.; Szarek, M.; et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: The IDEAL study: A randomized controlled trial. JAMA 2005, 294, 2437–2445. [Google Scholar] [CrossRef]
- LaRosa, J.C.; Grundy, S.M.; Waters, D.D.; Shear, C.; Barter, P.; Fruchart, J.C.; Gotto, A.M.; Greten, H.; Kastelein, J.J.; Shepherd, J.; et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N. Engl. J. Med. 2005, 352, 1425–1435. [Google Scholar] [CrossRef]
- Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group; Armitage, J.; Bowman, L.; Wallendszus, K.; Bulbulia, R.; Rahimi, K.; Haynes, R.; Parish, S.; Peto, R.; Collins, R. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: A double-blind randomised trial. Lancet 2010, 376, 1658–1669. [Google Scholar]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.N.; Hitman, G.A.; Neil, H.A.; Livingstone, S.J.; Thomason, M.J.; Mackness, M.I.; Charlton-Menys, V.; Fuller, J.H.; et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): Multicentre randomised placebo-controlled trial. Lancet 2004, 364, 685–696. [Google Scholar] [CrossRef]
- Knopp, R.H.; d’Emden, M.; Smilde, J.G.; Pocock, S.J. Efficacy and safety of atorvastatin in the prevention of cardiovascular end points in subjects with type 2 diabetes: The Atorvastatin Study for Prevention of Coronary Heart Disease Endpoints in non-insulin-dependent diabetes mellitus (ASPEN). Diabetes Care 2006, 29, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Armitage, J.; Parish, S.; Sleigh, P.; Peto, R.; Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: A randomised placebo-controlled trial. Lancet 2003, 361, 2005–2016. [Google Scholar] [PubMed]
- Sever, P.S.; Poulter, N.R.; Dahlöf, B.; Wedel, H.; Collins, R.; Beevers, G.; Caulfield, M.; Kjeldsen, S.E.; Kristinsson, A.; McInnes, G.T.; et al. Reduction in cardiovascular events with atorvastatin in 2,532 patients with type 2 diabetes: Anglo-Scandinavian Cardiac Outcomes Trial-lipid-lowering arm (ASCOT-LLA). Diabetes Care 2005, 28, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Holdaas, H.; Fellström, B.; Jardine, A.G.; Holme, I.; Nyberg, G.; Fauchald, P.; Grönhagen-Riska, C.; Madsen, S.; Neumayer, H.H.; Cole, E.; et al. Effect of fluvastatin on cardiac outcomes in renal transplant recipients: A multicenter, randomized, placebo-controlled trial. Lancet 2003, 361, 2024–2031. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Krane, V.; März, W.; Olschewski, M.; Mann, J.F.; Ruf, G.; Ritz, E.; German Diabetes and Dialysis Study Investigators. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N. Engl. J. Med. 2005, 353, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Fellström, B.C.; Jardine, A.G.; Schmieder, R.E.; Holdaas, H.; Bannister, K.; Beutler, J.; Chae, D.W.; Chevaile, A.; Cobbe, S.M.; Grönhagen-Riska, C.; et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N. Engl. J. Med. 2009, 360, 1395–1407. [Google Scholar] [CrossRef]
- Baigent, C.; Landray, M.J.; Reith, C.; Emberson, J.; Wheeler, D.C.; Tomson, C.; Wanner, C.; Krane, V.; Cass, A.; Craig, J.; et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): A randomised placebo-controlled trial. Lancet 2011, 377, 2181–2192. [Google Scholar] [CrossRef]
- Kjekshus, J.; Apetrei, E.; Barrios, V.; Böhm, M.; Cleland, J.G.; Cornel, J.H.; Dunselman, P.; Fonseca, C.; Goudev, A.; Grande, P.; et al. Rosuvastatin in older patients with systolic heart failure. N. Engl. J. Med. 2007, 357, 2248–2261. [Google Scholar] [CrossRef]
- Tavazzi, L.; Maggioni, A.P.; Marchioli, R.; Barlera, S.; Franzosi, M.G.; Latini, R.; Lucci, D.; Nicolosi, G.L.; Porcu, M.; Tognoni, G.; et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 1231–1239. [Google Scholar] [PubMed]
- Shepherd, J.; Blauw, G.J.; Murphy, M.B.; Bollen, E.L.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; Jukema, J.W.; et al. PROspective Study of Pravastatin in the Elderly at Risk. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): A randomised controlled trial. Lancet 2002, 360, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Grinspoon, S.K.; Fitch, K.V.; Zanni, M.V.; Fichtenbaum, C.J.; Umbleja, T.; Aberg, J.A.; Overton, E.T.; Malvestutto, C.D.; Bloomfield, G.S.; Currier, J.S.; et al. Pitavastatin to Prevent Cardiovascular Disease in HIV Infection. N. Engl. J. Med. 2023, 389, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [PubMed]
- Tsouka, A.N.; Tellis, C.C.; Tselepis, A.D. Pharmacology of PCSK9 Inhibitors: Current Status and Future Perspectives. Curr. Pharm. Des. 2018, 24, 3622–3633. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Kereiakes, D.J.; McKenney, J.M.; Roth, E.M.; Hanotin, C.; Gipe, D.; Du, Y.; Ferrand, A.C.; Ginsberg, H.N.; Stein, E.A. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am. J. Cardiol. 2014, 114, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Giugliano, R.P.; Wiviott, S.D.; Atar, D.; Keech, A.; Kuder, J.F.; Im, K.; Murphy, S.A.; Flores-Arredondo, J.H.; López, J.A.G.; et al. Long-Term Evolocumab in Patients With Established Atherosclerotic Cardiovascular Disease. Circulation 2022, 146, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhao, X.; Qi, L.; Li, X.; Huang, Z.; Yang, G.; Qian, L.; Deng, H.; Li, H.; Huo, Y. A potential long-acting LDL-cholesterol-lowering PCSK9 monoclonal antibody: Randomized, placebo-controlled phase 1 studies. JACC Asia 2021, 1, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Chen, B.; Lian, Q.; Wang, S.; Liu, L.; Lu, D.; Qu, Y.; Zheng, G.; Li, L.; Ji, Y.; et al. Tafolecimab in Chinese patients with non-familial hypercholesterolemia (CREDIT-1): A 48-week randomized, double-blind, placebo-controlled phase 3 trial. Lancet Reg. Health West. Pac. 2023, 41, 100907. [Google Scholar] [CrossRef] [PubMed]
- Chai, M.; He, Y.; Zhao, W.; Han, X.; Zhao, G.; Ma, X.; Qiao, P.; Shi, D.; Liu, Y.; Han, W.; et al. Efficacy and safety of tafolecimab in Chinese patients with heterozygous familial hypercholesterolemia: A randomized, double-blind, placebo-controlled phase 3 trial (CREDIT-2). BMC Med. 2023, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.; Toth, P.; Butcher, M.B.; Kereiakes, D.; Magnu, P.; Bays, H.; Zhou, R.; Turner, T.A. Safety, tolerability and LDL-C reduction with a novel anti-PCSK9 recombinant fusion protein (LIB003): Results of a randomized, double-blind, placebo controlled, phase 2 study [abstract]. Atherosclerosis 2019, 287, e7. [Google Scholar] [CrossRef]
- Stein, E.A.; Turner, T.; Kereiakes, D.J.; Butcher, B.; Mangu, P.; Safety, Z.R. Tolerability and LDL-C reduction with LIB003 a novel anti-PCSK9 recombinant fusion protein: Results of open-label extension phase 2B study [abstract]. Circulation 2019, 140, A17222. [Google Scholar]
- Raal, F.; Fourie, N.; Scott, R.; Blom, R.; De Vries Basson, M.; Kayikcioglu, M.; Caldwell, K.; Kallend, D.; Stein, E.; LIBerate-HeFH Investigators. Long-term efficacy and safety of lerodalcibep in heterozygous familial hypercholesterolaemia: The LIBerate-HeFH trial. Eur. Heart J. 2023, 44, 4272–4280. [Google Scholar] [CrossRef]
- Ridker, P.M.; Lei, L.; Louie, M.J.; Haddad, T.; Nicholls, S.J.; Lincoff, A.M.; Libby, P.; Nissen, S.E. Inflammation and Cholesterol as Predictors of Cardiovascular Events among 13970 Contemporary High-Risk Patients with Statin Intolerance. Circulation 2024, 149, 28–35. [Google Scholar] [CrossRef]
- Phan, B.A.P.; Dayspring, T.D.; Toth, P.P. Ezetimibe therapy: Mechanism of action and clinical update. Vasc. Health Risk Manag. 2012, 8, 415–427. [Google Scholar]
- Dujovne, C.A.; Ettinger, M.P.; McNeer, J.F.; Lipka, L.J.; LeBeaut, A.P.; Suresh, R.; Yang, B.; Veltri, E.P.; Ezetimibe Study Group. Efficacy and safety of a potential new selective cholesterol absorption inhibitor, ezetimibe, in patients with primary hypercholesterolemia. Am. J. Cardiol. 2002, 90, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.A.; Denke, M.A.; McBride, P.E.; Battisti, W.P.; Brady, W.E.; Palmisano, J. A community-based, randomized trial of ezetimibe added to statin therapy to attain NCEP ATP III goals for LDL cholesterol in hypercholesterolemic patients: The ezetimibe add-on to statin for effectiveness (EASE) trial. Mayo Clin. Proc. 2005, 80, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J.; Akdim, F.; Stroes, E.S.; Zwinderman, A.H.; Bots, M.L.; Stalenhoef, A.F.; Visseren, F.L.; Sijbrands, E.J.; Trip, M.D.; Stein, E.A.; et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N. Engl. J. Med. 2008, 358, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, N.; Kawada-Watanabe, E.; Koyanagi, R.; Arashi, H.; Yamaguchi, J.; Nakao, K.; Tobaru, T.; Tanaka, H.; Oka, T.; Endoh, Y.; et al. Low-density lipoprotein cholesterol targeting with pitavastatin + ezetimibe for patients with acute coronary syndrome and dyslipidemia: The HIJ-PROPER study, a prospective, open-label, randomized trial. Eur. Heart J. 2017, 38, 2264–2276. [Google Scholar] [CrossRef]
- Ouchi, Y.; Sasaki, J.; Arai, H.; Yokote, K.; Harada, K.; Katayama, Y.; Urabe, T.; Uchida, Y.; Hayashi, M.; Yokota, N.; et al. Ezetimibe Lipid-Lowering Trial on Prevention of Atherosclerotic Cardiovascular Disease in 75 or Older (EWTOPIA 75): A Randomized, Controlled Trial. Circulation 2019, 140, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Cho, J.Y.; You, S.C.; Lee, Y.H.; Yun, K.H.; Cho, Y.H.; Shin, W.Y.; Im, S.W.; Kang, W.C.; Park, Y.; et al. Moderate-intensity statin with ezetimibe vs. high-intensity statin in patients with diabetes and atherosclerotic cardiovascular disease in the RACING trial. Eur. Heart J. 2023, 44, 972–983. [Google Scholar] [CrossRef]
- Pinkosky, S.L.; Newton, R.S.; Day, E.A.; Ford, R.J.; Lhotak, S.; Austin, R.C.; Birch, C.M.; Smith, B.K.; Filippov, S.; Groot, P.H.E.; et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 2016, 7, 13457. [Google Scholar] [CrossRef]
- Nissen, S.E.; Lincoff, A.M.; Brennan, D.; Ray, K.K.; Mason, D.; Kastelein, J.J.P.; Thompson, P.D.; Libby, P.; Cho, L.; Plutzky, J.; et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N. Engl. J. Med. 2023, 388, 1353–1364. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Laufs, U.; Ray, K.K.; Leiter, L.A.; Bays, H.E.; Goldberg, A.C.; Stroes, E.S.; MacDougall, D.; Zhao, X.; Catapano, A.L. Bempedoic acid plus ezetimibe fixed-dose combination in patients with hypercholesterolemia and high CVD risk treated with maximally tolerated statin therapy. Eur. J. Prev. Cardiol. 2022, 27, 593–603. [Google Scholar] [CrossRef]
- Mutschlechner, D.; Tscharre, M.; Huber, K.; Gremmel, T. Cardiovascular events in patients treated with bempedoic acid vs. placebo: Systematic review and meta-analysis. Eur. Heart J. Cardiovasc. Pharmacother. 2023, 9, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Lei, L.; Ray, K.K.; Ballantyne, C.M.; Bradwin, G.; Rifai, N. Effects of bempedoic acid on CRP, IL-6, fibrinogen and lipoprotein(a) in patients with residual inflammatory risk: A secondary analysis of the CLEAR harmony trial. J. Clin. Lipidol. 2023, 17, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Berg, K. A new serum type system in man—The Lp system. Acta Pathol. Microbiol. Scand. 1963, 59, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S. A test in context: Lipoprotein (a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Everett, B.M.; Caulfield, M.P.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Mora, S. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: An analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2014, 129, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.H.; Ballantyne, C.M.; Jacobson, T.A.; Bittner, V.A.; Braun, L.T.; Brown, A.S.; Brown, W.V.; Cromwell, W.C.; Goldberg, R.B.; McKenney, J.M.; et al. Clinical utility of inflammatory markers and advanced lipoprotein testing: Advice from an expert panel of lipid specialists. J. Clin. Lipidol. 2011, 5, 338–367. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Graham, I.; Backer, G.D.; Wiklund, O.; Chapman, M.J.; Drexel, H.; Hoes, A.W.; Jennings, C.S.; Landmesser, U.; Pedersen, T.R.; et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidemias. Eur. Heart J. 2016, 37, 2999–3058. [Google Scholar] [CrossRef] [PubMed]
- Yeang, C.; Hung, M.Y.; Byun, Y.S.; Clopton, P.; Yang, X.; Witztum, J.L.; Tsimikas, S. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). J. Clin. Lipidol. 2016, 10, 594–603. [Google Scholar] [CrossRef]
- Yeang, C.; Witztum, J.L.; Tsimikas, S. ‘LDL-C’ = LDL-C + Lp(a)-C: Implications of achieved ultra-low LDL-C levels in the proprotein convertase subtilisin/kexin type 9 era of potent LDL-C lowering. Curr. Opin. Lipidol. 2015, 26, 169–178. [Google Scholar] [CrossRef]
- AIM-HIGH Investigators; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar]
- Shlipak, M.G.; Simon, J.A.; Vittinghoff, E.; Lin, F.; Barrett-Connor, E.; Knopp, R.H.; Levy, R.I.; Hulley, S.B. Estrogen and Progestin, Lipoprotein(a), and the Risk of Recurrent Coronary Heart Disease Events After Menopause. JAMA 2000, 283, 1845–1852. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Einarsson, K.; Ericsson, S.; Ewerth, S.; Reihner, E.; Rudling, M.; Stahlberg, D.; Angelin, B. Bile acid sequestrants: Mechanisms of action on bile acid and cholesterol metabolism. Eur. J. Clin. Pharmacol. 1991, 40, S53–S58. [Google Scholar] [CrossRef] [PubMed]
- The Lipid Research Clinics Coronary: Primary Prevention Trial Results. JAMA 1984, 251, 365–374. [CrossRef]
- Heel, R.C.; Brogden, R.N.; Pakes, G.E.; Speight, T.M.; Avery, G.S. Colestipol: A review of its pharmacological properties and therapeutic efficacy in patients with hypercholesterolaemia. Drugs 1980, 19, 161–180. [Google Scholar] [CrossRef]
- Davidson, M.H.; Dillon, M.A.; Gordon, B.; Jones, P.; Samuels, J.; Weiss, S.; Isaacsohn, J.; Toth, P.; Burke, S.K. Colesevelam hydrochloride (cholestagel): A new, potent bile acid sequestrant associated with a low incidence of gastrointestinal side effects. Arch. Intern. Med. 1999, 159, 1893–1900. [Google Scholar] [CrossRef]
- Insull, W., Jr.; Toth, P.; Mullican, W.; Hunninghake, D.; Burke, S.; Donovan, J.M.; Davidson, M.H. Effectiveness of colesevelam hydrochloride in decreasing LDL cholesterol in patients with primary hypercholesterolemia: A 24-week randomized controlled trial. Mayo Clin. Proc. 2001, 76, 971–982. [Google Scholar] [CrossRef]
- Ross, S.; D’Mello, M.; Anand, S.S.; Eikelboom, J.; Stewart, A.F.R.; Samani, N.J.; Roberts, R.; Paré, G. The Effect of Bile Acid Sequestrants on the Risk of Cardiovascular Events: A Mendelian Randomization Analysis. Circ. Cardiovasc. Genet. 2015, 8, 618–627. [Google Scholar] [CrossRef]
- Coronary Drug Project Research Group. Clofibrate and niacin in coronary heart disease. JAMA 1975, 231, 360–381. [Google Scholar] [CrossRef]
- Carlson, L.A.; Danielson, M.; Ekberg, I.; Klintemar, B.; Rosenhamer, G. Reduction of myocardial reinfarction by the combined treatment with clofibrate and nicotinic acid. Atherosclerosis 1977, 28, 81–86. [Google Scholar] [CrossRef]
- Blankenhorn, D.H.; Nessim, S.A.; Johnson, R.L.; Sanmarco, M.E.; Azen, S.P.; Cashin-Hemphill, L. Beneficial effects of combined colestipol-niacin therapy on coronary atherosclerosis and coronary venous bypass grafts. JAMA 1987, 257, 3233–3240. [Google Scholar] [CrossRef]
- Brown, B.G.; Zhao, X.Q.; Chait, A.; Fisher, L.D.; Cheung, M.C.; Morse, J.S.; Dowdy, A.A.; Marino, E.K.; Bolson, E.L.; Alaupovic, P.; et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N. Engl. J. Med. 2001, 345, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.J.; Sullenberger, L.E.; Lee, H.J.; Grace, K.A. Arterial biology for the investigation of the treatment effects of reducing cholesterol (ARBITER) 2: A double-blind, placebo-controlled study of extended release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation 2004, 110, 3512–3517. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.J.; Villines, T.C.; Stanek, E.J.; Devine, P.J.; Griffen, L.; Miller, M.; Weissman, N.J.; Turco, M. Extended-release niacin or ezetimibe and carotid intima-media thickness. N. Engl. J. Med. 2009, 361, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Whitney, E.J.; Krasuski, R.A.; Personius, B.E.; Michalek, J.E.; Maranian, A.M.; Kolasa, M.W.; Monick, E.; Brown, G.; Gotto, A.M., Jr. A randomized trial of a strategy for increasing high-density lipoprotein cholesterol levels: Effects on progression of coronary heart disease and clinical events. Ann. Intern. Med. 2005, 142, 95–104. [Google Scholar] [CrossRef] [PubMed]
- HPS2-THRIVE Collaborative Group; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [PubMed]
- D’Andrea, E.; Hey, S.P.; Ramirez, C.L.; Kesselheim, A.S. Assessment of the Role of Niacin in Managing Cardiovascular Disease Outcomes. JAMA Netw. Open 2019, 2, e192224. [Google Scholar] [CrossRef] [PubMed]
- Ronsein, G.E.; Vaisar, T.; Davidson, W.S.; Bornfeldt, K.E.; Probstfield, J.L.; O’Brien, K.D.; Zhao, X.Q.; Heinecke, J.W. Niacin Increases Atherogenic Proteins in High-Density Lipoprotein of Statin-Treated Subjects. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2330–2341. [Google Scholar] [CrossRef]
- Frick, M.H.; Elo, O.; Haapa, K.; Heinonen, O.P.; Heinsalmi, P.; Helo, P.; Huttunen, J.K.; Kaitaniemi, P.; Koskinen, P.; Manninen, V.; et al. Helsinki Heart Study: Primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N. Engl. J. Med. 1987, 317, 1237–1245. [Google Scholar] [CrossRef]
- Rubins, H.B.; Robins, S.J.; Collins, D.; Fye, C.L.; Anderson, J.W.; Elam, M.B.; Faas, F.H.; Linares, E.; Schaefer, E.J.; Schectman, G.; et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 1999, 341, 410–418. [Google Scholar] [CrossRef]
- Bezafibrate Infarction Prevention (BIP) Study. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation 2000, 102, 21–27. [Google Scholar] [CrossRef]
- Meade, T.; Zuhrie, R.; Cook, C.; Cooper, J. Bezafibrate in men with lower extremity arterial disease: Randomised controlled trial. BMJ 2002, 325, 1139. [Google Scholar] [CrossRef]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- The ACCORD Study Group. Effects of Combination Lipid Therapy in Type 2 Diabetes Mellitus. N. Engl. J. Med. 2010, 362, 1563–1574. [Google Scholar] [CrossRef]
- Kim, N.H.; Han, K.H.; Choi, J.; Lee, J.; Kim, S.G. Use of fenofibrate on cardiovascular outcomes in statin users with metabolic syndrome: Propensity matched cohort study. BMJ 2019, 366, l5125. [Google Scholar] [CrossRef]
- Das Pradhan, A.; Glynn, R.J.; Fruchart, J.-C.; MacFadyen, J.G.; Zaharris, E.S.; Everett, B.M.; Campbell, S.E.; Oshima, R.; Amarenco, P.; Blom, D.J.; et al. Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. N. Engl. J. Med. 2022, 387, 1923–1934. [Google Scholar] [CrossRef]
- Jakob, T.; Nordmann, A.J.; Schandelmaier, S.; Ferreira-González, I.; Briel, M. Fibrates for primary prevention of cardiovascular disease events. Cochrane Database Syst. Rev. 2016, 11, CD009753. [Google Scholar] [CrossRef]
- Millan, J.; Ointó, X.; Brea, A.; Blasco, M.; Hernández-Mijares, A.; Ascaso, J.; Diaz, A.; Mantilla, T.; Pedro-Botet, J. Fibrates in the secondary prevention of cardiovascular disease (infarction and stroke). Results of a systematic review and meta-analysis of the Cochrane collaboration. Clin. Investig. Arterioscler. 2018, 30, 30–35. [Google Scholar] [CrossRef]
- Le Jossic-Corcos, C.; Gonthier, C.; Zaghini, I.; Logette, E.; Shechter, I.; Bournot, P. Hepatic farnesyl diphosphate synthase expression is suppressed by polyunsaturated fatty acids. Biochem. J. 2005, 385 Pt 3, 787–794. [Google Scholar] [CrossRef]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomized open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- Rauch, B.; Schiele, R.; Schneider, S.; Diller, F.; Victor, N.; Gohlke, H.; Gottwik, M.; Steinbeck, G.; Del Castillo, U.; Sack, R.; et al. OMEGA, a randomized, placebo-controlled trial to test the effect of highly purified omega-3 fatty acids on top of modern guideline-adjusted therapy after myocardial infarction. Circulation 2010, 122, 2152–2159. [Google Scholar] [CrossRef]
- Manson, J.E.; Cook, N.R.; Lee, I.-M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Albert, C.M.; Gordon, D.; Copeland, T.; et al. Marine n-3 Fatty Acids and Prevention of Cardiovascular Disease and Cancer. N. Engl. J. Med. 2019, 380, 23–32. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; MacFadyen, J.; Glynn, R.J.; Jiao, L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Tardif, J.-C.; et al. Effects of randomized treatment with icosapent ethyl and a mineral oil comparator on interleukin-1β, interleukin-6, C-reactive protein, oxidized low-density lipoprotein cholesterol, homocysteine, lipoprotein(a), and lipoprotein-associated phospholipase A2: The REDUCE-IT biomarker substudy. Circulation 2022, 146, 372–379. [Google Scholar]
- Nicholls, S.J.; Lincoff, A.M.; Garcia, M.; Bash, D.; Ballantyne, C.M.; Barter, P.J.; Davidson, M.H.; Kastelein, J.J.P.; Koenig, W.; McGuire, D.K.; et al. Effect of High-Dose Omega-3 Fatty Acids vs Corn Oil on Major Adverse Cardiovascular Events in Patients at High Cardiovascular Risk. JAMA 2020, 324, 2268–2280. [Google Scholar] [CrossRef]
- Kalstad, A.A.; Myhre, P.L.; Laake, K.; Tvelt, S.H.; Schmidt, E.B.; Smith, P.; Nilsen, D.W.T.; Tveit, A.; Fagerland, M.W.; Solheim, S.; et al. Effects of n-3 Fatty Acid Supplements in Elderly Patients After Myocardial Infarction. Circulation 2021, 143, 528–539. [Google Scholar] [CrossRef]
- Khan, S.U.; Lone, A.N.; Khan, M.S.; Virani, S.S.; Blumenthal, R.S.; Nasir, K.; Miller, M.; Michos, E.D.; Ballantyne, C.M.; Boden, W.E.; et al. Effect of omega-3 fatty acids on cardiovascular outcomes: A systematic review and meta-analysis. EClinicalMedicine 2021, 38, 100997. [Google Scholar] [CrossRef]
- Hunt, J.A.; Lu, Z. Cholesteryl ester transfer protein (CETP) inhibitors. Curr. Top. Med. Chem. 2009, 9, 419–427. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef]
- HPS3/TIMI55–REVEAL Collaborative Group; Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ditmarsch, M.; Kastelein, J.J.; Rigby, S.P.; Kling, D.; Curcio, D.L.; Alp, N.J.; Davidson, M.H. Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: A randomized phase 2 trial. Nat. Med. 2022, 28, 1672–1678. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Ditmarsch, M.; Kastelein, J.J.; Nelson, A.J.; Kling, D.; Hsieh, A.; Curcio, D.L.; Maki, K.C.; Davidson, M.H.; Nicholls, S.J. Obicetrapib plus ezetimibe as an adjunct to high-intensity statin therapy: A randomized phase 2 trial. J. Clin. Lipidol. 2023, 17, 491–503. [Google Scholar] [CrossRef]
- Grossman, M.; Rader, D.J.; Muller, D.W.; Kolansky, D.M.; Kozarsky, K.; Clark, B.J., 3rd; Stein, E.A.; Lupien, P.J.; Brewer, H.B., Jr.; Raper, S.E.; et al. A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia. Nat. Med. 1995, 1, 1148–1154. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, L.Y.; Cheng, X.S. Novel Approaches for the Treatment of Familial Hypercholesterolemia: Current Status and Future Challenges. J. Atheroscler. Thromb. 2018, 25, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Martínez, T.; Jiménez, A.I.; Pañeda, C. Short-interference RNAs: Becoming medicines. EXCLI J. 2015, 14, 714–746. [Google Scholar]
- Nissen, S.E.; Linnebjerg, H.; Shen, X.; Wolski, K.; Ma, X.; Lim, S.; Michael, L.F.; Ruotolo, G.; Gribble, G.; Navar, A.M.; et al. Lepodisiran, an Extended-Duration Short Interfering RNA Targeting Lipoprotein(a): A Randomized Dose-Ascending Clinical Trial. JAMA 2023, 330, 2075–2083. [Google Scholar] [CrossRef]
- Khan, S.A.; Naz, A.; Masood, M.Q.; Shah, R. Meta-Analysis of Inclisiran for the Treatment of Hypercholesterolemia. Am. J. Cardiol. 2020, 134, 69–73. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Estrella, A.M.; Sourlas, A.; Silverio, D.; Hilario, E.; Montan, P.D.; Guzman, E. Inclisiran: A New Promising Agent in the Management of Hypercholesterolemia. Diseases 2018, 6, 63. [Google Scholar] [CrossRef]
- Fitzgerald, K.; Frank-Kamenetsky, M.; Shulga-Morskaya, S.; Liebow, A.; Bettencourt, B.R.; Sutherland, J.E.; Hutabarat, R.M.; Clausen, V.A.; Karsten, V.; Cehelsky, J.; et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: A randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 2014, 383, 60–68. [Google Scholar] [CrossRef]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.T.; Turner, T.; Visseren, F.L.J.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef]
- Ray, K.K.; Troquay, R.P.T.; Visseren, F.L.J.; Leiter, L.A.; Wright, R.S.; Vikarunnessa, S.; Talloczy, Z.; Zang, X.; Maheux, P.; Lesogor, A.; et al. Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION-3): Results from the 4-year open-label extension of the ORION-1 trial. Lancet Diabetes Endoncrinol. 2023, 11, 109–119. [Google Scholar] [CrossRef]
- Raal, F.; Durst, R.; Bi, R.; Talloczy, Z.; Maheux, P.; Lesogor, A.; Kastelein, J.J.P.; on behalf of the ORION-5 Study Investigators. Efficacy, Safety, and Tolerability of Inclisiran in Patients With Homozygous Familial Hypercholesterolemia: Results From the ORION-5 Randomized Clinical Trial. Circulation 2024, 149, 354–362. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Eng. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- Ray, K.K.; Phil, M.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Eng. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Koenig, W.; Conde, L.G.; Landmesser, U.; Leiter, L.A.; Ray, K.K.; Schwartz, G.G.; Wright, R.S.; Han, J.; Raal, F.J. Efficacy and Safety of Inclisiran in Patients with Polyvascular Disease: Pooled, Post Hoc Analysis of the ORION-9, ORION-10, and ORION-11 Phase 3 Randomized Controlled Trials. Cardiovasc. Drugs Ther. 2022. epub ahead of print. [Google Scholar] [CrossRef]
- Ray, K.K.; Raal, F.J.; Kallend, D.G.; Jaros, M.J.; Koenig, W.; Leiter, L.A.; Landmesser, U.; Schwartz, G.G.; Lawrence, D.; Friedman, A.; et al. Inclisiran and cardiovascular events: A patient-level analysis of phase III trials. Eur. Heart J. 2023, 44, 129–138. [Google Scholar] [CrossRef]
- Stoekenbroek, R.M.; Kallend, D.; Wijngaard, P.L.; Kastelein, J.J. Inclisiran for the treatment of cardiovascular disease: The ORION clinical development program. Future Cardiol. 2018, 14, 433–442. [Google Scholar] [CrossRef]
- Kersten, S. ANGPTL3 as therapeutic target. Curr. Opin. Lipidol. 2021, 32, 335–341. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Pottanat, T.G.; Siegel, R.W.; Ehsani, M.; Qian, Y.; Zhen, E.Y.; Regmi, A.; Roell, W.C.; Guo, H.; Luo, M.L.; et al. Angiopoietin-like protein 8 differentially regulates ANGPTL3 and ANGPTL4 during postprandial partitioning of fatty acids. J. Lipid. Res. 2020, 61, 1203–1220. [Google Scholar] [CrossRef]
- Oldoni, F.; Cheng, H.; Banfi, S.; Gusarova, V.; Cohen, J.C.; Hobbs, H.H. ANGPTL8 has both endocrine and autocrine effects on substrate utilization. JCI Insight 2020, 5, e138777. [Google Scholar] [CrossRef]
- Kovrov, O.; Kristensen, K.K.; Larsson, E.; Ploug, M.; Olivecrona, G. On the mechanism of angiopoietin-like protein 8 for control of lipoprotein lipase activity. J. Lipid Res. 2019, 60, 783–793. [Google Scholar] [CrossRef]
- Gusarova, V.; Alexa, C.A.; Wang, Y.; Rafique, A.; Kim, J.H.; Buckler, D.; Mintah, I.J.; Shihanian, L.M.; Cohen, J.C.; Hobbs, H.H.; et al. ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. J. Lipid Res. 2015, 56, 1308–1317. [Google Scholar] [CrossRef]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Hout, C.V.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef]
- Raal, F.J.; Rosenson, R.S.; Reeskamp, L.F.; Hovingh, G.K.; Kastelein, J.J.P.; Rubba, P.; Ali, S.; Banerjee, P.; Chan, K.C.; Gipe, D.A.; et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Eng. J. Med. 2020, 383, 711–720. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Burgess, L.J.; Ebenbichler, C.F.; Baum, S.J.; Stroes, E.S.G.; Ali, S.; Khilla, N.; Hamlin, R.; Pordy, R.; Dong, Y.; et al. Evinacumab in Patients with Refractory Hypercholesterolemia. N. Eng. J. Med. 2020, 383, 2307–2319. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, k.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- El-Mounadi, K.; Morales-Floriano, M.L.; Garcia-Ruiz, H. Principles, Applications, and Biosafety of Plant Genome Editing Using CRISPR-Cas9. Front. Plant. Sci. 2020, 11, 56. [Google Scholar] [CrossRef]
- Sun, J.; Wang, J.; Zheng, D.; Hu, X. Advances in therapeutic application of CRISPR-Cas9. Brief. Funct. Genomics 2020, 19, 164–174. [Google Scholar] [CrossRef]
- Liang, P.; Xu, Y.; Zhang, X.; Ding, C.; Huang, R.; Zhang, Z.; Lv, J.; Xie, X.; Chen, Y.; Li, Y.; et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 2015, 6, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, Y.; He, L.; Pu, W.; Yu, W.; Li, Y.; Wu, Y.-T.; Xu, C.; Wei, Y.; Ding, Q.; et al. In Vivo AAV-CRISPR/Cas9-Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation 2020, 141, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Muthuramu, I.; Somanathan, S.; Zhang, H.; Bell, P.; He, Z.; Yu, H.; Zhu, Y.; Tretiakova, A.P.; Wilson, J.M. Developing a second-generation clinical candidate AAV vector for gene therapy of familial hypercholesterolemia. Mol. Ther. Methods Clin. Dev. 2021, 22, 1–10. [Google Scholar] [CrossRef]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and safety of an investigational single-course CRISPR base-editing therapy targeting PCSK9 in nonhuman primate and mouse models. Circulation 2023, 147, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Duell, P.; Santos, R.; Kirwan, B.; Witztum, J.; Tsimikas, S.; Kastelein, J. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J. Clin. Lipidol. 2016, 10, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Astaneh, B.; Makhdami, N.; Astaneh, V.; Guyatt, G. The Effect of Mipomersen in the Management of Patients with Familial Hypercholesterolemia: A Systematic Review and Meta-Analysis of Clinical Trials. J. Cardiovasc. Dev. Dis. 2021, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Salinas, C.A.; Gómez-Díaz, R.A.; Corral, P. New Therapies for Primary Hyperlipidemia. J. Clin. Endocrinol. Metab. 2022, 107, 1216–1224. [Google Scholar] [CrossRef]
- Blom, D.J.; O’Dea, L.; Digenio, A.; Alexander, V.J.; Karwatowska-Prokopczuk, E.; Williams, K.R.; Hemphill, L.; Muñiz-Grijalvo, O.; Santos, R.D.; Baum, S.; et al. Characterizing familial chylomicronemia syndrome: Baseline data of the APPROACH study. J Clin Lipidol. 2018, 12, 1234–1243.e5. [Google Scholar] [CrossRef]
- Gouni-Berthold, I.; Alexander, V.J.; Yang, Q.; Hurh, E.; Steinhagen-Thiessen, E.; Moriarty, P.M.; Hughes, S.G.; Gaudet, D.; Hegele, R.A.; O’Dea, L.S.L.; et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronemia (COMPASS): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021, 9, 264–275. [Google Scholar] [CrossRef]
- Oral, E.A.; Garg, A.; Tami, J.; Huang, E.A.; O’Dea, L.S.L.; Schmidt, H.; Tiulpakov, A.; Mertens, A.; Alexander, V.J.; Watts, L.; et al. Assessment of efficacy and safety of volanesorsen for treatment of metabolic complications in patients with familial partial lipodystrophy: Results of the BROADEN study: Volanesorsen in FPLD; The BROADEN Study. J. Clin. Lipidol. 2022, 16, 833–849. [Google Scholar] [CrossRef]
- Tardif, J.C.; Karwatowska-Prokopczuk, E.; Amour, E.S.; Ballantyne, C.M.; Shapiro, M.D.; Moriarty, P.M.; Baum, S.J.; Hurh, E.; Bartlett, V.J.; Kingsbury, J.; et al. Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridemia and at high cardiovascular risk. Eur. Heart J. 2022, 43, 1401–1412. [Google Scholar] [CrossRef]
- Sniderman, A.D.; Navar, A.M.; Thanassoulis, G. Apolipoprotein B vs. low density lipoprotein cholesterol and non-high-density lipoprotein cholesterol as the primary measure of apolipoprotein B lipoprotein-related risk: The debate is over. JAMA Cardiol. 2022, 7, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.G.; Sanderson, E.; Palmer, T.M.; Ala-Korpela, M.; Ference, B.A.; Davey, S.G.; Holmes, M.V.; Rader, D.J. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020, 17, e1003062. [Google Scholar] [CrossRef] [PubMed]
- Zuber, V.; Gill, D.; Ala-Korpela, M.; Langenberg, C.; Butterworth, A.; Bottolo, L.; Burgess, S. High-throughput multivariable Mendelian randomization analysis prioritizes apolipoprotein B as key lipid risk factor for coronary artery disease. Int. J. Epidemiol. 2021, 50, 893–901. [Google Scholar] [CrossRef]
- Yuan, S.; Tang, B.; Zheng, J.; Larsson, S.C. Circulating lipoprotein lipids, apolipoproteins and ischemic stroke. Ann. Neurol. 2020, 88, 1229–1236. [Google Scholar] [CrossRef]
- Levin, M.G.; Zuber, V.; Walker, V.M.; Klarin, D.; Lynch, J.; Malik, R.; Aday, A.W.; Bottolo, L.; Pradhan, A.D.; Dichgans, M.; et al. Prioritizing the role of major lipoproteins and subfractions as risk factors for peripheral artery disease. Circulation 2021, 144, 353–364. [Google Scholar] [CrossRef]
- Richardson, T.G.; Wang, Q.; Sanderson, E.; Mahajan, A.; McCarthy, M.I.; Frayling, T.M.; Ala-Korpela, M.; Sniderman, A.; Smith, G.D.; Holmes, M.V. Effects of apolipoprotein B on lifespan and risks of major diseases including type 2 diabetes: A mendelian randomisation analysis using outcomes in first-degree relatives. Lancet Healthy Longev. 2021, 2, e317–e326. [Google Scholar] [CrossRef]
- Johannesen, C.D.; Mortensen, M.B.; Langsted, A.; Nordestgaard, B. Apolipoprotein B and Non-HDL-C better reflect residual risk than LDL cholesterol in statin-treated patients with atherosclerosis. J. Am. Coll. Cardiol. 2021, 77, 1439–1450. [Google Scholar] [CrossRef]
- Marston, N.A.; Giugliano, R.P.; Melloni, G.; Park, J.G.; Morrill, V.; Blazing, M.A.; Ference, G.A.; Stein, E.; Stroes, E.; Braunwald, E.; et al. Association of apolipoprotein-B-containing lipoproteins and risk of myocardial infarction in individuals with and without atherosclerosis: Distinguishing between particle concentration, Type and Content. JAMA Cardiol. 2022, 7, 250–256. [Google Scholar] [CrossRef]
- Kastelein, J.J.P.; Wedel, M.K.; Baker, B.F.; Su, J.; Bradley, J.D.; Yu, R.Z.; Chuang, E.; Graham, M.J.; Crooke, R.M. Potent Reduction of Apolipoprotein B and Low-Density Lipoprotein Cholesterol by Short-Term Administration of an Antisense Inhibitor of Apolipoprotein B. Circulation 2006, 114, 1729–1735. [Google Scholar] [CrossRef]
- Li, N.; Li, Q.; Tian, X.Q.; Qian, H.Y.; Yang, Y.J. Mipomersen is a Promising Therapy in the Management of Hypercholesterolemia: A Meta-Analysis of Randomized Controlled Trials. Am. J. Cardiovasc. Drugs 2014, 14, 367–376. [Google Scholar] [CrossRef]
- Fogacci, F.; Ferri, N.; Toth, P.P.; Ruscica, M.; Corsini, A.; Cicero, A.F.G. Efficacy and Safety of Mipomersen: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Drugs 2019, 79, 751–766. [Google Scholar] [CrossRef]
- Zelcer, N.; Hong, C.; Boyadjian, R.; Tontonoz, P. LXR Regulates Cholesterol Uptake through Idol-dependent Ubiquitination of the LDL Receptor. Science 2009, 325, 100–104. [Google Scholar] [CrossRef]
- Liang, C.; Wang, X.; Peng, K.; Lai, P.; Liu, Z.; Ma, J.; Chen, X.; Liu, G.; Zheng, M.; Wang, Y.; et al. Idol Depletion Protects against Spontaneous Atherosclerosis in a Hamster Model of Familial Hypercholesterolemia. Oxid. Med. Cell. Longev. 2022, 2022, 1889632. [Google Scholar] [CrossRef] [PubMed]
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271. [Google Scholar] [CrossRef]
- Waterworth, D.M.; Ricketts, S.L.; Song, K.; Chen, L.; Zhao, J.H.; Ripatti, S.; Aulchenko, Y.S.; Zhang, W.; Yuan, X.; Lim, N.; et al. Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2264–2276. [Google Scholar] [CrossRef]
- Blom, D.J.; Raal, F.J.; Santos, R.D.; Marais, A.D. Lomitapide and Mipomersen-Inhibiting Microsomal Triglyceride Transfer Protein (MTP) and apoB100 Synthesis. Curr. Atheroscler. Rep. 2019, 21, 48. [Google Scholar] [CrossRef]
- Hooper, A.J.; Burnett, J.R.; Watts, G.F. Contemporary aspects of the biology and therapeutic regulation of the microsomal triglyceride transfer protein. Circ. Res. 2015, 116, 193–205. [Google Scholar] [CrossRef]
- Cuchel, M.; Bloedon, L.T.; Szapary, P.O.; Kolansky, D.M.; Wolfe, M.L.; Sarkis, A.; Millar, J.S.; Ikewaki, K.; Siegelman, E.S.; Gregg, R.E. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N. Engl. J. Med. 2007, 356, 148–156. [Google Scholar] [CrossRef]
- Cuchel, M.; Meagher, E.A.; Theron, H.T.; Blom, D.J.; Marais, A.D.; Hegele, R.A.; Averna, M.R.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet 2013, 381, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Landlinger, C.; Pouwer, M.G.; Juno, C.; van der Hoorn, J.W.A.; Pieterman, E.J.; Jukema, J.W.; Staffler, G.; Princen, H.M.G.; Galabova, G. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE*3Leiden.CETP mice. Eur. Heart J. 2017, 38, 2499–2507. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Jaafari, M.R.; Badiee, A.; Banach, M.; Sahebkar, A. Therapeutic effect of nanoliposomal PCSK9 vaccine in a mouse model of atherosclerosis. BMC Med. 2019, 17, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Crossey, E.; Amar, M.J.A.; Sampson, M.; Peabody, J.; Schiller, J.T.; Chackerian, B.; Remaley, A.T. A cholesterol-lowering VLP vaccine that targets PCSK9. Vaccine 2015, 33, 5747–5755. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, R.; Nozato, Y.; Nakagami, H.; Ikeda, Y.; Shimamura, M.; Yoshida, S.; Sun, J.; Kawano, T.; Takami, Y.; Noma, T.; et al. Development of vaccine for dyslipidemia targeted to a proprotein convertase subtilisin/kexin type 9 (PCSK9) epitope in mice. PLoS ONE 2018, 13, e0191895. [Google Scholar] [CrossRef]
- Fattori, E.; Cappelletti, M.; Lo Surdo, P.; Calzetta, A.; Bendtsen, C.; Ni, Y.G.; Pandit, S.; Sitlani, A.; Mesiti, G.; Carfí, A.; et al. Immunization against proprotein convertase subtilisin-like/kexin type 9 lowers plasma LDL-cholesterol levels in mice. J. Lipid Res. 2012, 53, 1654–1661. [Google Scholar] [CrossRef]
- Pan, Y.; Zhou, Y.; Wu, H.; Chen, X.; Hu, X.; Zhang, H.; Zhou, Z.; Qiu, Z.; Liao, Y. A therapeutic peptide vaccine against PCSK9. Sci. Rep. 2017, 7, 12534. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Jaafari, M.R.; Afshar, M.; Banach, M.; Sahebkar, A. PCSK9 immunization using nanoliposomes: Preventive efficacy against hypercholesterolemia and atherosclerosis. Arch. Med. Sci. 2021, 17, 1365–1377. [Google Scholar] [CrossRef]
- Fowler, A.; Van Rompay, K.K.A.; Sampson, M.; Leo, J.; Watanabe, J.K.; Usachenko, J.L.; Immareddy, R.; Lovato, D.M.; Schiller, J.T.; Remaley, A.T.; et al. A Virus-like particle-based bivalent PCSK9 vaccine lowers LDL-cholesterol levels in Non-Human Primates. npj Vaccines 2023, 8, 142. [Google Scholar] [CrossRef]
- King, M.E.; Breslow, J.L.; Lees, R.S. Plasma-exchange therapy of homozygous familial hypercholesterolemia. N. Engl. J. Med. 1980, 302, 1457–1459. [Google Scholar] [CrossRef]
- Taylan, C.; Weber, L.T. An update on lipid apheresis for familial hypercholesterolemia. Pediatr. Nephrol. 2023, 38, 371–382. [Google Scholar] [CrossRef]
- Albayrak, M.; Yıldız, A.; Ateş, N.; Pala, Ç. The efficacy of double filtration plasmapheresis in the treatment of homozygous familial hypercholesterolemia: A single-center experience. Transfus. Apher. Sci. 2019, 58, 61–64. [Google Scholar] [CrossRef]
- Cao, L.; Chen, Y.; Liu, S.; Huang, W.; Wu, D.; Hong, D.; Wang, Z.; Sun, Y.; Qin, K.; Guo, F.; et al. Chinese Acute Pancreatitis Clinical Trials Group (CAPCTG). Early Plasmapheresis Among Patients With Hypertriglyceridemia-Associated Acute Pancreatitis. JAMA Netw. Open 2023, 6, e2320802. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Shi, D.; Cui, Q.; Yu, S.; Yang, J.; Song, P.; Walline, J.; Xu, J.; Zhu, H.; Yu, X. Intensive insulin therapy versus plasmapheresis in the management of hypertriglyceridemia-induced acute pancreatitis (Bi-TPAI trial): Study protocol for a randomized controlled trial. Trials 2019, 20, 365. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Allen, T.M.; Cullis, P.R. Lipid nanoparticle delivery systems for siRNA-based therapeutics. Drug Deliv. Transl. Res. 2014, 4, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Bu, H.; Gao, Z.; Huang, Y.; Gao, F.; Li, F. The characteristics and mechanism of simvastatin loaded lipid nanoparticles to increase oral bioavailability in rats. Int. J. Pharm. 2010, 394, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Rakshit, M.; Darwitan, A.; Muktabar, A.; Das, P.; Nguyen, L.T.H.; Cao, Y.; Vizetto-Duarte, C.; Tang, J.K.; Wong, Y.S.; Venkatraman, S.; et al. Anti-inflammatory potential of simvastatin loaded nanoliposomes in 2D and 3D foam cell models. Nanomedicine 2021, 37, 102434. [Google Scholar] [CrossRef]
- Salaheldin, T.A.; Godugu, K.; Bharali, D.J.; Fujioka, K.; Elshourbagy, N.; Mousa, S.A. Novel oral nano-hepatic targeted anti-PCSK9 in hypercholesterolemia. Nanomedicine 2022, 40, 102480. [Google Scholar] [CrossRef]


| Study | Sample Size | Characteristics of Patients | Comparison Groups | Follow-Up | LDL-C Reduction | CV Effects |
|---|---|---|---|---|---|---|
| FOURIER (2017) [41] | 27,564 | Documented atherosclerosis + LDL-C > 70 mg/dL + on statin therapy | Evolocumab (140 mg every 2 weeks or 420 mg monthly) vs. placebo | 2.2 years | 59% | Reduction in CV death, MI, stroke, UA hospitalization, or coronary revascularization (HR 0.85, 95% CI 0.79 to 0.92, NNT 67) |
| ODYSSEY OUTCOMES (2018) [42] | 18,924 | ACS < 1–12 months + LDL-C > 70 mg/dL + on high-intensity statin or maximum tolerated dose | Alirocumab 75 mg vs. placebo | 2.8 years | 54.7% | Reduction in coronary death, MI, stroke, or UA hospitalization (HR 0.85, 95% CI 0.78 to 0.93, NNT 63) |
| Study | Sample Size | Characteristics of Patients | Comparison Groups | Follow-Up | LDL-C Reduction | CV Effects |
|---|---|---|---|---|---|---|
| EASE (2005) [53] | 3030 | LDL-C > NCEP ATP III goals + TGs < 350 mg/dL + on statin | Ezetimibe 10 mg vs. placebo | 6 weeks | 25.8% | NE |
| ENHANCE (2008) [54] | 720 | FH | Simvastatin 80 mg vs. simvastatin 80 mg + ezetimibe 10 mg | 2 years | 55.6% | Reduction in carotid artery intima-media thickness |
| IMPROVE-IT (2015) [55] | 18,144 | ACS < 10 days + LDL-C 50–125 mg/dL (50–100 mg/dL if prior lipid-lowering therapy) | Simvastatin 40 mg vs. simvastatin 40 mg + ezetimibe 10 mg | 6 years | 25.7% vs. 43.4% | Reduction in CV events (HR 0.93, 95% CI 0.89 to 0.99, NNT 50) |
| HIJ-PROPER (2017) [56] | 1734 | ACS + LDL-C > 100 mg/dL + TGs < 400 mg/dL | Pitavastatin (1–4 mg) vs. pitavastatin (1–4 mg) + ezetimibe 10 mg | 3.8 years | 37.6% vs. 51.7% | No significant differences in CV events |
| EWTOPIA 75 (2019) [57] | 3411 | ≥75 years + LDL-C > 140 mg/dL + ≥1 high risk factor | Ezetimibe 10 mg vs. control | 4.1 years | 25.9% | Reduction in SCD, MI, PCI, or CABG, or stroke (HR 0.66 95% CI 0.50 to 0.86, NNT 38.5) |
| RACING (2023) [58] | 3780 | Documented atherosclerotic disease | Rosuvastatin 10 mg + ezetimibe 10 mg vs. rosuvastatin 20 mg | 3 years | 58% vs. 72% | No significant differences in CV events |
| Study | Sample Size | Characteristics of Patients | Comparison Groups | Follow-Up | Lipids Effect | CV Effects |
|---|---|---|---|---|---|---|
| CDP (1975) [81] | 8341 | Men with previous MI | Niacin (1 g 3×) vs. placebo | 6 years | Reduction TC 9.9% | Reduction in nonfatal MI 27% Reduction in cerebrovascular events 24% |
| Stockholm trial (1977) [82] | 558 | ACS + aged <70 years | Clofibrate (1 g 2×) + niacin (1 g 3×) vs. placebo | 5 years | Reduction TC 26% | Reduction in CV events (HR 0.59, 95% CI 0.41 to 0.84) |
| CLAS (1987) [83] | 162 | Men aged 40–59 years + previous CABG | Niacin (3–12 g) + colestipol (30 g) vs. placebo | 2 years | Reduction LDL-C 43% Increase HDL-C 37% | Reduction in atherosclerotic regression (16.2% vs. 2.4%, p = 0.002) |
| HATS (2001) [84] | 160 | CAD + LDL-C < 140 mg/dL + TGs < 400 mg/dL | Simvastatin (10–20 mg) + niacin (250–1000 mg 2×) vs. antioxidant vs. simvastatin + niacin + antioxidant vs. placebo | 3 years | Reduction LDL-C 42% Increase HDL-C 26% | Reduction in death, MI, stroke, or revascularization |
| ARBITER-2 (2004) [85] | 149 | CAD + on statin therapy | ER-niacin (1000 mg 1×) vs. placebo | 1 year | Increase HDL-C 21% | Reduction in progression of carotid intima-media thickness |
| ARBITER-6 (2009) [86] | 208 | CAD or CAD risk equivalent on statin therapy | ER-niacin (2 g 1×) vs. ezetimibe (10 mg) | 1.2 years | Reduction LDL-C (20% vs. 12%) Increase HDL-C (+18% vs. −7%) | Reduction in progression of carotid intima-media thickness |
| AFREGS (2005) [87] | 143 | CAD + LDL-C < 160 mg/dL + HDL-C < 40 mg/dL | Niacin (240–3000 mg) + gemfibrozil (600 mg 2×) + cholestyramine (2–16 g 1×) vs. placebo | 2.5 years | Reduction LDL-C 24% Increase HDL-C 38% | Reduction in angiographic progression (34.72% vs. 36.02%, p = 0.0002) |
| AIM-HIGH (2011) [71] | 3414 | Aged >45 years + CV disease | ER-niacin (1.5–2 g 1×) vs. placebo | 3 years | Reduction LDL-C 14% Increase HDL-C 25% | No significant differences in CV events |
| HPS2-THRIVE (2014) [88] | 25,673 | Aged >50 years + CV disease | ER-niacin (2 g 1×)/laropiprant (40 mg) vs. placebo | 3.9 years | Reduction LDL-C 10% Increase HDL-C 6% | No significant differences in CV events |
| Study | Sample Size | Characteristics of Patients | Comparison Groups | Follow-Up | Lipids Effect | CV Effects |
|---|---|---|---|---|---|---|
| HHS (1987) [91] | 4081 | Men with non-HDL-C > 200 mg/dL | Gemfibrozil (1200 mg) vs. placebo | 5 years | Increase HDL-C 11% Reduction LDL-C 11% Reduction TGs 35% | Reduction in incidence of CAD |
| VA-HIT (1999) [92] | 2531 | Documented CAD + LDL < 140 mg/dL | Gemfibrozil (1200 mg) vs. placebo | 5.1 years | Reduction TGs 31% Increase HDL-C 6% | Reduction in MI or coronary death |
| BIP (2000) [93] | 3090 | Previous MI or stable angina + LDL-C < 180 mg/dL | Bezafibrate (400 mg) vs. placebo | 6.2 years | Reduction TGs 21% Increase HDL-C 18% | No significant differences in CV events |
| LEADER (2002) [94] | 1568 | Men with lower PAD | Bezafibrate (400 mg) vs. placebo | 4.6 years | NE | No significant differences in CV events |
| FIELD (2005) [95] | 9795 | T2DM | Fenofibrate (200 mg) vs. placebo | 5 years | Reduction TGs 29% Increase HDL-C 5% Reduction LDL-C 12% | No significant differences in CV events |
| ACCORD-Lipid (2010) [96] | 5518 | T2DM + CV risk factor or documented CV disease | Fenofibrate (160 mg) vs. placebo | 4.7 years | Increase HDL-C 9% Reduction TGs 23% | No significant differences in CV events |
| ECLIPSE-REAL (2019) [97] | 10,705 | Metabolic syndrome | Statin vs. statin + fenofibrate | 6 years | Reduction TGs with statin + fenofibrate | Reduction in CAD, stroke, or CV death with statin + fenofibrate (HR 0.74, 95% CI 0.58 to 0.93) |
| PROMINENT (2022) [98] | 10,497 | T2DM + TGs 200–499 mg/dL + HDL < 40 mg/dL | Pemafibrate (0.4 mg) vs. placebo | 3.4 years | Reduction TGs 31.1% | No significant differences in CV events |
| Study | Sample Size | Characteristics of Patients | Comparison Groups | Follow-Up | Lipids Effect | CV Effects |
|---|---|---|---|---|---|---|
| ILLUMINATE (2007) [111] | 15,067 | CV disease | Torcetrapib (60 mg/d) vs. placebo | 1 year | Increase HDL-C 72.1% Reduction LDL-C 24.9% | Reduction in CV events (HR 1.25, 95% CI 1.09 to 1.44) |
| dal-OUTCOMES (2012) [112] | 15,871 | ACS | Dalcetrapib (600 mg/d) vs. placebo | 31 months | Increase HDL-C 30% | No significant differences in CV events |
| ACCELERATE (2017) [113] | 12,092 | CV disease | Evacetrapib (130 mg/d) vs. placebo | 26 months | Increase HDL-C 133.2% Reduction LDL-C 31.1% | No significant differences in CV events |
| REVEAL (2017) [114] | 30,449 | Atherosclerotic disease + on atorvastatin therapy + mean LDL-C 61 mg/dL | Anacetrapib (100 mg/d) vs. placebo | 4.1 years | Reduction non-HDL-C 18% | Reduction in coronary death, MI, or coronary revascularization (HR 0.91, 95% CI 0.85 to 0.97, NNT 100) |
| ROSE (2022) [115] | 120 | Dyslipidemia on statin treatment | Obicetrapib (10 mg/d) vs. placebo | 8 weeks | Reduction LDL-C 51% | NE |
| Study | Sample Size | Characteristics of Patients | Comparison Groups | Follow-Up | LDL-C Reduction | CV Effects |
|---|---|---|---|---|---|---|
| ORION-1 (2017) [124] | 501 | Patients at high risk for CV disease and elevated LDL-C | Different doses of inclisiran vs. placebo | 180 days | 27.9% (200 mg inclisiran); 38.4% (300 mg inclisiran); 41.9% (500 mg inclisiran); 35.5% (double dose 100 mg inclisiran); 44.9% (double dose 200 mg inclisiran); 52.6% (double dose 300 mg inclisiran) | NE |
| ORION-3 (2023) [125] | 382 | Patients at high risk for CV disease and elevated LDL-C | Inclisiran-only (patients who already received inclisiran continued to receive it) vs. switching-arm (patients who received placebo in ORION-1 first received evolocumab for 1 year and then switched to inclisiran) | 4 years | 44.2% | NE |
| ORION-5 (2023) [126] | 56 | HoFH | Inclisiran (300 mg) vs. placebo | 150 days | 1.68% | NE |
| ORION-9 (2020) [127] | 482 | HeFH | Inclisiran (300 mg) vs. placebo | 510 days | 39.7% | NE |
| ORION-10 and ORION-11 (2020) [128] | 1561 (ORION-10) and 1617 (ORION-11) | ASCVD (ORION-10 trial) and ASCVD-risk equivalent (ORION-11 trial) and elevated LDL-C on maximum tolerated dose of statin | Inclisiran (284 mg) vs. placebo | 510 days | 52.3% (ORION-10) 49.9% (ORION-11) | NE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira, L.L.H.; de Assis, A.C.R.; Giraldez, V.Z.R.; Scudeler, T.L.; Soares, P.R. Dyslipidemia: A Narrative Review on Pharmacotherapy. Pharmaceuticals 2024, 17, 289. https://doi.org/10.3390/ph17030289
de Oliveira LLH, de Assis ACR, Giraldez VZR, Scudeler TL, Soares PR. Dyslipidemia: A Narrative Review on Pharmacotherapy. Pharmaceuticals. 2024; 17(3):289. https://doi.org/10.3390/ph17030289
Chicago/Turabian Stylede Oliveira, Lucas Lentini Herling, Arthur Cicupira Rodrigues de Assis, Viviane Zorzanelli Rocha Giraldez, Thiago Luis Scudeler, and Paulo Rogério Soares. 2024. "Dyslipidemia: A Narrative Review on Pharmacotherapy" Pharmaceuticals 17, no. 3: 289. https://doi.org/10.3390/ph17030289