
The Effects of CYP2C19 Genotype on Proxies of SSRI Antidepressant Response in the UK Biobank

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. Study Demographics and CYP2C19 Metaboliser Phenotype Prevalence
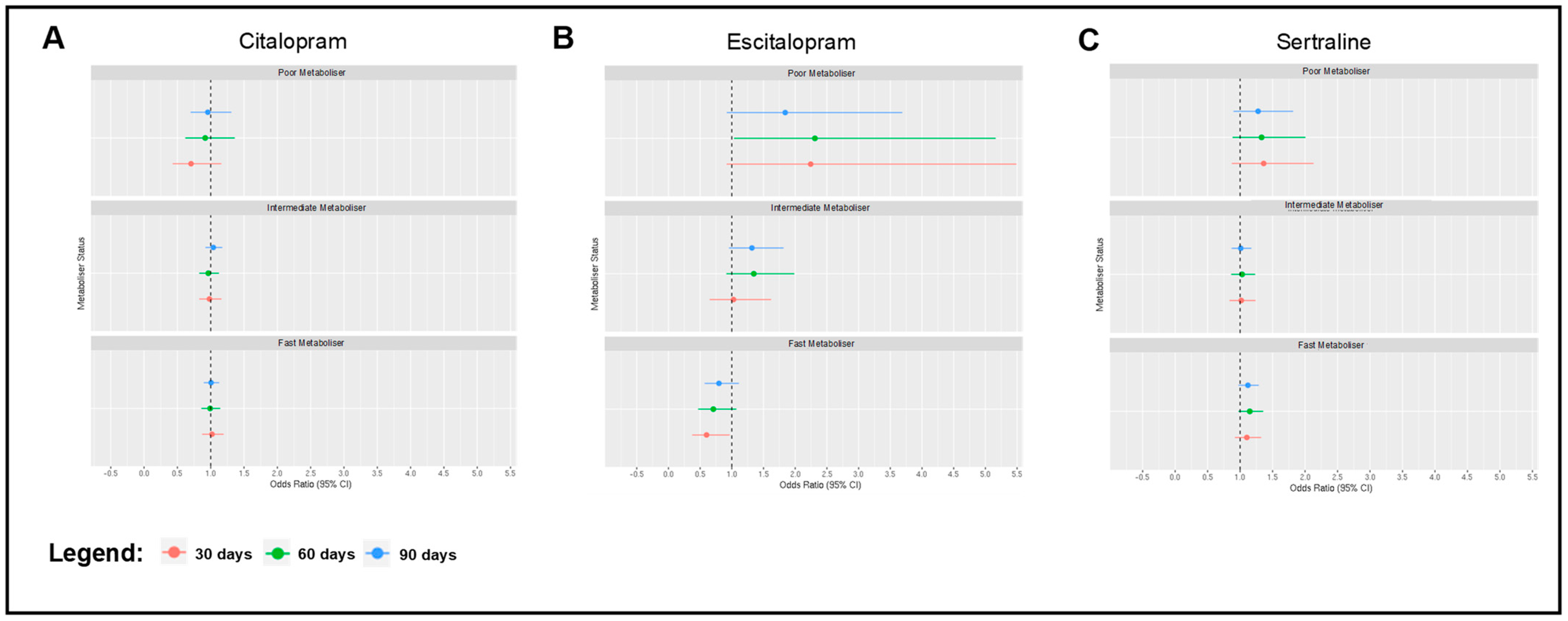
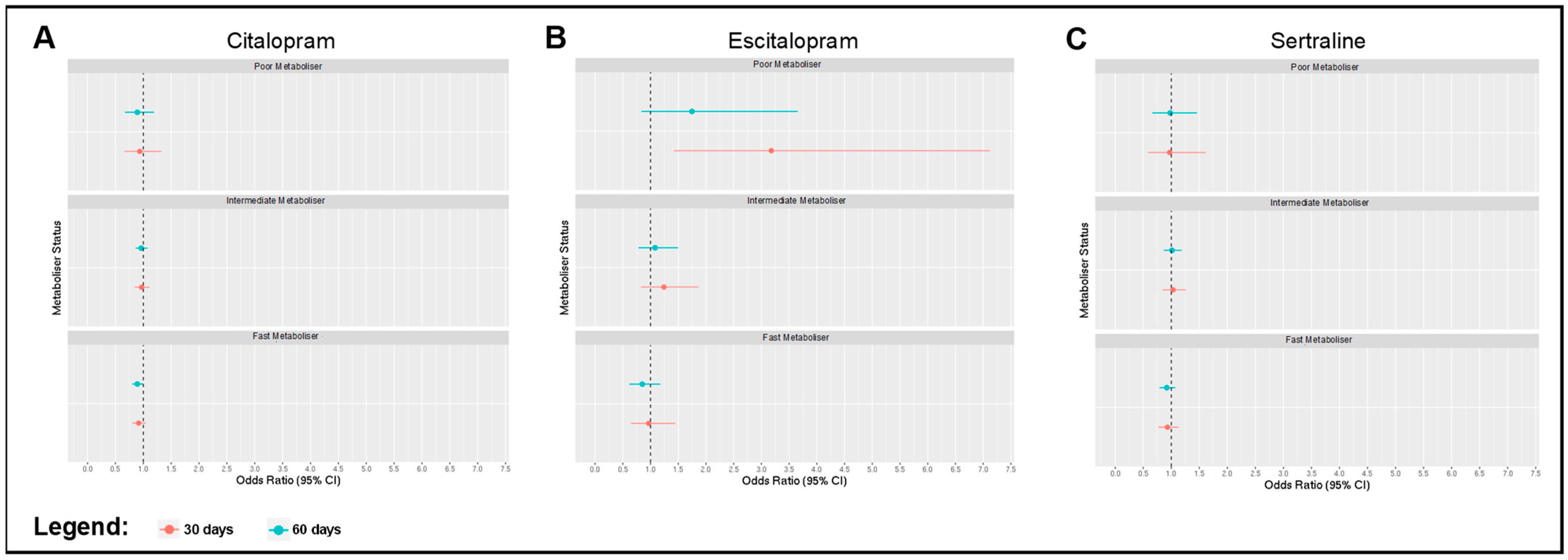
2.2. Associations between Metaboliser Phenotype and Antidepressant Switching
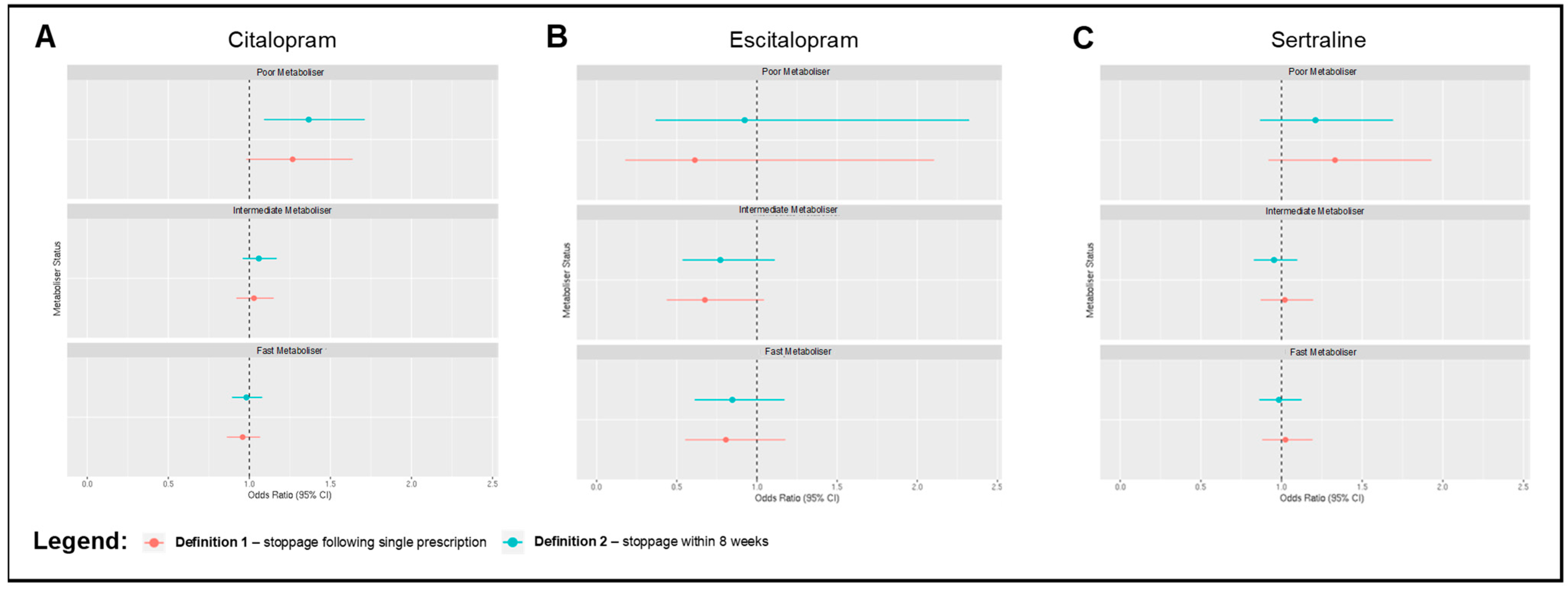
2.3. Associations between Metaboliser Phenotype and Antidepressant Discontinuation
2.4. Associations between Metaboliser Phenotype and Antidepressant Duration
2.5. Associations between Metaboliser Phenotype and Side Effects
2.6. Sensitivity Analyses
3. Discussion
4. Methods
4.1. Study Population
4.2. CYP2C19 Genotyping and Metaboliser Phenotype
4.3. Outcomes
4.4. Statistical Analyses
4.5. Sensitivity Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- COVID-19 Mental Disorders Collaborators. Global prevalence and burden of depressive and anxiety disorders in 204 countries and territories in 2020 due to the COVID-19 pandemic. Lancet 2021, 398, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Lepine, J.P.; Briley, M. The increasing burden of depression. Neuropsychiatr. Dis. Treat. 2011, 7 (Suppl. S1), 3–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; He, H.; Yang, J.; Feng, X.; Zhao, F.; Lyu, J. Changes in the global burden of depression from 1990 to 2017: Findings from the Global Burden of Disease study. J. Psychiatr. Res. 2020, 126, 134–140. [Google Scholar] [CrossRef]
- Dupuy, J.M.; Ostacher, M.J.; Huffman, J.; Perlis, R.H.; Nierenberg, A.A. A critical review of pharmacotherapy for major depressive disorder. Int. J. Neuropsychopharmacol. 2011, 14, 1417–1431. [Google Scholar] [CrossRef]
- Karrouri, R.; Hammani, Z.; Benjelloun, R.; Otheman, Y. Major depressive disorder: Validated treatments and future challenges. World J. Clin. Cases 2021, 9, 9350–9367. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.M. SSRI Antidepressant Medications: Adverse Effects and Tolerability. Prim. Care Companion J. Clin. Psychiatry 2001, 3, 22–27. [Google Scholar] [CrossRef]
- Hirschfeld, R.M. Efficacy of SSRIs and newer antidepressants in severe depression: Comparison with TCAs. J. Clin. Psychiatry 1999, 60, 326–335. [Google Scholar] [CrossRef]
- Kornstein, S.G.; Schneider, R.K. Clinical features of treatment-resistant depression. J. Clin. Psychiatry 2001, 62 (Suppl. S16), 18–25. [Google Scholar] [PubMed]
- Trivedi, M.H.; Lin, E.H.; Katon, W.J. Consensus recommendations for improving adherence, self-management, and outcomes in patients with depression. CNS Spectr. 2007, 12 (Suppl. S13), 1–27. [Google Scholar]
- Buch, A.M.; Liston, C. Dissecting diagnostic heterogeneity in depression by integrating neuroimaging and genetics. Neuropsychopharmacology 2021, 46, 156–175. [Google Scholar] [CrossRef] [PubMed]
- Pain, O.; Hodgson, K.; Trubetskoy, V.; Ripke, S.; Marshe, V.S.; Adams, M.J.; Byrne, E.M.; Campos, A.I.; Carrillo-Roa, T.; Cattaneo, A.; et al. Identifying the Common Genetic Basis of Antidepressant Response. Biol. Psychiatry Glob. Open Sci. 2022, 2, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Tansey, K.E.; Guipponi, M.; Hu, X.; Domenici, E.; Lewis, G.; Malafosse, A.; Wendland, J.R.; Lewis, C.M.; McGuffin, P.; Uher, R. Contribution of common genetic variants to antidepressant response. Biol. Psychiatry 2013, 73, 679–682. [Google Scholar] [CrossRef]
- McDonnell, A.M.; Dang, C.H. Basic review of the cytochrome p450 system. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar] [CrossRef]
- Desta, Z.; Zhao, X.; Shin, J.G.; Flockhart, D.A. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin. Pharmacokinet. 2002, 41, 913–958. [Google Scholar] [CrossRef]
- de Leon, J.; Armstrong, S.C.; Cozza, K.L. Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006, 47, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Fricke-Galindo, I.; Cespedes-Garro, C.; Rodrigues-Soares, F.; Naranjo, M.E.; Delgado, A.; de Andres, F.; Lopez-Lopez, M.; Penas-Lledo, E.; LLerena, A. Interethnic variation of CYP2C19 alleles, ‘predicted’ phenotypes and ‘measured’ metabolic phenotypes across world populations. Pharmacogenom. J. 2016, 16, 113–123. [Google Scholar] [CrossRef]
- Braten, L.S.; Haslemo, T.; Jukic, M.M.; Ingelman-Sundberg, M.; Molden, E.; Kringen, M.K. Impact of CYP2C19 genotype on sertraline exposure in 1200 Scandinavian patients. Neuropsychopharmacology 2020, 45, 570–576. [Google Scholar] [CrossRef]
- Jukic, M.M.; Haslemo, T.; Molden, E.; Ingelman-Sundberg, M. Impact of CYP2C19 Genotype on Escitalopram Exposure and Therapeutic Failure: A Retrospective Study Based on 2087 Patients. Am. J. Psychiatry 2018, 175, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Milosavljevic, F.; Bukvic, N.; Pavlovic, Z.; Miljevic, C.; Pesic, V.; Molden, E.; Ingelman-Sundberg, M.; Leucht, S.; Jukic, M.M. Association of CYP2C19 and CYP2D6 Poor and Intermediate Metabolizer Status With Antidepressant and Antipsychotic Exposure: A Systematic Review and Meta-analysis. JAMA Psychiatry 2021, 78, 270–280. [Google Scholar] [CrossRef]
- Brouwer, J.; Nijenhuis, M.; Soree, B.; Guchelaar, H.J.; Swen, J.J.; van Schaik, R.H.N.; Weide, J.V.; Rongen, G.; Buunk, A.M.; de Boer-Veger, N.J.; et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction between CYP2C19 and CYP2D6 and SSRIs. Eur. J. Hum. Genet. 2022, 30, 1114–1120. [Google Scholar] [CrossRef]
- van Westrhenen, R.; Aitchison, K.J.; Ingelman-Sundberg, M.; Jukic, M.M. Pharmacogenomics of Antidepressant and Antipsychotic Treatment: How Far Have We Got and Where Are We Going? Front. Psychiatry 2020, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- van Westrhenen, R.; van Schaik, R.H.N.; van Gelder, T.; Birkenhager, T.K.; Bakker, P.R.; Houwink, E.J.F.; Bet, P.M.; Hoogendijk, W.J.G.; van Weelden-Hulshof, M.J.M. Policy and Practice Review: A First Guideline on the Use of Pharmacogenetics in Clinical Psychiatric Practice. Front. Pharmacol. 2021, 12, 640032. [Google Scholar] [CrossRef]
- Hicks, J.K.; Sangkuhl, K.; Swen, J.J.; Ellingrod, V.L.; Muller, D.J.; Shimoda, K.; Bishop, J.R.; Kharasch, E.D.; Skaar, T.C.; Gaedigk, A.; et al. Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin. Pharmacol. Ther. 2017, 102, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, C.; Tansey, K.E.; Perlis, R.H.; Hauser, J.; Henigsberg, N.; Maier, W.; Mors, O.; Placentino, A.; Rietschel, M.; Souery, D.; et al. Effect of cytochrome CYP2C19 metabolizing activity on antidepressant response and side effects: Meta-analysis of data from genome-wide association studies. Eur. Neuropsychopharmacol. 2018, 28, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Mrazek, D.A.; Biernacka, J.M.; O’Kane, D.J.; Black, J.L.; Cunningham, J.M.; Drews, M.S.; Snyder, K.A.; Stevens, S.R.; Rush, A.J.; Weinshilboum, R.M. CYP2C19 variation and citalopram response. Pharmacogenet. Genom. 2011, 21, 1–9. [Google Scholar] [CrossRef]
- Peters, E.J.; Slager, S.L.; Kraft, J.B.; Jenkins, G.D.; Reinalda, M.S.; McGrath, P.J.; Hamilton, S.P. Pharmacokinetic genes do not influence response or tolerance to citalopram in the STAR*D sample. PLoS ONE 2008, 3, e1872. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef]
- Ionova, Y.; Ashenhurst, J.; Zhan, J.; Nhan, H.; Kosinski, C.; Tamraz, B.; Chubb, A. CYP2C19 Allele Frequencies in Over 2.2 Million Direct-to-Consumer Genetics Research Participants and the Potential Implication for Prescriptions in a Large Health System. Clin. Transl. Sci. 2020, 13, 1298–1306. [Google Scholar] [CrossRef]
- Tsai, M.H.; Lin, K.M.; Hsiao, M.C.; Shen, W.W.; Lu, M.L.; Tang, H.S.; Fang, C.K.; Wu, C.S.; Lu, S.C.; Liu, S.C.; et al. Genetic polymorphisms of cytochrome P450 enzymes influence metabolism of the antidepressant escitalopram and treatment response. Pharmacogenomics 2010, 11, 537–546. [Google Scholar] [CrossRef]
- Tsuchimine, S.; Ochi, S.; Tajiri, M.; Suzuki, Y.; Sugawara, N.; Inoue, Y.; Yasui-Furukori, N. Effects of Cytochrome P450 (CYP) 2C19 Genotypes on Steady-State Plasma Concentrations of Escitalopram and its Desmethyl Metabolite in Japanese Patients With Depression. Ther. Drug Monit. 2018, 40, 356–361. [Google Scholar] [CrossRef]
- Chang, M.; Tybring, G.; Dahl, M.L.; Lindh, J.D. Impact of cytochrome P450 2C19 polymorphisms on citalopram/escitalopram exposure: A systematic review and meta-analysis. Clin. Pharmacokinet. 2014, 53, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Rudberg, I.; Hendset, M.; Uthus, L.H.; Molden, E.; Refsum, H. Heterozygous mutation in CYP2C19 significantly increases the concentration/dose ratio of racemic citalopram and escitalopram (S-citalopram). Ther. Drug Monit. 2006, 28, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, S.L.; Poweleit, E.A.; Prows, C.A.; Martin, L.J.; Strawn, J.R.; Ramsey, L.B. Influence of CYP2C19 Metabolizer Status on Escitalopram/Citalopram Tolerability and Response in Youth With Anxiety and Depressive Disorders. Front. Pharmacol. 2019, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry 1960, 23, 56–62. [Google Scholar] [CrossRef]
- Zastrozhin, M.S.; Skryabin, V.Y.; Petukhov, A.E.; Torrado, M.V.; Pankratenko, E.P.; Zastrozhina, A.K.; Grishina, E.A.; Ryzhikova, K.A.; Shipitsyn, V.V.; Bryun, E.A.; et al. Effects of CYP2C19 genetic polymorphism on the steady-state concentration of citalopram in patients with major depressive disorder. Pharmacogenom. J. 2021, 21, 435–439. [Google Scholar] [CrossRef]
- Jokovic, D.; Milosavljevic, F.; Stojanovic, Z.; Supic, G.; Vojvodic, D.; Uzelac, B.; Jukic, M.M.; Petkovic Curcin, A. CYP2C19 slow metabolizer phenotype is associated with lower antidepressant efficacy and tolerability. Psychiatry Res. 2022, 312, 114535. [Google Scholar] [CrossRef]
- Bradley, P.; Shiekh, M.; Mehra, V.; Vrbicky, K.; Layle, S.; Olson, M.C.; Maciel, A.; Cullors, A.; Garces, J.A.; Lukowiak, A.A. Improved efficacy with targeted pharmacogenetic-guided treatment of patients with depression and anxiety: A randomized clinical trial demonstrating clinical utility. J. Psychiatr. Res. 2018, 96, 100–107. [Google Scholar] [CrossRef]
- Hall-Flavin, D.K.; Winner, J.G.; Allen, J.D.; Carhart, J.M.; Proctor, B.; Snyder, K.A.; Drews, M.S.; Eisterhold, L.L.; Geske, J.; Mrazek, D.A. Utility of integrated pharmacogenomic testing to support the treatment of major depressive disorder in a psychiatric outpatient setting. Pharmacogenet. Genom. 2013, 23, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Hall-Flavin, D.K.; Winner, J.G.; Allen, J.D.; Jordan, J.J.; Nesheim, R.S.; Snyder, K.A.; Drews, M.S.; Eisterhold, L.L.; Biernacka, J.M.; Mrazek, D.A. Using a pharmacogenomic algorithm to guide the treatment of depression. Transl. Psychiatry 2012, 2, e172. [Google Scholar] [CrossRef]
- Perez, V.; Salavert, A.; Espadaler, J.; Tuson, M.; Saiz-Ruiz, J.; Saez-Navarro, C.; Bobes, J.; Baca-Garcia, E.; Vieta, E.; Olivares, J.M.; et al. Efficacy of prospective pharmacogenetic testing in the treatment of major depressive disorder: Results of a randomized, double-blind clinical trial. BMC Psychiatry 2017, 17, 250. [Google Scholar] [CrossRef]
- Singh, A.B. Improved Antidepressant Remission in Major Depression via a Pharmacokinetic Pathway Polygene Pharmacogenetic Report. Clin. Psychopharmacol. Neurosci. 2015, 13, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Winner, J.; Allen, J.D.; Altar, C.A.; Spahic-Mihajlovic, A. Psychiatric pharmacogenomics predicts health resource utilization of outpatients with anxiety and depression. Transl. Psychiatry 2013, 3, e242. [Google Scholar] [CrossRef]
- McInnes, G.; Altman, R.B. Drug Response Pharmacogenetics for 200,000 UK Biobank Participants. Pac. Symp. Biocomput. 2021, 26, 184–195. [Google Scholar] [PubMed]
- Feng, C.; Le, D.; McCoy, A.B. Using Electronic Health Records to Identify Adverse Drug Events in Ambulatory Care: A Systematic Review. Appl. Clin. Inform. 2019, 10, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, E.; Mallah, R.; Jackson, R.G.; Ball, M.; Ibrahim, Z.M.; Broadbent, M.; Dzahini, O.; Stewart, R.; Johnston, C.; Dobson, R.J. Identification of Adverse Drug Events from Free Text Electronic Patient Records and Information in a Large Mental Health Case Register. PLoS ONE 2015, 10, e0134208. [Google Scholar] [CrossRef]
- Campos, A.I.; Byrne, E.M.; Mitchell, B.L.; Wray, N.R.; Lind, P.A.; Licinio, J.; Medland, S.E.; Martin, N.G.; Hickie, I.B.; Renteria, M.E. Impact of CYP2C19 metaboliser status on SSRI response: A retrospective study of 9500 participants of the Australian Genetics of Depression Study. Pharmacogenom. J. 2022, 22, 130–135. [Google Scholar] [CrossRef]
- Bender, R.; Lange, S. Adjusting for multiple testing--when and how? J. Clin. Epidemiol. 2001, 54, 343–349. [Google Scholar] [CrossRef]
- Streiner, D.L. Best (but oft-forgotten) practices: The multiple problems of multiplicity-whether and how to correct for many statistical tests. Am. J. Clin. Nutr. 2015, 102, 721–728. [Google Scholar] [CrossRef]
- Armstrong, R.A. When to use the Bonferroni correction. Ophthalmic Physiol. Opt. 2014, 34, 502–508. [Google Scholar] [CrossRef]
- Bousman, C.A.; Stevenson, J.M.; Ramsey, L.B.; Sangkuhl, K.; Hicks, J.K.; Strawn, J.R.; Singh, A.B.; Ruano, G.; Mueller, D.J.; Tsermpini, E.E.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6, CYP2C19, CYP2B6, SLC6A4, and HTR2A Genotypes and Serotonin Reuptake Inhibitor Antidepressants. Clin. Pharmacol. Ther. 2023, 114, 51–68. [Google Scholar] [CrossRef]
- Rutherford, B.R.; Roose, S.P. A model of placebo response in antidepressant clinical trials. Am. J. Psychiatry 2013, 170, 723–733. [Google Scholar] [CrossRef]
- Wang, S.M.; Han, C.; Lee, S.J.; Jun, T.Y.; Patkar, A.A.; Masand, P.S.; Pae, C.U. Efficacy of antidepressants: Bias in randomized clinical trials and related issues. Expert Rev. Clin. Pharmacol. 2018, 11, 15–25. [Google Scholar] [CrossRef]
- Howard, D.M.; Adams, M.J.; Shirali, M.; Clarke, T.K.; Marioni, R.E.; Davies, G.; Coleman, J.R.I.; Alloza, C.; Shen, X.; Barbu, M.C.; et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat. Commun. 2018, 9, 1470. [Google Scholar] [CrossRef]
- Davis, K.A.S.; Coleman, J.R.I.; Adams, M.; Allen, N.; Breen, G.; Cullen, B.; Dickens, C.; Fox, E.; Graham, N.; Holliday, J.; et al. Mental health in UK Biobank-development, implementation and results from an online questionnaire completed by 157 366 participants: A reanalysis. BJPsych Open 2020, 6, e18. [Google Scholar] [CrossRef] [PubMed]
- de Jong, J.C.; van den Berg, P.B.; Tobi, H.; de Jong-van den Berg, L.T. Combined use of SSRIs and NSAIDs increases the risk of gastrointestinal adverse effects. Br. J. Clin. Pharmacol. 2003, 55, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Hemeryck, A.; Belpaire, F.M. Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: An update. Curr. Drug Metab. 2002, 3, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef]
- Genotyping and Quality Control of UK Biobank, a Large-Scale, Extensively Phenotyped Prospective Resource. Available online: https://biobank.ctsu.ox.ac.uk/crystal/crystal/docs/genotyping_qc.pdf (accessed on 1 June 2023).
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [CrossRef]
- UK Biobank Primary Care Linked Data. Available online: https://biobank.ndph.ox.ac.uk/showcase/showcase/docs/primary_care_data.pdf (accessed on 15 June 2023).
- McInnes, G.; Lavertu, A.; Sangkuhl, K.; Klein, T.E.; Whirl-Carrillo, M.; Altman, R.B. Pharmacogenetics at Scale: An Analysis of the UK Biobank. Clin. Pharmacol. Ther. 2021, 109, 1528–1537. [Google Scholar] [CrossRef]
- Sangkuhl, K.; Whirl-Carrillo, M.; Whaley, R.M.; Woon, M.; Lavertu, A.; Altman, R.B.; Carter, L.; Verma, A.; Ritchie, M.D.; Klein, T.E. Pharmacogenomics Clinical Annotation Tool (PharmCAT). Clin. Pharmacol. Ther. 2020, 107, 203–210. [Google Scholar] [CrossRef]
- Deshpande, N.; Sharanya, V.; VV, R.K.; Murthy, H.V.V.; Sasikala, M.; Banerjee, R.; Tandan, M.; Reddy, N. Rapid and ultra-rapid metabolizers with CYP2C19*17 polymorphism do not respond to standard therapy with proton pump inhibitors. Meta Gene 2016, 9, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Lingjaerde, O.; Ahlfors, U.G.; Bech, P.; Dencker, S.J.; Elgen, K. The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr. Scand. Suppl. 1987, 334, 1–100. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- RStudio Team. RStudio: Integrated Development Environment for R. RStudio; PBC: Boston, MA, USA, 2022. [Google Scholar]
- King’s College London. King’s Computational Research, Engineering and Technology Environment (CREATE). 2022. Available online: https://docs.er.kcl.ac.uk/ (accessed on 15 June 2023).
- Hanscombe, K. ukbkings: KCL Interface to UKB Project Data on Rosalind/CREATE HPC. 2022. Available online: https://kenhanscombe.github.io/ukbkings/ (accessed on 15 June 2023).
- Hanscombe, K.B.; Coleman, J.R.I.; Traylor, M.; Lewis, C.M. ukbtools: An R package to manage and query UK Biobank data. PLoS ONE 2019, 14, e0214311. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Francois, R.; Henry, L.; Muller, K. dplyr: A Grammar of Data Manipulation. 2021. Available online: https://dplyr.tidyverse.org/ (accessed on 15 June 2023).
- Wickham, H. Stringr: Simple, Consistent Wrappers for Common String Operations. 2019. Available online: https://stringr.tidyverse.org/ (accessed on 15 June 2023).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Dodgen, T.M.; Hochfeld, W.E.; Fickl, H.; Asfaha, S.M.; Durandt, C.; Rheeder, P.; Drogemoller, B.I.; Wright, G.E.; Warnich, L.; Labuschagne, C.D.; et al. Introduction of the AmpliChip CYP450 Test to a South African cohort: A platform comparative prospective cohort study. BMC Med. Genet. 2013, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, J.; Brockmoller, J.; Steimer, W.; Pischa, K.; Leucht, S.; Kullmann, M.; Jensen, O.; Ouethy, T.; Tzvetkov, M.V.; Rafehi, M. Effects of Genetic Polymorphism in CYP2D6, CYP2C19, and the Organic Cation Transporter OCT1 on Amitriptyline Pharmacokinetics in Healthy Volunteers and Depressive Disorder Patients. Front. Pharmacol. 2021, 12, 688950. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Citalopram | Escitalopram | Sertraline | Total Sample | |
|---|---|---|---|---|
| Metaboliser phenotype | ||||
| Poor, N (%) | 675 (2.7%) | 75 (2.5%) | 349 (2.8%) | 905 (2.7%) |
| Intermediate, N (%) | 6468 (26.2%) | 793 (26.3%) | 3306 (26.4%) | 8619 (26.0%) |
| Normal, N (%) | 9731 (39.3%) | 1197 (39.7%) | 4965 (39.6%) | 13,098 (39.6%) |
| Fast, N (%) | 7855 (31.7%) | 947 (31.4%) | 3924 (31.3%) | 10,444 (31.6%) |
| Total number of individuals prescribed SSRI | ||||
| N | 24,729 | 3012 | 12,544 | 33,094 |
| Demographics | ||||
| Age (mean ± SD) | 55.0 (8.1) | 55.2 (7.9) | 55.4 (8.2) | 55.2 ± 8.1 |
| Sex (% Female) | 66.9% | 68.1% | 65.6% | 66.2% |
| Citalopram | ||||||
|---|---|---|---|---|---|---|
| Metaboliser Phenotype | Count-Based Definition | Weeks-Based Definition | ||||
| Mean (SD) | β (SE) | p | Mean (SD) | β (SE) | p | |
| Poor metaboliser | 21.3 (33.4) | −0.068 (0.060) | 0.255 | 99.8 (146.1) | −0.082 (0.063) | 0.191 |
| Intermediate metaboliser | 21.4 (34.0) | −0.048 (0.024) | 0.045 | 98.7 (143.3) | −0.051 (0.025) | 0.043 |
| Normal metaboliser | 22.0 (34.1) | - | - | 101.8 (143.7) | - | - |
| Fast metaboliser | 21.6 (33.3) | −0.014 (0.023) | 0.531 | 99.7 (141.6) | −0.015 (0.024) | 0.525 |
| Escitalopram | ||||||
| Metaboliser phenotype | Count-based definition | Weeks-based definition | ||||
| Mean (SD) | β (SE) | p | Mean (SD) | β (SE) | p | |
| Poor metaboliser | 10.5 (17.6) | −0.351 (0.174) | 0.043 | 53.3 (90.1) | −0.369 (0.185) | 0.046 |
| Intermediate metaboliser | 17.1 (28.4) | −0.073 (0.067) | 0.274 | 81.9 (130.2) | −0.079 (0.071) | 0.265 |
| Normal metaboliser | 18.6 (36.3) | - | - | 87.8 (134.5) | - | - |
| Fast metaboliser | 19.1 (32.0) | 0.074 (0.063) | 0.244 | 89.1 (136.8) | 0.066 (0.068) | 0.326 |
| Sertraline | ||||||
| Metaboliser phenotype | Count-based definition | Weeks-based definition | ||||
| Mean (SD) | β (SE) | p | Mean (SD) | β (SE) | p | |
| Poor metaboliser | 18.7 (35.7) | −0.123 (0.081) | 0.128 | 82.4 (138.6) | −0.148 (0.084) | 0.079 |
| Intermediate metaboliser | 18.2 (33.0) | −0.050 (0.033) | 0.126 | 84.7 (141.9) | −0.057 (0.034) | 0.095 |
| Normal metaboliser | 19.2 (37.6) | - | - | 81.0 (137.9) | - | - |
| Fast metaboliser | 17.9 (32.4) | −0.060 (0.031) | 0.056 | 78.3 (130.5) | −0.071 (0.032) | 0.029 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, W.L.E.; Fabbri, C.; Laplace, B.; Li, D.; van Westrhenen, R.; Lewis, C.M.; Dawe, G.S.; Young, A.H. The Effects of CYP2C19 Genotype on Proxies of SSRI Antidepressant Response in the UK Biobank. Pharmaceuticals 2023, 16, 1277. https://doi.org/10.3390/ph16091277
Wong WLE, Fabbri C, Laplace B, Li D, van Westrhenen R, Lewis CM, Dawe GS, Young AH. The Effects of CYP2C19 Genotype on Proxies of SSRI Antidepressant Response in the UK Biobank. Pharmaceuticals. 2023; 16(9):1277. https://doi.org/10.3390/ph16091277
Chicago/Turabian StyleWong, Win Lee Edwin, Chiara Fabbri, Benjamin Laplace, Danyang Li, Roos van Westrhenen, Cathryn M. Lewis, Gavin Stewart Dawe, and Allan H. Young. 2023. "The Effects of CYP2C19 Genotype on Proxies of SSRI Antidepressant Response in the UK Biobank" Pharmaceuticals 16, no. 9: 1277. https://doi.org/10.3390/ph16091277