
Age of Antibiotic Resistance in MDR/XDR Clinical Pathogen of Pseudomonas aeruginosa
 , , , and
, , , and
Abstract
:
1. Introduction
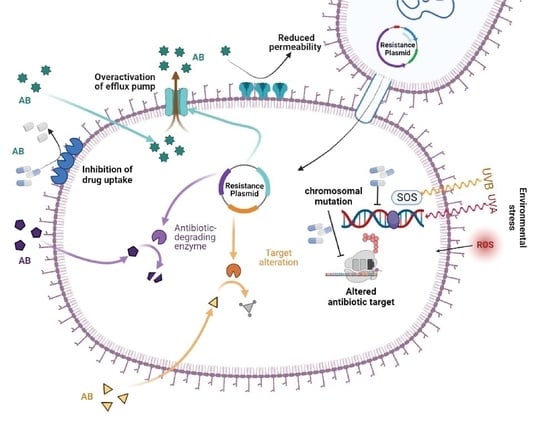
2. Molecular Mechanism of Drug Resistance in P. aeruginosa
2.1. Inherent Resistome
2.2. Mutational Resistome
2.3. Horizontally Acquired Resistome
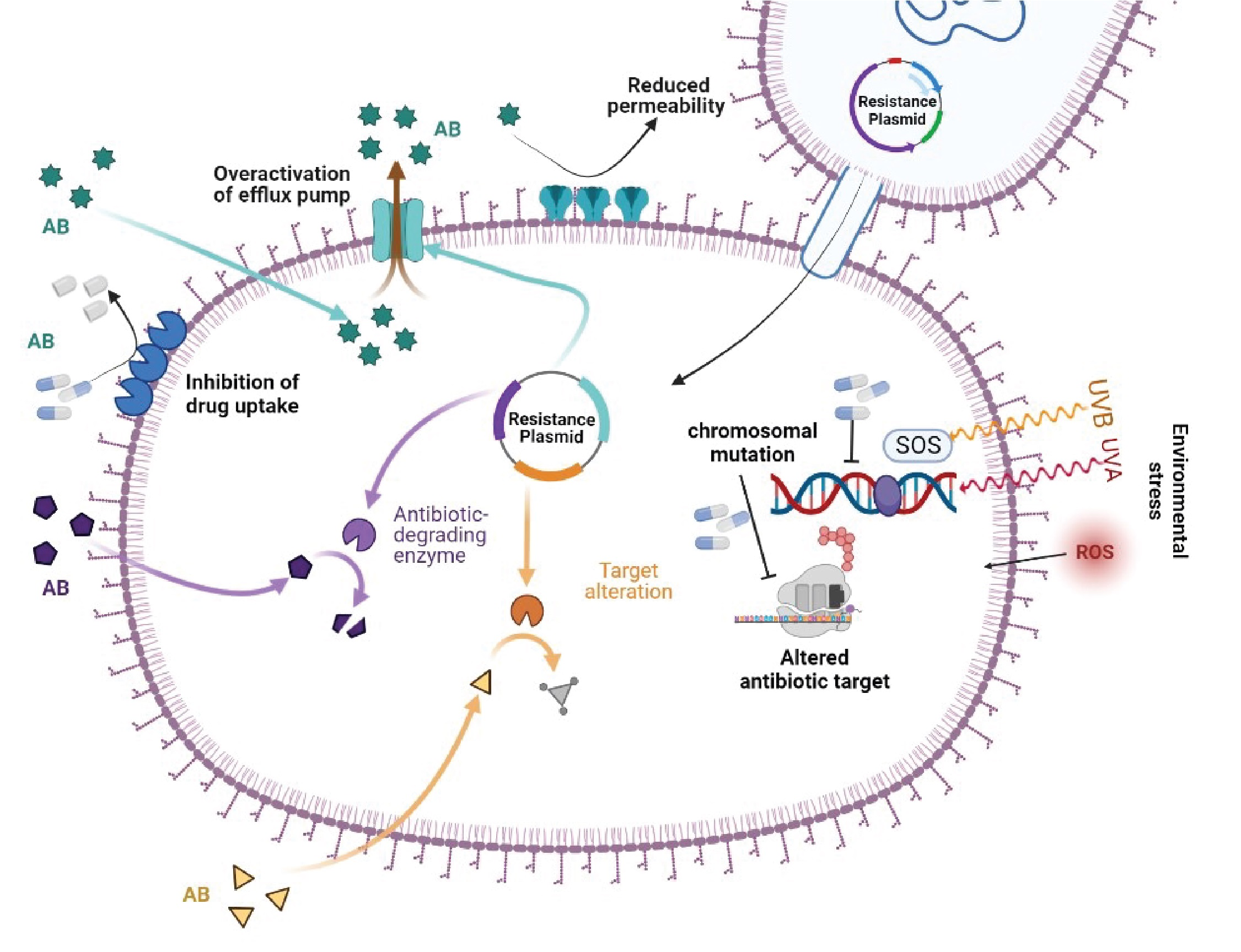
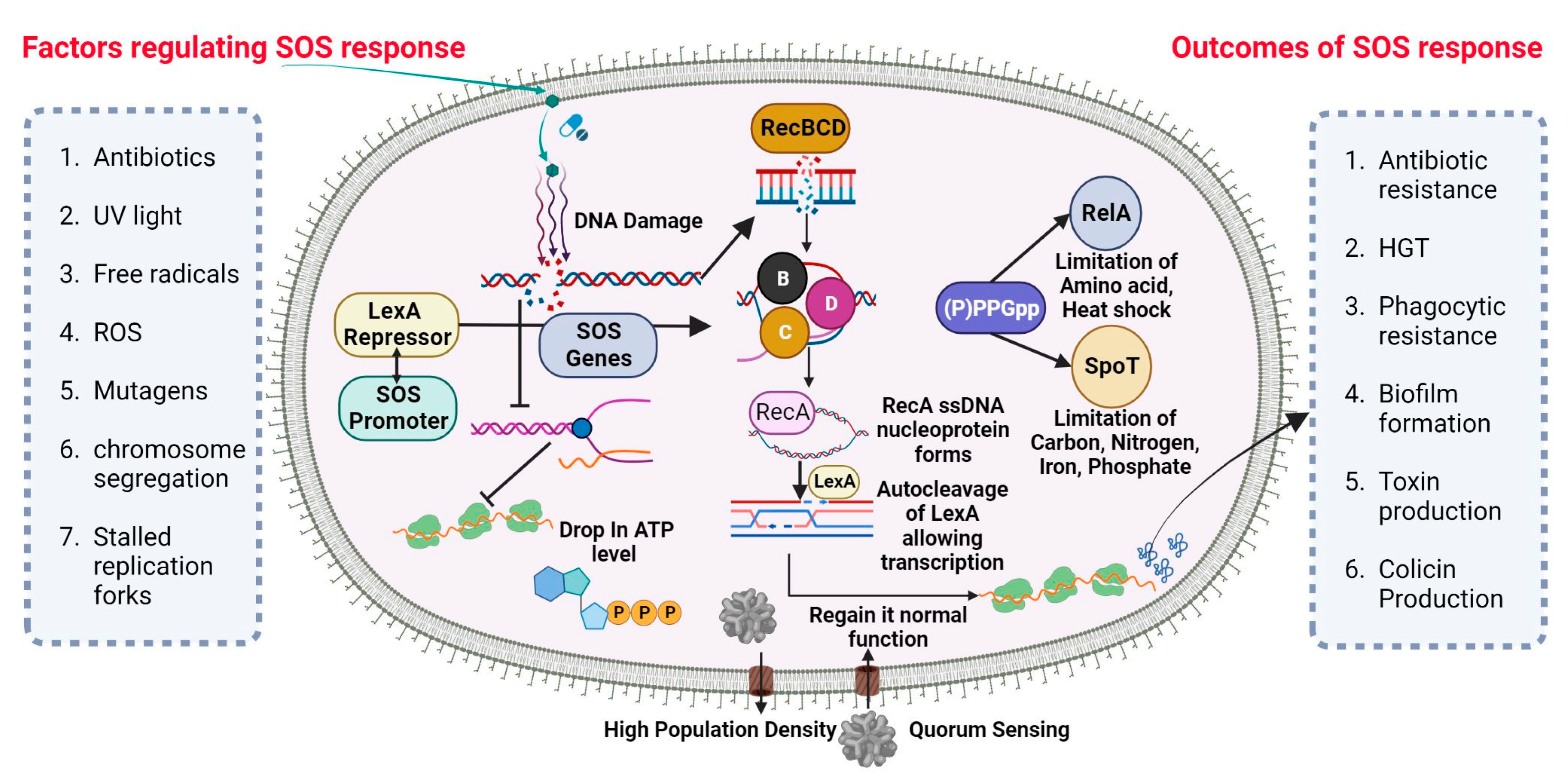
2.4. Antibiotic Resistance by SOS Response
SOS-Dependent Mutagenesis and Resistance
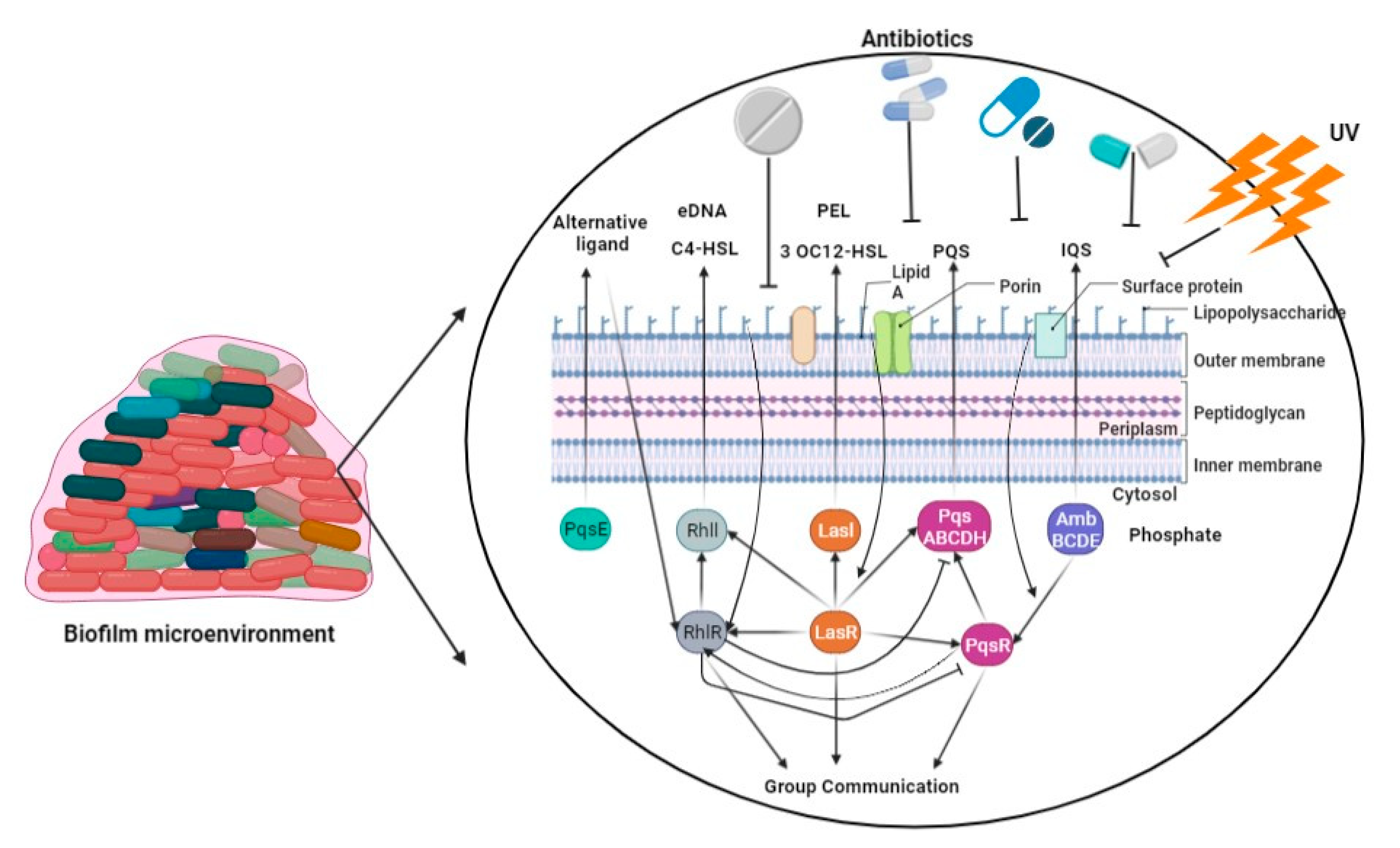
2.5. Biofilm-Mediated Resistome
3. MDR/XDR P. aeruginosa Epidemiology
4. Clinical Impact of MDR P. aeruginosa
5. Antibiotic Agents for of MDR P. aeruginosa
5.1. Polymyxins
5.2. Carbapenems
5.3. Antipseudomonal β-Lactams
5.4. Aminoglycosides
5.5. Fosfomycin
5.6. Newly Discovered Antimicrobial Agents
6. Therapeutic Strategies for P. aeruginosa Treatment
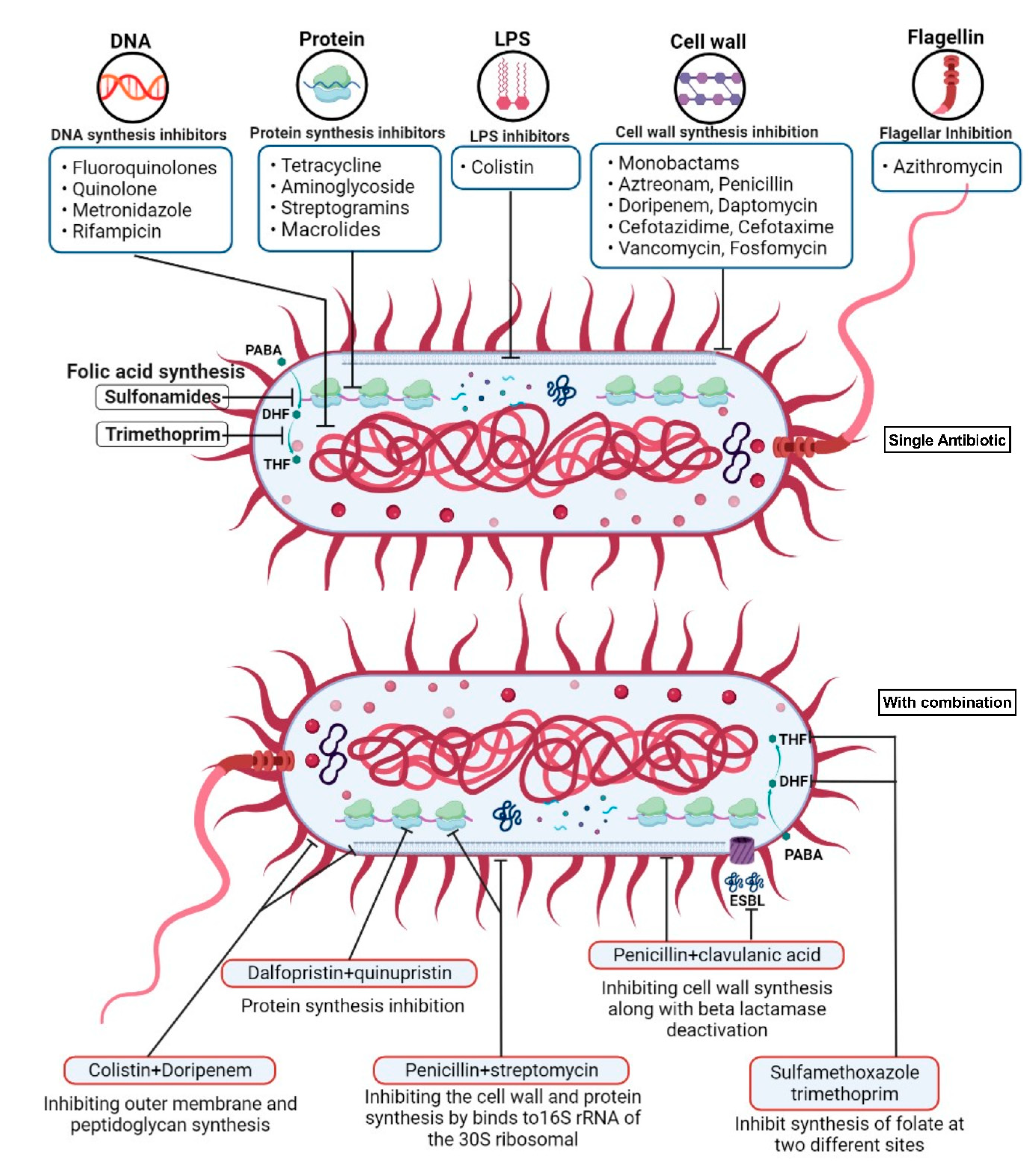
6.1. Combinatory Therapy
6.2. Antimicrobial Peptides
6.3. Phage Therapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Breidenstein, E.B.; de la Fuente-Núñez, C.; Hancock, R.E. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef]
- Oliver, A.; Mulet, X.; López-Causapé, C.; Juan, C. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist. Updat. 2015, 21–22, 41–59. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Peña, C.; Cabot, G.; Gómez-Zorrilla, S.; Zamorano, L.; Ocampo-Sosa, A.; Murillas, J.; Almirante, B.; Pomar, V.; Aguilar, M.; Granados, A.; et al. Influence of Virulence Genotype and Resistance Profile in the Mortality of Pseudomonas aeruginosa Bloodstream Infections. Clin. Infect. Dis. 2015, 60, 539–548. [Google Scholar] [CrossRef]
- Walkty, A.; Lagace-Wiens, P.; Adam, H.; Baxter, M.; Karlowsky, J.; Mulvey, M.R.; McCracken, M.; Zhanel, G.G. Antimicrobial susceptibility of 2906 Pseudomonas aeruginosa clinical isolates obtained from patients in Canadian hospitals over a period of 8 years: Results of the Canadian Ward surveillance study (CANWARD), 2008–2015. Diagn. Microbiol. Infect. Dis. 2017, 87, 60–63. [Google Scholar] [CrossRef]
- Solomon, S.L.; Oliver, K.B. Antibiotic resistance threats in the United States: Stepping back from the brink. Am. Fam. Physician 2014, 89, 938–941. [Google Scholar]
- Pogue, J.M.; Kaye, K.S.; Veve, M.P.; Patel, T.S.; Gerlach, A.T.; Davis, S.L.; Puzniak, L.A.; File, T.M.; Olson, S.; Dhar, S.; et al. Ceftolozane/Tazobactam vs Polymyxin or Aminoglycoside-based Regimens for the Treatment of Drug-resistant Pseudomonas aeruginosa. Clin. Infect. Dis. 2020, 71, 304–310. [Google Scholar] [CrossRef]
- Sader, H.S.; Carvalhaes, C.G.; Shortridge, D.; Castanheira, M. Comparative activity of newer β-lactam/β-lactamase inhibitor combinations against Pseudomonas aeruginosa from patients hospitalized with pneumonia in European medical centers in 2020. Eur. J. Clin. Microbiol. Infect. Dis. 2022, 41, 319–324. [Google Scholar] [CrossRef]
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-Resistant Pseudomonas aeruginosa: Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef]
- Skiada, A.; Markogiannakis, A.; Plachouras, D.; Daikos, G.L. Adaptive resistance to cationic compounds in Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2011, 37, 187–193. [Google Scholar] [CrossRef]
- Muller, C.; Plésiat, P.; Jeannot, K. A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and β-lactams in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2011, 55, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.; Torrens, G.; González-Nicolau, M.; Oliver, A. Diversity and regulation of intrinsic β-lactamases from non-fermenting and other Gram-negative opportunistic pathogens. FEMS Microbiol. Rev. 2017, 41, 781–815. [Google Scholar] [CrossRef]
- Horcajada, J.P.; Montero, M.; Oliver, A.; Sorlí, L.; Luque, S.; Gómez-Zorrilla, S.; Benito, N.; Grau, S. Epidemiology and Treatment of Multidrug-Resistant and Extensively Drug-Resistant Pseudomonas aeruginosa Infections. Clin. Microbiol. Rev. 2019, 32, e00031-19. [Google Scholar] [CrossRef] [PubMed]
- Girlich, D.; Naas, T.; Nordmann, P. Biochemical Characterization of the Naturally Occurring Oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2004, 48, 2043–2048. [Google Scholar] [CrossRef]
- Fajardo, A.; Hernando-Amado, S.; Oliver, A.; Ball, G.; Filloux, A.; Martinez, J.L. Characterization of a novel Zn2+-dependent intrinsic imipenemase from Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2014, 69, 2972–2978. [Google Scholar] [CrossRef]
- Li, X.-Z.; Plésiat, P.; Nikaido, H. The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef]
- Cabot, G.; Ocampo-Sosa, A.A.; Tubau, F.; Macia, M.D.; Rodríguez, C.; Moya, B.; Zamorano, L.; Suárez, C.; Peña, C.; Martínez-Martínez, L.; et al. Overexpression of AmpC and Efflux Pumps in Pseudomonas aeruginosa Isolates from Bloodstream Infections: Prevalence and Impact on Resistance in a Spanish Multicenter Study. Antimicrob. Agents Chemother. 2011, 55, 1906–1911. [Google Scholar] [CrossRef]
- Moya, B.; Dötsch, A.; Juan, C.; Blázquez, J.; Zamorano, L.; Haussler, S.; Oliver, A. Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 2009, 5, e1000353. [Google Scholar] [CrossRef]
- Cabot, G.; Bruchmann, S.; Mulet, X.; Zamorano, L.; Moyà, B.; Juan, C.; Haussler, S.; Oliver, A. Pseudomonas aeruginosa Ceftolozane-Tazobactam Resistance Development Requires Multiple Mutations Leading to Overexpression and Structural Modification of AmpC. Antimicrob. Agents Chemother. 2014, 58, 3091–3099. [Google Scholar] [CrossRef]
- Fraile-Ribot, P.A.; Cabot, G.; Mulet, X.; Periañez, L.; Martín-Pena, M.L.; Juan, C.; Pérez, J.L.; Oliver, A. Mechanisms leading to in vivo ceftolozane/tazobactam resistance development during the treatment of infections caused by MDR Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 658–663. [Google Scholar] [CrossRef]
- Lahiri, S.D.; Alm, R.A. Identification of Novel VEB β-Lactamase Enzymes and Their Impact on Avibactam Inhibition. Antimicrob. Agents Chemother. 2016, 60, 3183–3186. [Google Scholar] [CrossRef] [PubMed]
- Haidar, G.; Philips, N.J.; Shields, R.K.; Snyder, D.; Cheng, S.; Potoski, B.A.; Doi, Y.; Hao, B.; Press, E.G.; Cooper, V.S.; et al. Ceftolozane-Tazobactam for the Treatment of Multi-drug-Resistant Pseudomonas aeruginosa Infections: Clinical Effectiveness and Evolution of Resistance. Clin. Infect. Dis. 2017, 65, 110–120. [Google Scholar] [CrossRef]
- Berrazeg, M.; Jeannot, K.; Enguéné, V.Y.N.; Broutin, I.; Loeffert, S.; Fournier, D.; Plésiat, P. Mutations in β-Lactamase AmpC Increase Resistance of Pseudomonas aeruginosa Isolates to Antipseudomonal Cephalosporins. Antimicrob. Agents Chemother. 2015, 59, 6248–6255. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J.D.; Clark, S.T.; Coburn, B.; Zhang, Y.; Wang, P.W.; Donaldson, S.L.; Tullis, D.E.; Yau, Y.C.W.; Waters, V.J.; Hwang, D.M.; et al. Selective Sweeps and Parallel Pathoadaptation Drive Pseudomonas aeruginosa Evolution in the Cystic Fibrosis Lung. mBio 2015, 6, e00981-15. [Google Scholar] [CrossRef] [PubMed]
- López-Causapé, C.; Sommer, L.M.; Cabot, G.; Rubio, R.; Ocampo-Sosa, A.A.; Johansen, H.K.; Figuerola, J.; Cantón, R.; Kidd, T.J.; Molin, S.; et al. Evolution of the Pseudomonas aeru-ginosa mutational resistome in an international Cystic Fibrosis clone. Sci. Rep. 2017, 7, 5555. [Google Scholar] [CrossRef] [PubMed]
- Cabot, G.; López-Causapé, C.; Ocampo-Sosa, A.A.; Sommer, L.M.; Domínguez, M.; Zamorano, L.; Juan, C.; Tubau, F.; Rodríguez, C.; Moyà, B.; et al. Deciphering the Resistome of the Widespread Pseudomonas aeruginosa Sequence Type 175 International High-Risk Clone through Whole-Genome Sequencing. Antimicrob. Agents Chemother. 2016, 60, 7415–7423. [Google Scholar] [CrossRef]
- Del Barrio-Tofiño, E.; López-Causapé, C.; Cabot, G.; Rivera, A.; Benito, N.; Segura, C.; Montero, M.M.; Sorlí, L.; Tubau, F.; Gómez-Zorrilla, S.; et al. Genomics and Susceptibility Profiles of Extensively Drug-Resistant Pseudomonas aeruginosa Isolates from Spain. Antimicrob. Agents Chemother. 2017, 61, e01589-17. [Google Scholar] [CrossRef]
- Cabot, G.; Zamorano, L.; Moyà, B.; Juan, C.; Navas, A.; Blázquez, J.; Oliver, A. Evolution of Pseudomonas aeruginosa Antimicrobial Resistance and Fitness under Low and High Mutation Rates. Antimicrob. Agents Chemother. 2016, 60, 1767–1778. [Google Scholar] [CrossRef]
- Cabot, G.; Florit-Mendoza, L.; Sánchez-Diener, I.; Zamorano, L.; Oliver, A. Deciphering β-lactamase-independent β-lactam resistance evolution trajectories in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 3322–3331. [Google Scholar] [CrossRef]
- Han, S.; Zaniewski, R.P.; Marr, E.S.; Lacey, B.M.; Tomaras, A.P.; Evdokimov, A.; Miller, J.R.; Shanmugasundaram, V. Structural basis for effectiveness of sidero-phore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2010, 107, 22002–22007. [Google Scholar] [CrossRef]
- Moyá, B.; Beceiro, A.; Cabot, G.; Juan, C.; Zamorano, L.; Alberti, S.; Oliver, A. Pan-β-lactam resistance development in Pseudomonas aeruginosa clinical strains: Molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob. Agents Chemother. 2012, 56, 4771–4778. [Google Scholar] [CrossRef] [PubMed]
- Hocquet, D.; Berthelot, P.; Roussel-Delvallez, M.; Favre, R.; Jeannot, K.; Bajolet, O.; Marty, N.; Grattard, F.; Mariani-Kurkdjian, P.; Bingen, E.; et al. Pseudomonas aeruginosa may accumulate drug resistance mechanisms without losing its ability to cause bloodstream infections. Antimicrob. Agents Chemother. 2007, 51, 3531–3536. [Google Scholar] [CrossRef] [PubMed]
- Solé, M.; Fabrega, A.; Cobos-Trigueros, N.; Zamorano, L.; Ferrer-Navarro, M.; Ballesté-Delpierre, C.; Reustle, A.; Castro, P.; Nicolás, J.M.; Oliver, A.; et al. In vivo evolution of resistance of Pseudomonas aeruginosa strains isolated from patients admitted to an intensive care unit: Mechanisms of resistance and antimicrobial exposure. J. Antimicrob. Chemother. 2015, 70, 3004–3013. [Google Scholar] [CrossRef] [PubMed]
- Riera, E.; Cabot, G.; Mulet, X.; García-Castillo, M.; del Campo, R.; Juan, C.; Cantón, R.; Oliver, A. Pseudomonas aeruginosa carbapenem resistance mechanisms in Spain: Impact on the activity of imipenem, meropenem and doripenem. J. Antimicrob. Chemother. 2011, 66, 2022–2027. [Google Scholar] [CrossRef] [PubMed]
- Guénard, S.; Muller, C.; Monlezun, L.; Benas, P.; Broutin, I.; Jeannot, K.; Plésiat, P. Multiple mutations lead to MexXY-OprM-dependent aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Köhler, T.; Epp, S.F.; Curty, L.K.; Pechère, J.C. Characterization of MexT, the regulator of the MexE-MexF-OprN multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 6300–6305. [Google Scholar] [CrossRef] [PubMed]
- Mulet, X.; Moyá, B.; Juan, C.; Macià, M.D.; Pérez, J.L.; Blázquez, J.; Oliver, A. Antagonistic Interactions of Pseudomonas aeruginosa Antibiotic Resistance Mechanisms in Planktonic but Not Biofilm Growth. Antimicrob. Agents Chemother. 2011, 55, 4560–4568. [Google Scholar] [CrossRef]
- Bruchmann, S.; Dötsch, A.; Nouri, B.; Chaberny, I.F.; Häussler, S. Quantitative Contributions of Target Alteration and Decreased Drug Accumulation to Pseudomonas aeruginosa Fluoroquinolone Resistance. Antimicrob. Agents Chemother. 2013, 57, 1361–1368. [Google Scholar] [CrossRef]
- Feng, Y.; Jonker, M.J.; Moustakas, I.; Brul, S.; ter Kuile, B.H. Dynamics of Mutations during Development of Resistance by Pseudomonas aeruginosa against Five Antibiotics. Antimicrob. Agents Chemother. 2016, 60, 4229–4236. [Google Scholar] [CrossRef]
- López-Causapé, C.; Cabot, G.; del Barrio-Tofiño, E.; Oliver, A. The Versatile Mutational Resistome of Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 685. [Google Scholar] [CrossRef]
- Greipel, L.; Fischer, S.; Klockgether, J.; Dorda, M.; Mielke, S.; Wiehlmann, L.; Cramer, N.; Tümmler, B. Molecular Epidemiology of Mutations in Antimicrobial Resistance Loci of Pseudomonas aeruginosa Isolates from Airways of Cystic Fibrosis Patients. Antimicrob. Agents Chemother. 2016, 60, 6726–6734. [Google Scholar] [CrossRef]
- Olaitan, A.O.; Morand, S.; Rolain, J.-M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef] [PubMed]
- Gutu, A.D.; Sgambati, N.; Strasbourger, P.; Brannon, M.K.; Jacobs, M.A.; Haugen, E.; Kaul, R.K.; Johansen, H.K.; Høiby, N.; Moskowitz, S.M. Polymyxin Resistance of Pseudomonas aeruginosa phoQ Mutants Is Dependent on Additional Two-Component Regulatory Systems. Antimicrob. Agents Chemother. 2013, 57, 2204–2215. [Google Scholar] [CrossRef] [PubMed]
- Cirz, R.T.; Chin, J.K.; Andes, D.R.; de Crécy-Lagard, V.; Craig, W.A.; Romesberg, F.E. Inhibition of Mutation and Combating the Evolution of Antibiotic Resistance. PLoS Biol. 2005, 3, e176. [Google Scholar] [CrossRef]
- McKenzie, G.J.; Harris, R.S.; Lee, P.L.; Rosenberg, S.M. The SOS response regulates adaptive mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 6646–6651. [Google Scholar] [CrossRef]
- Panda, A.; Drancourt, M.; Tuller, T.; Pontarotti, P. Genome-wide analysis of horizontally acquired genes in the genus Mycobacterium. Sci. Rep. 2018, 8, 14817. [Google Scholar] [CrossRef] [PubMed]
- Prudhomme, M.; Attaiech, L.; Sanchez, G.; Martin, B.; Claverys, J.P. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 2006, 313, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Kothari, A.; Kumar, P.; Gaurav, A.; Kaushal, K.; Pandey, A.; Yadav, S.R.M.; Jain, N.; Omar, B.J. Association of antibiotics and heavy metal arsenic to horizontal gene transfer from multidrug-resistant clinical strains to antibiotic-sensitive environmental strains. J. Hazard Mater. 2023, 443, 130260. [Google Scholar] [CrossRef]
- Halary, S.; Leigh, J.W.; Cheaib, B.; Lopez, P.; Bapteste, E. Network analyses structure genetic diversity in independent genetic worlds. Proc. Natl. Acad. Sci. USA 2010, 107, 127–132. [Google Scholar] [CrossRef]
- Patel, G.; Bonomo, R.A. Status report on carbapenemases: Challenges and prospects. Expert Rev. Anti-Infect. Ther. 2011, 9, 555–570. [Google Scholar] [CrossRef]
- Juan, C.; Conejo, M.C.; Tormo, N.; Gimeno, C.; Pascual, Á.; Oliver, A. Challenges for accurate susceptibility testing, detection and interpretation of β-lactam resistance phenotypes in Pseudomonas aeruginosa: Results from a Spanish multicentre study. J. Anti-Microb. Chemother. 2013, 68, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Botelho, J.; Grosso, F.; Quinteira, S.; Mabrouk, A.; Peixe, L. The complete nucleotide sequence of an IncP-2 megaplasmid unveils a mosaic architecture comprising a putative novel blaVIM-2-harbouring transposon in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2017, 72, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- Van der Zee, A.; Kraak, W.B.; Burggraaf, A.; Goessens, W.H.F.; Pirovano, W.; Ossewaarde, J.M.; Tommassen, J. Spread of Carbapenem Resistance by Transposition and Conjugation Among Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 2057. [Google Scholar] [PubMed]
- Botelho, J.; Grosso, F.; Peixe, L. Unravelling the genome of a Pseudomonas aeruginosa isolate belonging to the high-risk clone ST235 reveals an integrative conjugative element housing a blaGES-6 carbapenemase. J. Antimicrob. Chemother. 2018, 73, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Potron, A.; Poirel, L.; Nordmann, P. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: Mechanisms and epidemiology. Int. J. Antimicrob. Agents 2015, 45, 568–585. [Google Scholar]
- Del Barrio-Tofiño, E.; Zamorano, L.; Cortes-Lara, S.; López-Causapé, C.; Sánchez-Diener, I.; Cabot, G.; Bou, G.; Martínez-Martínez, L.; Oliver, A.; Galán, F.; et al. Spanish nationwide survey on Pseudomonas aeruginosa antimicrobial resistance mechanisms and epidemiology. J. Antimicrob. Chemother. 2019, 74, 1825–1835. [Google Scholar]
- Chávez-Jacobo, V.M.; Hernández-Ramírez, K.C.; Romo-Rodríguez, P.; Pérez-Gallardo, R.V.; Campos-García, J.; Gutiérrez-Corona, J.F.; García-Merinos, J.P.; Meza-Carmen, V.; Silva-Sánchez, J.; Ramírez-Díaz, M.I. CrpP Is a Novel Ciprofloxacin-Modifying Enzyme Encoded by the Pseudomonas aeruginosa pUM505 Plasmid. Antimicrob. Agents Chemother. 2018, 62, e02629-17. [Google Scholar] [CrossRef]
- Moya, B.; Zamorano, L.; Juan, C.; Pérez, J.L.; Ge, Y.; Oliver, A. Activity of a new cephalosporin, CXA-101 (FR264205), against be-ta-lactam-resistant Pseudomonas aeruginosa mutants selected in vitro and after antipseudomonal treatment of intensive care unit patients. Antimicrob. Agents Chemother. 2010, 54, 1213–1217. [Google Scholar] [CrossRef]
- Torrens, G.; Cabot, G.; Ocampo-Sosa, A.A.; Conejo, M.C.; Zamorano, L.; Navarro, F.; Pascual, Á.; Martínez-Martínez, L.; Oliver, A. Activity of Ceftazidime-Avibactam against Clinical and Isogenic Laboratory Pseudomonas aeruginosa Isolates Expressing Combinations of Most Relevant β-Lactam Resistance Mechanisms. Antimicrob. Agents Chemother. 2016, 60, 6407–6410. [Google Scholar] [CrossRef]
- Sanz-García, F.; Hernando-Amado, S.; Martínez, J.L. Mutation-Driven Evolution of Pseudomonas aeruginosa in the Presence of either Ceftazidime or Ceftazidime-Avibactam. Antimicrob. Agents Chemother. 2018, 62, e01379-18. [Google Scholar] [CrossRef]
- Recio, R.; Villa, J.; Viedma, E.; Orellana, M.; Lora-Tamayo, J.; Chaves, F. Bacteraemia due to extensively drug-resistant Pseudomonas aeruginosa sequence type 235 high-risk clone: Facing the perfect storm. Int. J. Antimicrob. Agents 2018, 52, 172–179. [Google Scholar] [CrossRef]
- Díaz-Cañestro, M.; Periañez, L.; Mulet, X.; Martin-Pena, M.L.; Fraile-Ribot, P.A.; Ayestarán, I.; Colomar, A.; Nuñez, B.; Maciá, M.; Novo, A.; et al. Ceftolozane/tazobactam for the treatment of multidrug resistant Pseudomonas aeruginosa: Experience from the Balearic Islands. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 2191–2200. [Google Scholar] [PubMed]
- Fraile-Ribot, P.A.; Mulet, X.; Cabot, G.; del Barrio-Tofiño, E.; Juan, C.; Pérez, J.L.; Oliver, A. In Vivo Emergence of Resistance to Novel Cephalosporin–β-Lactamase Inhibitor Combinations through the Duplication of Amino Acid D149 from OXA-2 β-Lactamase (OXA-539) in Sequence Type 235 Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e01117-17. [Google Scholar] [PubMed]
- Watson, M.E.; Burns, J.L.; Smith, A.L. Hypermutable Haemophilus influenzae with mutations in mutS are found in cystic fibrosis sputum. Microbiology 2004, 150, 2947–2958. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.; Cantón, R.; Campo, P.; Baquero, F.; Blázquez, J. High Frequency of Hypermutable Pseudomonas aeruginosa in Cystic Fibrosis Lung Infection. Science 2000, 288, 1251–1253. [Google Scholar] [CrossRef]
- Hocquet, D.; Llanes, C.; Thouverez, M.; Kulasekara, H.D.; Bertrand, X.; Plésiat, P.; Mazel, D.; Miller, S.I. Evidence for induction of integron-based an-tibiotic resistance by the SOS response in a clinical setting. PLoS Pathog. 2012, 8, e1002778. [Google Scholar] [CrossRef]
- Breidenstein, E.B.M.; Bains, M.; Hancock, R.E.W. Involvement of the Lon Protease in the SOS Response Triggered by Ciprofloxacin in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 2012, 56, 2879–2887. [Google Scholar]
- Rossi, E.; La Rosa, R.; Bartell, J.A.; Marvig, R.L.; Haagensen, J.A.J.; Sommer, L.M.; Molin, S.; Johansen, H.K. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat. Rev. Microbiol. 2020, 19, 331–342. [Google Scholar]
- Smith, P.A.; Romesberg, F.E. Combating bacteria and drug resistance by inhibiting mechanisms of persistence and adaptation. Nat. Chem. Biol. 2007, 3, 549–556. [Google Scholar] [CrossRef]
- Horii, T.; Ogawa, T.; Nakatani, T.; Hase, T.; Matsubara, H.; Ogawa, H. Regulation of SOS functions: Purification of E. coli LexA protein and determination of its specific site cleaved by the RecA protein. Cell 1981, 27, 515–522. [Google Scholar] [CrossRef]
- Courcelle, J.; Khodursky, A.; Peter, B.; Brown, P.O.; Hanawalt, P.C. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 2001, 158, 41–64. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.D.; Smith, B.T.; Godoy, V.G.; Walker, G.C. The SOS response: Recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu. Rev. Genet. 2000, 34, 479–497. [Google Scholar] [PubMed]
- Gruber, A.J.; Erdem, A.L.; Sabat, G.; Karata, K.; Jaszczur, M.M.; Vo, D.D.; Olsen, T.M.; Woodgate, R.; Goodman, M.F.; Cox, M.M. A RecA Protein Surface Required for Activation of DNA Polymerase V. PLoS Genet. 2015, 11, e1005066. [Google Scholar] [CrossRef]
- Naiman, K.; Pagès, V.; Fuchs, R.P. A defect in homologous recombination leads to increased translesion synthesis in E. coli. Nucleic Acids Res. 2016, 44, 7691–7699. [Google Scholar] [CrossRef]
- Yakimov, A.; Bakhlanova, I.; Baitin, D. Targeting evolution of antibiotic resistance by SOS response inhibition. Comput. Struct. Biotechnol. J. 2021, 19, 777–783. [Google Scholar]
- Sniegowski, P.D.; Gerrish, P.J.; Lenski, R.E. Evolution of high mutation rates in experimental populations of E. coli. Nature 1997, 387, 703–705. [Google Scholar] [CrossRef]
- Pomerantz, R.T.; Kurth, I.; Goodman, M.F.; O’Donnell, M.E. Preferential D-loop extension by a translesion DNA polymerase underlies error-prone recombination. Nat. Struct. Mol. Biol. 2013, 20, 748–755. [Google Scholar]
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; et al. The Y-family of DNA polymerases. Mol. Cell 2001, 8, 7–8. [Google Scholar] [CrossRef]
- Boshoff, H.I.M.; Reed, M.B.; Barry, C.E.; Mizrahi, V. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 2003, 113, 183–193. [Google Scholar] [CrossRef]
- Stewart, P.S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358, 135–138. [Google Scholar] [CrossRef]
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.C.; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.K.; Yeung, A.T.; Hancock, R.E. Antibiotic resistance in Pseudomonas aeruginosa biofilms: Towards the development of novel anti-biofilm therapies. J. Biotechnol. 2014, 191, 121–130. [Google Scholar] [CrossRef]
- Rasamiravaka, T.; Labtani, Q.; Duez, P.; El Jaziri, M. The Formation of Biofilms by Pseudomonas aeruginosa: A Review of the Natural and Synthetic Compounds Interfering with Control Mechanisms. Biomed Res. Int. 2015, 2015, 759348. [Google Scholar] [CrossRef]
- Miller, M.B.; Bassler, B.L. Quorum Sensing in Bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef]
- Kang, D.; Turner, K.E.; Kirienko, N.V. PqsA Promotes Pyoverdine Production via Biofilm Formation. Pathogens 2017, 7, 3. [Google Scholar] [CrossRef]
- Parkins, M.D.; Ceri, H.; Storey, D.G. Pseudomonas aeruginosa GacA, a factor in multihost virulence, is also essential for biofilm for-mation. Mol. Microbiol. 2001, 40, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Wilton, M.; Charron-Mazenod, L.; Moore, R.; Lewenza, S. Extracellular DNA Acidifies Biofilms and Induces Aminoglycoside Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 544–553. [Google Scholar] [CrossRef]
- Hengge, R. Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 2009, 7, 263–273. [Google Scholar] [CrossRef]
- Ha, D.-G.; O’Toole, G.A.; Ghannoum, M.; Parsek, M.; Whiteley, M.; Mukherjee, P.K. c-di-GMP and its Effects on Biofilm Formation and Dispersion: A Pseudomonas aeruginosa Review. Microbiol. Spectr. 2015, 3, MB-0003-2014. [Google Scholar] [CrossRef]
- Drenkard, E. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 2003, 5, 1213–1219. [Google Scholar] [CrossRef]
- Pritt, B.; O’Brien, L.; Winn, W. Mucoid Pseudomonas in cystic fibrosis. Am. J. Clin. Pathol. 2007, 128, 32–34. [Google Scholar] [CrossRef]
- Mah, T.-F.; Pitts, B.; Pellock, B.; Walker, G.C.; Stewart, P.S.; O’Toole, G.A. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 2003, 426, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mah, T.-F. Involvement of a Novel Efflux System in Biofilm-Specific Resistance to Antibiotics. J. Bacteriol. 2008, 190, 4447–4452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hinz, A.J.; Nadeau, J.P.; Mah, T.F. Pseudomonas aeruginosa tssC1 links type VI secretion and biofilm-specific antibiotic resistance. J. Bacteriol. 2011, 193, 5510–5513. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Castanheira, M.; Duncan, L.R.; Flamm, R.K. Antimicrobial Susceptibility of Enterobacteriaceae and Pseudomonas aeruginosa Isolates from United States Medical Centers Stratified by Infection Type: Results from the International Network for Optimal Resistance Monitoring (INFORM) Surveillance Program, 2015–2016. Diagn. Microbiol. Infect. Dis. 2018, 92, 69–74. [Google Scholar]
- Souli, M.; Galani, I.; Giamarellou, H. Emergence of extensively drug-resistant and pandrug-resistant Gram-negative bacilli in Europe. Eurosurveillance 2008, 13, 19045. [Google Scholar] [CrossRef]
- Viedma, E.; Juan, C.; Acosta, J.; Zamorano, L.; Otero, J.R.; Sanz, F.; Chaves, F.; Oliver, A. Nosocomial spread of colistin-only-sensitive sequence type 235 Pseudomonas aeruginosa isolates producing the extended-spectrum beta-lactamases GES-1 and GES-5 in Spain. Antimicrob. Agents Chemother. 2009, 53, 4930–4933. [Google Scholar] [CrossRef]
- Flamm, R.K.; Farrell, D.J.; Sader, H.S.; Jones, R.N. Ceftazidime/avibactam activity tested against Gram-negative bacteria isolated from bloodstream, pneumonia, intra-abdominal and urinary tract infections in US medical centres (2012). J. Antimicrob. Chemother. 2014, 69, 1589–1598. [Google Scholar] [CrossRef]
- Giani, T.; Arena, F.; Pollini, S.; Di Pilato, V.; D’Andrea, M.M.; Henrici De Angelis, L.; Bassetti, M.; Rossolini, G.M. Italian nationwide survey on Pseudomonas aeruginosa from invasive infections: Activity of ceftolozane/tazobactam and comparators, and molecular epidemiology of car-bapenemase producers. J. Antimicrob. Chemother. 2018, 73, 664–671. [Google Scholar] [CrossRef]
- Livermore, D.M.; Meunier, D.; Hopkins, K.L.; Doumith, M.; Hill, R.; Pike, R.; Staves, P.; Woodford, N. Activity of ceftazidime/avibactam against problem Enterobacteriaceae and Pseudomonas aeruginosa in the UK, 2015–2016. J. Antimicrob. Chemother. 2017, 73, 648–657. [Google Scholar] [CrossRef]
- Paterson, D.L.; Rice, L.B. Empirical antibiotic choice for the seriously ill patient: Are minimization of selection of resistant organisms and maximization of individual outcome mutually exclusive? Clin. Infect. Dis. 2003, 36, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Guillamet, C.V.; Vazquez, R.; Noe, J.; Micek, S.T.; Kollef, M.H. A cohort study of bacteremic pneumonia: The importance of antibiotic resistance and appropriate initial therapy? Medicine 2016, 95, e4708. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Kim, S.; Kim, H.; Park, S.; Choe, Y.; Oh, M.; Kim, E.; Choe, K. Pseudomonas aeruginosa Bacteremia: Risk Factors for Mortality and Influence of Delayed Receipt of Effective Antimicrobial Therapy on Clinical Outcome. Clin. Infect. Dis. 2003, 37, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Micek, S.T.; Lloyd, A.E.; Ritchie, D.J.; Reichley, R.M.; Fraser, V.J.; Kollef, M.H. Pseudomonas aeruginosa Bloodstream Infection: Importance of Appropriate Initial Antimicrobial Treatment. Antimicrob. Agents Chemother. 2005, 49, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Park, H.J.; Moon, S.M.; Park, K.H.; Chong, Y.P.; Kim, M.N.; Kim, S.H.; Lee, S.O.; Kim, Y.S.; Woo, J.H.; et al. Impact of adequate empirical combination therapy on mortality from bacteremic Pseudomonas aeruginosa pneumonia. BMC Infect. Dis. 2012, 12, 308. [Google Scholar] [CrossRef]
- Hirsch, E.B.; Tam, V.H. Impact of multidrug-resistant Pseudomonas aeruginosa infection on patient outcomes. Expert Rev. Phar-macoecon. Outcomes Res. 2010, 10, 441–451. [Google Scholar] [CrossRef]
- Gómez-Zorrilla, S.; Morandeira, F.; Castro, M.J.; Tubau, F.; Periche, E.; Cañizares, R.; Dominguez, M.A.; Ariza, J.; Peña, C. Acute Inflammatory Response of Patients with Pseudomonas aeruginosa Infections: A Prospective Study. Microb. Drug Resist. 2017, 23, 523–530. [Google Scholar] [CrossRef]
- Planquette, B.; Timsit, J.F.; Misset, B.Y.; Schwebel, C.; Azoulay, E.; Adrie, C.; Vesin, A.; Jamali, S.; Zahar, J.R.; Allaouchiche, B.; et al. Pseudomonas aeruginosa ventilator-associated pneu-monia. predictive factors of treatment failure. Am. J. Respir. Crit. Care Med. 2013, 188, 69–76. [Google Scholar] [CrossRef]
- Kaminski, C.; Timsit, J.-F.; Dubois, Y.; Zahar, J.-R.; Garrouste-Orgeas, M.; Vesin, A.; Azoulay, E.; Feger, C.; Dumenil, A.-S.; Adrie, C.; et al. Impact of ureido/carboxypenicillin resistance on the prognosis of ventilator-associated pneumonia due to Pseudomonas aeruginosa. Crit. Care 2011, 15, R112. [Google Scholar] [CrossRef]
- Juan, C.; Peña, C.; Oliver, A. Host and Pathogen Biomarkers for Severe Pseudomonas aeruginosa Infections. J. Infect. Dis. 2017, 215 (Suppl. S1), S44–S51. [Google Scholar] [CrossRef]
- McCarthy, K.; Paterson, D. Long-term mortality following Pseudomonas aeruginosa bloodstream infection. J. Hosp. Infect. 2017, 95, 292–299. [Google Scholar] [CrossRef]
- Gómez-Zorrilla, S.; Camoez, M.; Tubau, F.; Periche, E.; Cañizares, R.; Dominguez, M.A.; Ariza, J.; Peña, C. Antibiotic Pressure Is a Major Risk Factor for Rectal Colonization by Multidrug-Resistant Pseudomonas aeruginosa in Critically Ill Patients. Antimicrob. Agents Chemother. 2014, 58, 5863–5870. [Google Scholar] [CrossRef] [PubMed]
- Blot, S.; Vandewoude, K.; De Bacquer, D.; Colardyn, F. Nosocomial Bacteremia Caused by Antibiotic-Resistant Gram-Negative Bacteria in Critically Ill Patients: Clinical Outcome and Length of Hospitalization. Clin. Infect. Dis. 2002, 34, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Olivares Pacheco, J.; Alvarez-Ortega, C.; Alcalde Rico, M.; Martínez, J.L. Metabolic Compensation of Fitness Costs Is a General Outcome for Antibiotic-Resistant Pseudomonas aeruginosa Mutants Overexpressing Efflux Pumps. mBio 2017, 8, e00500-17. [Google Scholar] [CrossRef]
- Skurnik, D.; Roux, D.; Cattoir, V.; Danilchanka, O.; Lu, X.; Yoder-Himes, D.R.; Han, K.; Guillard, T.; Jiang, D.; Gaultier, C.; et al. Enhanced in vivo fitness of carbapenem-resistant oprD mutants of Pseudomonas aeruginosa revealed through high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 20747–20752. [Google Scholar] [CrossRef]
- Andersson, D.I. The biological cost of mutational antibiotic resistance: Any practical conclusions? Curr. Opin. Microbiol. 2006, 9, 461–465. [Google Scholar] [CrossRef]
- McCarthy, K.; Wailan, A.; Jennison, A.; Kidd, T.; Paterson, D.P. aeruginosa blood stream infection isolates: A “full house” of virulence genes in isolates associated with rapid patient death and patient survival. Microb. Pathog. 2018, 119, 81–85. [Google Scholar] [CrossRef]
- Cuevas, D.A.; Edirisinghe, J.; Henry, C.S.; Overbeek, R.; O’connell, T.G.; Edwards, R.A. From DNA to FBA: How to Build Your Own Genome-Scale Metabolic Model. Front. Microbiol. 2016, 7, 907. [Google Scholar] [CrossRef]
- Engel, J.; Balachandran, P. Role of Pseudomonas aeruginosa type III effectors in disease. Curr. Opin. Microbiol. 2009, 12, 61–66. [Google Scholar] [CrossRef]
- El-Solh, A.A.; Hattemer, A.B.; Hauser, A.R.; Alhajhusain, A.; Vora, H. Clinical outcomes of type III Pseudomonas aeruginosa bacteremia. Crit. Care Med. 2012, 40, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.R. Pseudomonas aeruginosa virulence and antimicrobial resistance: Two sides of the same coin? Crit. Care Med. 2014, 42, 201–202. [Google Scholar] [CrossRef] [PubMed]
- Reboud, E.; Elsen, S.; Bouillot, S.; Golovkine, G.; Basso, P.; Jeannot, K.; Attrée, I.; Huber, P. Phenotype and toxicity of the recently discovered exlA-positive Pseudomonas aeruginosa strains collected worldwide. Environ. Microbiol. 2016, 18, 3425–3439. [Google Scholar] [CrossRef]
- Gómez-Zorrilla, S.; Camoez, M.; Tubau, F.; Cañizares, R.; Periche, E.; Dominguez, M.A.; Ariza, J.; Peña, C. Prospective Observational Study of Prior Rectal Colonization Status as a Predictor for Subsequent Development of Pseudomonas aeruginosa Clinical Infections. Antimicrob. Agents Chemother. 2015, 59, 5213–5219. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Zorrilla, S.; Juan, C.; Cabot, G.; Camoez, M.; Tubau, F.; Oliver, A.; Dominguez, M.A.; Ariza, J.; Peña, C. Impact of multidrug resistance on the pathogenicity of Pseudomonas aeruginosa: In vitro and in vivo studies. Int. J. Antimicrob. Agents 2016, 47, 368–374. [Google Scholar] [CrossRef]
- Kothari, A.; Kumar, S.K.; Singh, V.; Kumar, P.; Kaushal, K.; Pandey, A.; Jain, N.; Omar, B.J. Association of multidrug resistance behavior of clinical Pseudomonas aeruginosa to pigment coloration. Eur. J. Med. Res. 2022, 27, 120. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Koussoulas, V.; Panagou, C.; Adamis, T.; Baziaka, F.; Skiadas, I.; Perrea, D.; Dionyssiou-Asteriou, A.; Giamarellou, H. Experimental sepsis using Pseu-domonas aeruginosa: The significance of multi-drug resistance. Int. J. Antimicrob. Agents 2004, 24, 357–361. [Google Scholar] [CrossRef]
- Nation, R.L.; Li, J.; Cars, O.; Couet, W.; Dudley, M.N.; Kaye, K.S.; Mouton, J.W.; Paterson, D.L.; Tam, V.H.; Theuretzbacher, U.; et al. Framework for optimisation of the clinical use of colistin and polymyxin B: The Prato polymyxin consensus. Lancet Infect. Dis. 2015, 15, 225–234. [Google Scholar] [CrossRef]
- Garonzik, S.M.; Li, J.; Thamlikitkul, V.; Paterson, D.L.; Shoham, S.; Jacob, J.; Silveira, F.P.; Forrest, A.; Nation, R.L. Population pharmacokinetics of colistin methanesul-fonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob. Agents Chemother. 2011, 55, 3284–3294. [Google Scholar] [CrossRef]
- Nation, R.L.; Garonzik, S.M.; Thamlikitkul, V.; Giamarellos-Bourboulis, E.J.; Forrest, A.; Paterson, D.L.; Li, J.; Silveira, F.P. Dosing guidance for in-travenous colistin in critically-ill patients. Clin. Infect. Dis. 2017, 64, 565–571. [Google Scholar]
- Nation, R.L.; Garonzik, S.M.; Li, J.; Thamlikitkul, V.; Giamarellos-Bourboulis, E.J.; Paterson, D.L.; Turnidge, J.D.; Forrest, A.; Silveira, F.P. Updated US and European Dose Recommendations for Intravenous Colistin: How Do They Perform? Clin. Infect. Dis. 2016, 62, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Velkov, T.; Roberts, K.D.; Nation, R.L.; Thompson, P.E.; Li, J. Pharmacology of polymyxins: New insights into an “old” class of anti-biotics. Future Microbiol. 2013, 8, 711–724. [Google Scholar] [CrossRef]
- Poirel, L.; Jayol, A.; Nordmann, P. Polymyxins: Antibacterial Activity, Susceptibility Testing, and Resistance Mechanisms Encoded by Plasmids or Chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef] [PubMed]
- Nation, R.L.; Velkov, T.; Li, J. Colistin and polymyxin B: Peas in a pod, or chalk and cheese? Clin Infect Dis. 2014, 59, 88–94. [Google Scholar] [CrossRef]
- Zhang, L.; Dhillon, P.; Yan, H.; Farmer, S.; Hancock, R.E.W. Interactions of Bacterial Cationic Peptide Antibiotics with Outer and Cytoplasmic Membranes of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3317–3321. [Google Scholar] [CrossRef] [PubMed]
- Alhanout, K.; Malesinki, S.; Vidal, N.; Peyrot, V.; Rolain, J.M.; Brunel, J.M. New insights into the antibacterial mechanism of action of squalamine. J. Antimicrob. Chemother. 2010, 65, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, N.H.; Cushnie, T.P.T.; Matthews, K.H.; Lamb, A.J. Colistin causes profound morphological alteration but minimal cyto-plasmic membrane perforation in populations of Escherichia coli and Pseudomonas aeruginosa. Arch. Microbiol. 2018, 200, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Bergen, P.J.; Li, J.; Rayner, C.R.; Nation, R.L. Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas ae-ruginosa. Antimicrob. Agents Chemother. 2006, 50, 1953–1958. [Google Scholar] [CrossRef]
- Dalfino, L.; Puntillo, F.; Mosca, A.; Monno, R.; Spada, M.L.; Coppolecchia, S.; Miragliotta, G.; Bruno, F.; Brienza, N. High-dose, extended-interval colistin administration in critically ill patients: Is this the right dosing strategy? A preliminary study. Clin. Infect. Dis. 2012, 54, 1720–1726. [Google Scholar] [CrossRef]
- Paul, M.; Daikos, G.L.; Durante-Mangoni, E.; Yahav, D.; Carmeli, Y.; Benattar, Y.D.; Skiada, A.; Andini, R.; Eliakim-Raz, N.; Nutman, A.; et al. Colistin alone versus colistin plus meropenem for treatment of severe infections caused by carbapenem-resistant Gram-negative bacteria: An open-label, randomised controlled trial. Lancet Infect. Dis. 2018, 18, 391–400. [Google Scholar] [CrossRef]
- Sorlí, L.; Luque, S.; Segura, C.; Campillo, N.; Montero, M.; Esteve, E.; Herrera, S.; Benito, N.; Alvarez-Lerma, F.; Grau, S.; et al. Impact of colistin plasma levels on the clinical outcome of patients with infections caused by extremely drug-resistant Pseudomonas aeruginosa. BMC Infect. Dis. 2017, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, B.T.; Pogue, J.M.; Zavascki, A.P.; Paul, M.; Daikos, G.L.; Forrest, A.; Giacobbe, D.R.; Viscoli, C.; Giamarellou, H.; Karaiskos, I.; et al. International Consensus Guidelines for the Optimal Use of the Polymyxins: Endorsed by the American College of Clinical Pharmacy (ACCP), European Society of Clinical Microbiology and Infectious Diseases (ESCMID), Infectious Diseases Society of America (IDSA), International Society for Anti-infective Pharmacology (ISAP), Society of Critical Care Medicine (SCCM), and Society of Infectious Diseases Pharmacists (SIDP). Pharmacother. J. Hum. Pharmacol. Drug Ther. 2019, 39, 10–39. [Google Scholar]
- Montero, M.; Horcajada, J.P.; Sorlí, L.; Alvarez-Lerma, F.; Grau, S.; Riu, M.; Sala, M.; Knobel, H. Effectiveness and safety of colistin for the treatment of multidrug-resistant Pseudomonas aeruginosa infections. Infection 2009, 37, 461–465. [Google Scholar] [CrossRef]
- Sorlí, L.; Luque, S.; Grau, S.; Berenguer, N.; Segura, C.; Montero, M.M.; Álvarez-Lerma, F.; Knobel, H.; Benito, N.; Horcajada, J.P. Trough colistin plasma level is an independent risk factor for nephrotoxicity: A prospective observational cohort study. BMC Infect. Dis. 2013, 13, 380. [Google Scholar] [CrossRef]
- Kothari, A.; Jain, N.; Kumar, S.K.; Kumar, A.; Kaushal, K.; Kaur, S.; Pandey, A.; Gaurav, A.; Omar, B.J. Potential Synergistic Antibiotic Combinations against Fluoroquinolone-Resistant Pseudomonas aeruginosa. Pharmaceuticals 2022, 15, 243. [Google Scholar] [CrossRef]
- French, S.; Farha, M.; Ellis, M.J.; Sameer, Z.; Côté, J.P.; Cotroneo, N.; Lister, T.; Rubio, A.; Brown, E.D. Potentiation of Antibiotics against Gram-Negative Bacteria by Polymyxin B Analogue SPR741 from Unique Perturbation of the Outer Membrane. ACS Infect Dis. 2020, 6, 1405–1412. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Lister, T.; Walpole, S.; Keutzer, T.; Utley, L.; Tomayko, J.; Kopp, E.; Farinola, N.; Coleman, S. Safety, Tolerability, Pharmacokinetics, and Drug Interaction Potential of SPR741, an Intravenous Potentiator, after Single and Multiple Ascending Doses and When Combined with β-Lactam Antibiotics in Healthy Subjects. Antimicrob. Agents Chemother. 2019, 63, 10–1128. [Google Scholar] [CrossRef]
- Kuti, J.L.; Dandekar, P.K.; Nightingale, C.H.; Nicolau, D.P. Use of Monte Carlo Simulation to Design an Optimized Pharmacodynamic Dosing Strategy for Meropenem. J. Clin. Pharmacol. 2003, 43, 1116–1123. [Google Scholar] [CrossRef]
- Yadav, R.; Bulitta, J.B.; Wang, J.; Nation, R.L.; Landersdorfer, C.B. Evaluation of Pharmacokinetic/Pharmacodynamic Model-Based Optimized Combination Regimens against Multidrug-Resistant Pseudomonas aeruginosa in a Murine Thigh Infection Model by Using Humanized Dosing Schemes. Antimicrob. Agents Chemother. 2017, 61, e01268-17. [Google Scholar] [CrossRef]
- Lim, T.-P.; Wang, R.; Poh, G.Q.; Koh, T.-H.; Tan, T.-Y.; Lee, W.; Teo, J.Q.-M.; Cai, Y.; Tan, T.-T.; Ee, P.L.R.; et al. Integrated pharmacokinetic–pharmacodynamic modeling to evaluate empiric carbapenem therapy in bloodstream infections. Infect. Drug Resist. 2018, 11, 1591–1596. [Google Scholar] [CrossRef]
- Domenig, C.; Traunmüller, F.; Kozek, S.; Wisser, W.; Klepetko, W.; Steininger, R.; Spiss, C.; Thalhammer, F. Continuous beta-lactam antibiotic therapy in a double-lung transplanted patient with a multidrug-resistant Pseudomonas aeruginosa infection. Transplantation 2001, 71, 744–745. [Google Scholar] [CrossRef] [PubMed]
- Zavascki, A.P.; Barth, A.L.; Goldani, L.Z. Nosocomial bloodstream infections due to metallo-beta-lactamase-producing Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2008, 61, 1183–1185. [Google Scholar] [CrossRef]
- Sabuda, D.M.; Laupland, K.; Pitout, J.; Dalton, B.; Rabin, H.; Louie, T.; Conly, J. Utilization of Colistin for Treatment of Multidrug-Resistant Pseudomonas aeruginosa. Can. J. Infect. Dis. Med. Microbiol. 2008, 19, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, B.; Henning, S.A.; Childs, R.; Holland, S.M.; Anderson, V.L.; Morris, J.C.; Wilson, W.H.; Drusano, G.L.; Walsh, T.J. High-dose continuous infusion beta-lactam antibiotics for the treatment of resistant Pseudomonas aeruginosa infections in immunocompromised patients. Ann. Pharmacother. 2010, 44, 929–935. [Google Scholar] [CrossRef]
- Apisarnthanarak, A.; Mundy, L.M. Carbapenem-resistant Pseudomonas aeruginosa pneumonia with intermediate minimum in-hibitory concentrations to doripenem: Combination therapy with high-dose, 4-h infusion of doripenem plus fosfomycin versus intravenous colistin plus fosfomycin. Int. J. Antimicrob. Agents 2012, 39, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Drusano, G.L.; Bonomo, R.A.; Bahniuk, N.; Bulitta, J.B.; VanScoy, B.; DeFiglio, H.; Fikes, S.; Brown, D.; Drawz, S.M.; Kulawy, R.; et al. Resistance Emergence Mechanism and Mechanism of Resistance Suppression by Tobramycin for Cefepime for Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2012, 56, 231–242. [Google Scholar] [CrossRef]
- Lister, P.D.; Sanders, W.E.; Sanders, C.C. Cefepime-aztreonam: A unique double beta-lactam combination for Pseudomonas aeru-ginosa. Antimicrob. Agents Chemother. 1998, 42, 1610–1619. [Google Scholar] [CrossRef]
- Walkty, A.; Adam, H.; Baxter, M.; Lagacé-Wiens, P.; Karlowsky, J.A.; Hoban, D.J.; Zhanel, G.G. In vitro activity of ceftolozane/tazobactam versus antimicrobial non-susceptible Pseudomonas aeruginosa clinical isolates including MDR and XDR isolates obtained from across Canada as part of the CANWARD study, 2008–2016. J. Antimicrob. Chemother. 2018, 73, 703–708. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Shortridge, D.; Sader, H.S.; Castanheira, M.; Flamm, R.K. Ceftolozane/tazobactam activity against drug-resistant Enterobacteriaceae and Pseudomonas aeruginosa causing healthcare-associated infections in the Asia-Pacific region (minus China, Australia and New Zealand): Report from an Antimicrobial Surveillance Programme (2013–2015). Int. J. Antimicrob. Agents. 2018, 51, 181–189. [Google Scholar] [CrossRef]
- Buehrle, D.J.; Shields, R.K.; Chen, L.; Hao, B.; Press, E.G.; Alkrouk, A.; Potoski, B.A.; Kreiswirth, B.N.; Clancy, C.J.; Nguyen, M.H. Evaluation of the In Vitro Activity of Ceftazidime-Avibactam and Ceftolozane-Tazobactam against Meropenem-Resistant Pseudomonas aeruginosa Isolates. Antimicrob. Agents Chemother. 2016, 60, 3227–3231. [Google Scholar] [CrossRef]
- Craig, W.A.; Andes, D.R. In Vivo Activities of Ceftolozane, a New Cephalosporin, with and without Tazobactam against Pseudomonas aeruginosa and Enterobacteriaceae, Including Strains with Extended-Spectrum β-Lactamases, in the Thighs of Neutropenic Mice. Antimicrob. Agents Chemother. 2013, 57, 1577–1582. [Google Scholar] [CrossRef] [PubMed]
- Farrell, D.J.; Flamm, R.K.; Sader, H.S.; Jones, R.N. Antimicrobial Activity of Ceftolozane-Tazobactam Tested against Enterobacteriaceae and Pseudomonas aeruginosa with Various Resistance Patterns Isolated in U.S. Hospitals (2011–2012). Antimicrob. Agents Chemother. 2013, 57, 6305–6310. [Google Scholar] [CrossRef]
- Molinaro, M.; Morelli, P.; De Gregori, M.; De Gregori, S.; Giardini, I.; Tordato, F.; Monzillo, V.; Pocaterra, D.; Casari, E. Efficacy of intraventricular amikacin treatment in pan-resistant Pseudomonas aeruginosa postsurgical meningitis. Infect. Drug Resist. 2018, 11, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, B.; Rodulfo, H.; Carreño, N.; Guzmán, M.; Salazar, E.; DE Donato, M. Aminoglycoside resistance genes in Pseudomonas aeruginosa isolates from cumana, venezuela. Rev. Inst. De Med. Trop. De São Paulo 2016, 58, 13. [Google Scholar] [CrossRef]
- Jones, R.N.; Guzman-Blanco, M.; Gales, A.C.; Gallegos, B.; Castro, A.L.L.; Martino, M.D.V.; Vega, S.; Zurita, J.; Cepparulo, M.; Castanheira, M. Susceptibility rates in Latin American nations: Report from a regional resistance surveillance program (2011). Braz. J. Infect. Dis. 2013, 17, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Kastoris, A.C.; Karageorgopoulos, D.E.; Rafailidis, P.I. Fosfomycin for the treatment of infections caused by multi-drug-resistant non-fermenting Gram-negative bacilli: A systematic review of microbiological, animal and clinical studies. Int. J. Antimicrob. Agents 2009, 34, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, A.S.; Livaditis, I.G.; Gougoutas, V. The revival of fosfomycin. Int. J. Infect. Dis. 2011, 15, e732–e739. [Google Scholar] [CrossRef]
- Samonis, G.; Maraki, S.; Karageorgopoulos, D.E.; Vouloumanou, E.K.; Falagas, M.E. Synergy of fosfomycin with carbapenems, colistin, netilmicin, and tigecycline against multidrug-resistant Klebsiella pneumoniae, Escherichia coli, and Pseudomonas aeruginosa clinical isolates. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 695–701. [Google Scholar] [CrossRef]
- Frattari, A.; Savini, V.; Polilli, E.; Cibelli, D.; Talamazzi, S.; Bosco, D.; Consorte, A.; Fazii, P.; Parruti, G. Ceftolozane-tazobactam and Fosfomycin for rescue treatment of otogenous meningitis caused by XDR Pseudomonas aeruginosa: Case report and review of the literature. IDCases 2018, 14, e00451. [Google Scholar] [CrossRef]
- Asuphon, O.; Montakantikul, P.; Houngsaitong, J.; Kiratisin, P.; Sonthisombat, P. Optimizing intravenous fosfomycin dosing in combination with carbapenems for treatment of Pseudomonas aeruginosa infections in critically ill patients based on pharmaco-kinetic/pharmacodynamic (PK/PD) simulation. Int. J. Infect. Dis. 2016, 50, 23–29. [Google Scholar] [CrossRef]
- Hurley, M.N.; Cámara, M.; Smyth, A.R. Novel approaches to the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. Eur. Respir. J. 2012, 40, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Dale, G.E.; Rhomberg, P.R.; Flamm, R.K. Antimicrobial Activity of Murepavadin Tested against Clinical Isolates of Pseudomonas aeruginosa from the United States, Europe, and China. Antimicrob. Agents Chemother. 2018, 62, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Qadri, H.; Shah, A.H.; Mir, M. Novel Strategies to Combat the Emerging Drug Resistance in Human Pathogenic Microbes. Curr. Drug Targets 2021, 22, 1424–1436. [Google Scholar]
- Zhao, X.-L.; Chen, Z.-G.; Yang, T.-C.; Jiang, M.; Wang, J.; Cheng, Z.-X.; Yang, M.-J.; Zhu, J.-X.; Zhang, T.-T.; Li, H.; et al. Glutamine promotes antibiotic uptake to kill multidrug-resistant uropathogenic bacteria. Sci. Transl. Med. 2021, 13, eabj0716. [Google Scholar] [CrossRef]
- Wang, L.; Di Luca, M.; Tkhilaishvili, T.; Trampuz, A.; Moreno, M.G. Synergistic Activity of Fosfomycin, Ciprofloxacin, and Gentamicin Against Escherichia coli and Pseudomonas aeruginosa Biofilms. Front. Microbiol. 2019, 10, 2522. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef]
- Martinez, M.; Gonçalves, S.; Felício, M.R.; Maturana, P.; Santos, N.C.; Semorile, L.; Hollmann, A.; Maffía, P.C. Synergistic and antibiofilm activity of the an-timicrobial peptide P5 against carbapenem-resistant Pseudomonas aeruginosa. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1329–1337. [Google Scholar] [CrossRef]
- Vandenheuvel, D.; Lavigne, R.; Brüssow, H. Bacteriophage Therapy: Advances in Formulation Strategies and Human Clinical Trials. Annu. Rev. Virol. 2015, 2, 599–618. [Google Scholar] [CrossRef]
- Pei, R.; Lamas-Samanamud, G.R. Inhibition of Biofilm Formation by T7 Bacteriophages Producing Quorum-Quenching Enzymes. Appl. Environ. Microbiol. 2014, 80, 5340–5348. [Google Scholar] [CrossRef]
- Giacobbe, D.R.; Bassetti, M.; De Rosa, F.G.; Del Bono, V.; Grossi, P.A.; Menichetti, F.; Pea, F.; Rossolini, G.M.; Tumbarello, M.; Viale, P.; et al. Ceftolozane/tazobactam: Place in therapy. Expert Rev. Anti-Infect. Ther. 2018, 16, 307–320. [Google Scholar] [CrossRef]
- Gomis-Font, M.A.; Pitart, C.; del Barrio-Tofiño, E.; Zboromyrska, Y.; Cortes-Lara, S.; Mulet, X.; Marco, F.; Vila, J.; López-Causapé, C.; Oliver, A. Emergence of Resistance to Novel Cephalosporin-β-Lactamase Inhibitor Combinations through the Modification of the Pseudomonas aeruginosa MexCD-OprJ Efflux Pump. Antimicrob. Agents Chemother. 2021, 65, e0008921. [Google Scholar] [CrossRef] [PubMed]
- Balandin, B.; Ballesteros, D.; de Luna, R.R.; López-Vergara, L.; Pintado, V.; Sancho-González, M.; Soriano-Cuesta, C.; Pérez-Pedrero, M.J.; Asensio-Martín, M.J.; Fernández-Simón, I.; et al. Multicenter study of ceftolozane/tazobactam for treatment of Pseudomonas aeruginosa infections in critically ill patients. Int. J. Antimicrob. Agents 2021, 57, 106270. [Google Scholar] [CrossRef]
- Fernández-Cruz, A.; Alba, N.; Semiglia-Chong, M.A.; Padilla, B.; Rodríguez-Macías, G.; Kwon, M.; Cercenado, E.; Chamorro-de-Vega, E.; Machado, M.; Pérez-Lago, L.; et al. A Case-Control Study of Re-al-Life Experience with Ceftolozane-Tazobactam in Patients with Hematologic Malignancy and Pseudomonas aeruginosa Infection. Antimicrob, Agents Chemother. 2019, 63, e02340-18. [Google Scholar] [CrossRef]
- Solomkin, J.; Hershberger, E.; Miller, B.; Popejoy, M.; Friedland, I.; Steenbergen, J.; Yoon, M.; Collins, S.; Yuan, G.; Barie, P.S.; et al. Ceftolozane/Tazobactam Plus Metronidazole for Complicated Intra-abdominal Infections in an Era of Multidrug Resistance: Results from a Randomized, Double-Blind, Phase 3 Trial (ASPECT-cIAI). Clin. Infect. Dis. 2015, 60, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Tamma, P.D.; Aitken, S.L.; Bonomo, R.A.; Mathers, A.J.; van Duin, D.; Clancy, C.J. Infectious Diseases Society of America Guidance on the Treatment of Extended-Spectrum β-lactamase Producing Enterobacterales (ESBL-E), Carbapenem-Resistant Enterobacterales (CRE), and Pseudomonas aeruginosa with Difficult-to-Treat Resistance (DTR-). Clin. Infect. Dis. 2021, 72, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, M.; Castaldo, N.; Cattelan, A.; Mussini, C.; Righi, E.; Tascini, C.; Menichetti, F.; Mastroianni, C.M.; Tumbarello, M.; Grossi, P.; et al. Ceftolozane/tazobactam for the treatment of serious Pseudomonas aeruginosa infections: A multicentre nationwide clinical experience. Int. J. Antimicrob. Agents 2019, 53, 408–415. [Google Scholar] [CrossRef]
- De Jonge, M.C.; Kornelis, J.A.; van den Berg, J.W. Electrically induced evacuation of the neurogenic bladder. Acute and long-term experiments (2 years) with paraplegic dogs. Acta Neurol. Scand. 1966, 42, 155–173. [Google Scholar] [CrossRef]
- Losito, A.R.; Raffaelli, F.; Del Giacomo, P.; Tumbarello, M. New Drugs for the Treatment of Pseudomonas aeruginosa Infections with Limited Treatment Options: A Narrative Review. Antibiotics 2022, 11, 579. [Google Scholar] [CrossRef]
- Lob, S.H.; Karlowsky, J.A.; Young, K.; Motyl, M.R.; Hawser, S.; Kothari, N.D.; Sahm, D.F. In vitro activity of imipenem-relebactam against resistant phenotypes of Enterobacteriaceae and Pseudomonas aeruginosa isolated from intraabdominal and urinary tract infection samples—SMART Surveillance Europe 2015–2017. J. Med. Microbiol. 2020, 69, 207–217. [Google Scholar] [CrossRef]
- Lob, S.H.; DePestel, D.D.; DeRyke, C.A.; Kazmierczak, K.M.; Young, K.; Motyl, M.R.; Sahm, D.F. Ceftolozane/tazobactam and imipenem/relebactam cross-susceptibility among clinical isolates of Pseudomonas aeruginosa from patients with respiratory tract infections in ICU and non-ICU wards—SMART United States 2017–2019. Open Forum Infect. Dis. 2021, 8, ofab320. [Google Scholar] [CrossRef]
- Noval, M.; Banoub, M.; Claeys, K.C.; Heil, E. The Battle Is on: New Beta-Lactams for the Treatment of Multidrug-Resistant Gram-Negative Organisms. Curr. Infect. Dis. Rep. 2020, 22, 1. [Google Scholar] [CrossRef]
- Shortridge, D.; Carvalhaes, C.; Deshpande, L.; Castanheira, M. Activity of meropenem/vaborbactam and comparators against Gram-negative isolates from Eastern and Western European patients hospitalized with pneumonia including ventila-tor-associated pneumonia (2014-19). J. Antimicrob. Chemother. 2021, 76, 2600–2605. [Google Scholar] [CrossRef]
- Dowell, J.A.; Marbury, T.C.; Smith, W.B.; Henkel, T. Safety and Pharmacokinetics of Taniborbactam (VNRX-5133) with Cefepime in Subjects with Various Degrees of Renal Impairment. Antimicrob. Agents Chemother. 2022, 66, e0025322. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Han, R.; Jiang, B.; Ding, L.; Yang, F.; Zheng, B.; Yang, Y.; Wu, S.; Yin, D.; Zhu, D.; et al. In Vitro Activity of New β-Lactam–β-Lactamase Inhibitor Combinations and Comparators against Clinical Isolates of Gram-Negative Bacilli: Results from the China Antimicrobial Surveillance Network (CHINET) in 2019. Microbiol. Spectr. 2022, 10, e0185422. [Google Scholar] [CrossRef] [PubMed]
- Wagenlehner, F.M.; Umeh, O.; Steenbergen, J.; Yuan, G.; Darouiche, R. Ceftolozane-tazobactam compared with levofloxacin in the treatment of complicated urinary-tract infections, including pyelonephritis: A randomised, double-blind, phase 3 trial (ASPECT-cUTI). Lancet 2015, 385, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, C.M.; Nunes, L.d.S.; Martins, A.F.; Barth, A.L. In vitro antimicrobial activity of imipenem plus amikacin or polymyxin B against carbapenem-resistant Pseudomonas aeruginosa isolates. Diagn. Microbiol. Infect. Dis. 2018, 92, 152–154. [Google Scholar] [CrossRef] [PubMed]
- McGrath, B.J.; Lamp, K.C.; Rybak, M.J. Pharmacodynamic effects of extended dosing intervals of imipenem alone and in combination with amikacin against Pseudomonas aeruginosa in an in vitro model. Antimicrob. Agents Chemother. 1993, 37, 1931–1937. [Google Scholar] [CrossRef]
- Yadav, R.; Bulitta, J.B.; Nation, R.L.; Landersdorfer, C.B. Optimization of Synergistic Combination Regimens against Carbapenem- and Aminoglycoside-Resistant Clinical Pseudomonas aeruginosa Isolates via Mechanism-Based Pharmacokinetic/Pharmacodynamic Modeling. Antimicrob. Agents Chemother. 2017, 61, 10–1128. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Grecka, P.; Giamarellou, H. Comparative in vitro interactions of ceftazidime, meropenem, and imipenem with amikacin on multiresistant Pseudomonas aeruginosa. Diagn. Microbiol. Infect. Dis. 1997, 29, 81–86. [Google Scholar] [CrossRef]
- Lister, P.D.; Wolter, D.J.; Wickman, P.A.; Reisbig, M.D. Levofloxacin/imipenem prevents the emergence of high-level resistance among Pseudomonas aeruginosa strains already lacking susceptibility to one or both drugs. J. Antimicrob. Chemother. 2006, 57, 999–1003. [Google Scholar] [CrossRef]
- Gunderson, B.W.; Ibrahim, K.H.; Hovde, L.B.; Fromm, T.L.; Reed, M.D.; Rotschafer, J.C. Synergistic Activity of Colistin and Ceftazidime against Multiantibiotic-Resistant Pseudomonas aeruginosa in an In Vitro Pharmacodynamic Model. Antimicrob. Agents Chemother. 2003, 47, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Bergen, P.J.; Forrest, A.; Bulitta, J.B.; Tsuji, B.T.; Sidjabat, H.E.; Paterson, D.L.; Li, J.; Nation, R.L. Clinically Relevant Plasma Concentrations of Colistin in Combination with Imipenem Enhance Pharmacodynamic Activity against Multidrug-Resistant Pseudomonas aeruginosa at Multiple Inocula. Antimicrob. Agents Chemother. 2011, 55, 5134–5142. [Google Scholar] [CrossRef] [PubMed]
- Bergen, P.J.; Tsuji, B.T.; Bulitta, J.B.; Forrest, A.; Jacob, J.; Sidjabat, H.E.; Paterson, D.L.; Nation, R.L.; Li, J. Synergistic killing of multidrug-resistant Pseudomonas aeru-ginosa at multiple inocula by colistin combined with doripenem in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob. Agents Chemother. 2011, 55, 5685–5695. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Junyent, J.; Benavent, E.; Sierra, Y.; El Haj, C.; Soldevila, L.; Torrejón, B.; Rigo-Bonnin, R.; Tubau, F.; Ariza, J.; Murillo, O. Efficacy of ceftolozane/tazobactam, alone and in combination with colistin, against multidrug-resistant Pseudomonas aeruginosa in an in vitro biofilm pharmacodynamic model. Int. J. Antimicrob. Agents 2019, 53, 612–619. [Google Scholar] [CrossRef]
- Montero, M.M.; Domene Ochoa, S.; López-Causapé, C.; Luque, S.; Sorlí, L.; Campillo, N.; López Montesinos, I.; Padilla, E.; Prim, N.; Angulo-Brunet, A.; et al. Time-Kill Evaluation of Antibiotic Combinations Containing Ceftazidime-Avibactam against Extensively Drug-Resistant Pseudomonas aeruginosa and Their Potential Role against Ceftazidime-Avibactam-Resistant Isolates. Microbiol. Spectr. 2021, 9, e0058521. [Google Scholar] [CrossRef] [PubMed]
- Scudeller, L.; Righi, E.; Chiamenti, M.; Bragantini, D.; Menchinelli, G.; Cattaneo, P.; Giske, C.G.; Lodise, T.; Sanguinetti, M.; Piddock, L.J.; et al. Systematic review and meta-analysis of in vitro efficacy of antibiotic combination therapy against carbapenem-resistant Gram-negative bacilli. Int. J. Antimicrob. Agents 2021, 57, 106344. [Google Scholar] [CrossRef]
- Galani, I.; Papoutsaki, V.; Karantani, I.; Karaiskos, I.; Galani, L.; Adamou, P.; Deliolanis, I.; Kodonaki, A.; Papadogeorgaki, E.; Markopoulou, M.; et al. In vitro activity of ceftolozane/tazobactam alone and in combination with amikacin against MDR/XDR Pseudomonas aeruginosa isolates from Greece. J. Antimicrob. Chemother. 2020, 75, 2164–2172. [Google Scholar] [CrossRef]
- Monogue, M.L.; Nicolau, D.P. Antibacterial activity of ceftolozane/tazobactam alone and in combination with other antimicrobial agents against MDR Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2017, 73, 942–952. [Google Scholar] [CrossRef]
- Sader, H.S.; Rhomberg, P.R.; Jones, R.N. In vitro activity of beta-lactam antimicrobial agents in combination with aztreonam tested against metallo-beta-lactamase-producing Pseudomonas aeruginosa and Acinetobacter baumannii. J. Chemother. 2005, 17, 622–627. [Google Scholar] [CrossRef]
- Gaurav, A.; Kothari, A.; Omar, B.J.; Pathania, R. Assessment of polymyxin B-doxycycline in combination against Pseudomonas aeru-ginosa in vitro and in a mouse model of acute pneumonia. Int. J. Antimicrob. Agents 2020, 56, 106022. [Google Scholar] [CrossRef]
- Saini, M.; Gaurav, A.; Kothari, A.; Omar, B.J.; Gupta, V.; Bhattacharjee, A.; Pathania, R. Small Molecule IITR00693 (2-Aminoperimidine) Synergizes Polymyxin B Activity against Staphylococcus aureus and Pseudomonas aeruginosa. ACS Infect Dis. 2023, 9, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Ridyard, K.E.; Elsawy, M.; Mattrasingh, D.; Klein, D.; Strehmel, J.; Beaulieu, C.; Wong, A.; Overhage, J. Synergy between Human Peptide LL-37 and Polymyxin B against Planktonic and Biofilm Cells of Escherichia coli and Pseudomonas aeruginosa. Antibiotics 2023, 12, 389. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Database-Guided Discovery of Potent Peptides to Combat HIV-1 or Superbugs. Pharmaceuticals 2013, 6, 728–758. [Google Scholar] [CrossRef]
- Aoki, W.; Ueda, M. Characterization of Antimicrobial Peptides toward the Development of Novel Antibiotics. Pharmaceuticals 2013, 6, 1055–1081. [Google Scholar] [CrossRef]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Chung, P.Y.; Khanum, R. Antimicrobial peptides as potential anti-biofilm agents against multidrug-resistant bacteria. J. Microbiol. Immunol. Infect. 2017, 50, 405–410. [Google Scholar] [CrossRef] [PubMed]
- León-Buitimea, A.; Garza-Cárdenas, C.R.; Garza-Cervantes, J.A.; Lerma-Escalera, J.A.; Morones-Ramírez, J.R. The Demand for New Antibiotics: Antimicrobial Peptides, Nanoparticles, and Combinatorial Therapies as Future Strategies in Antibacterial Agent Design. Front. Microbiol. 2020, 11, 1669. [Google Scholar] [CrossRef]
- Kim, S.; Rahman, M.; Seol, S.Y.; Yoon, S.S.; Kim, J. Pseudomonas aeruginosa Bacteriophage PA1Ø Requires Type IV Pili for Infection and Shows Broad Bactericidal and Biofilm Removal Activities. Appl. Environ. Microbiol. 2012, 78, 6380–6385. [Google Scholar] [CrossRef]
- Liu, P.V. Extracellular toxins of Pseudomonas aeruginosa. J. Infect. Dis. 1974, 130, S94–S99. [Google Scholar] [CrossRef]
- Shiau, J.-W.; Tang, T.-K.; Shih, Y.-L.; Tai, C.; Sung, Y.-Y.; Huang, J.-L.; Yang, H.-L. Mice immunized with DNA encoding a modified Pseudomonas aeruginosa exotoxin A develop protective immunity against exotoxin intoxication. Vaccine 2000, 19, 1106–1112. [Google Scholar] [CrossRef]
- Chang, C.; Yu, X.; Guo, W.; Guo, C.; Guo, X.; Li, Q.; Zhu, Y. Bacteriophage-Mediated Control of Biofilm: A Promising New Dawn for the Future. Front. Microbiol. 2022, 13, 825828. [Google Scholar] [CrossRef] [PubMed]
- Burrows, L.L. The Therapeutic Pipeline for Pseudomonas aeruginosa Infections. ACS Infect. Dis. 2018, 4, 1041–1047. [Google Scholar] [CrossRef]
- Patil, A.; Banerji, R.; Kanojiya, P.; Koratkar, S.; Saroj, S. Bacteriophages for ESKAPE: Role in pathogenicity and measures of control. Expert Rev. Anti-Infect. Ther. 2021, 19, 845–865. [Google Scholar] [CrossRef] [PubMed]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Responsible Genes | Mechanism of Resistance | Associated Antibiotics |
|---|---|---|
| gyrA | Target modification of Quinolones (DNA gyrase) | Fluoroquinolones |
| gyrB | Target modification of Quinolones (DNA gyrase) | Fluoroquinolones |
| parC | Target modification of Quinolones (DNA topoisomerase IV) | Fluoroquinolones |
| parE | Target modification of Quinolones (DNA topoisomerase IV) | Fluoroquinolones |
| phoQ, cprS, colR, colS, pmrA, pmrB | Modification of Lipopolysaccharide (addition, 4-amino-4-deoxy-L-arabinose moiety to the lipid A portion) | Polymyxins |
| parR | Modification of Lipopolysaccharide (addition, 4-amino-4-deoxy-L-arabinose moiety to the lipid A portion) | Polymyxins |
| Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones | |
| Downregulation of OprD | Imipenem, meropenem | |
| Hyperproduction of efflux-mediated genes (MexXY) | Aminoglycosides, cefepime | |
| parS | Modification of Lipopolysaccharide (addition, 4-amino-4-deoxy-L-arabinose moiety to the lipid A portion) | Polymyxins |
| Downregulation of OprD | Imipenem, meropenem | |
| Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones | |
| Hyperproduction of efflux-mediated genes (MexXY) | Fluoroquinolones | |
| mexR | Hyperproduction of efflux-mediated genes (MexAB-OprM) | Fluoroquinolones |
| nfxB | Hyperproduction of efflux-mediated genes (MexCD-OprJ) | Fluoroquinolones, cefepime |
| mexS | Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones |
| Downregulation of OprD | Imipenem, meropenem | |
| mexT | Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones |
| cmrA, mvaT, PA3271 | Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones |
| mexZ, PA5471.1, amgS | Hyperproduction of efflux-mediated genes (MexXY) | MexXY hyperproduction |
| oprD | Inactivation of Porin channels | Imipenem, meropenem |
| ampD, ampDh2, ampDh3, ampR, dacB, mpl | Hyperproduction of AmpC | Ceftazidime, cefepime, piperacillin–tazobactam |
| ftsI | Target modification (PBP3) | Ceftazidime, cefepime, piperacillin–tazobactam |
| fusA1 | Target modification of Aminoglycoside (elongation factor G) | Aminoglycosides |
| glpT | Transporter protein inactivation GlpT | Fosfomycin |
| rpoB | Rifampin target modification, RNA polymerase β-chain | Rifampin |
| Treatment Options | MDRPA (Multidrug Resistant P. aeruginosa) | DTRPA (Difficult-to-Treat Resistant P. aeruginosa) | XDRPA (Extensively Drug Resistant P. aeruginosa) | CRPA (Carbapenem Resistant P. aeruginosa) | MBL-Producing-CRPA (Metallo-β-lactamase) | P. aeruginosa Resistant to Ceftolozane–tazobactam Combination |
|---|---|---|---|---|---|---|
| COMBINATORY THERAPY | ||||||
| Ceftolozane–tazobactam | √ | √ | √ (High, immediate dose) | √ (1st line drug) | ||
| Ceftazidime–avibactam | √ | √ | √ | |||
| Imipenem–Cilastin–Relebactam | √ | √ | √ | |||
| Meropenem–Vaborbactam | √ | √ | ||||
| Cefepime–Zidebactam | √ | √ | ||||
| Cefepime–Taniborbactam | √ | √ | ||||
| Fosfomycin (along with other combinations) | √ | √ | ||||
| NEWER DRUGS | ||||||
| Cefiderocol | √ | √ | √ | √ | √ | |
| ANTIMICROBIAL PEPTIDES (AMPs) | ||||||
| AMPs–colistin | √ | |||||
| Therapeutic Strategies | Mechanisms | Advantages | Reference |
|---|---|---|---|
| Antibiotic combinations | Combinations with antibiotic to antibiotic or other substances to destroy biofilms and prevent the antibiotic resistance. | combinations provide an excellent solution to the resistance to antimicrobials as it explores the usage of multi-targeted attack, synergism, and reduced dosage and side effects | [174,175] |
| Antimicrobial peptides (AMPs) | Induce membrane disruption, leading to cell lysis and death. AMPs enter into the cells without membrane disruption and inhibiting intracellular function by binding to nucleic acid. | Inhibits the quorum-sensing system rapid killing kinetics, low levels of induced resistance, low toxicity to host | [176,177] |
| Phage therapy | Viral assembly, destroy extracellular matrix by encode enzymes. | Highly specific, replication at infection site without effects commensal flora. Easy administration, easy delivery. | [178,179] |
| SR NO. | Drug Combination | Numder of PA Strains | Synergy | Source | |
|---|---|---|---|---|---|
| 1. | Carbapenems + Aminoglycosides | Imipenem + Amikacin | 27 87 | Low (TK) High (PK/PD) | [196,197] |
| Imipenem + Tobramycin | 3 | Moderate (TK) | [198] | ||
| Meropenem + Amikacin | 63 3 | Moderate (TK) | [130,199] | ||
| 2. | Carbapenems + Fluoroquinolones | Imipenem + Levofloxacin | 5 | Moderate | [200] |
| Meropenem + Ciprofloxacin | 17 | Low | [201] | ||
| 3. | Fluoroquinolones + Polymyxins | Ciprofloxacin + Colistin | 2 17 | No synergy (PD) High | [145,201] |
| 4. | Polymyxins + Carbapenems | Colistin + Doripenem | 3 | Moderate | [202,203] |
| Colistin + Imipenem | 2 | Moderate (TK) | [202] | ||
| Colistin + Meropenem | 7 17 | Moderate Low | [145,204] | ||
| 5. | Cephalosporin + Aminoglycoside | Ceftazidime/avibactam + Amikacin | 3 21 | Low Moderate (TK) | [205] |
| Ceftolozane/tazobactam + Amikacin | 4 20 | No synergy Moderate (TK) | [206,207] | ||
| 6. | Cephalosporin + Polymyxin | Ceftolozane/tazobactam + Colistin | 4 | Moderate | [201,208] |
| Ceftazidime + Colistin | 2 | Moderate (PD) | |||
| 7. | Cephalosporin + Monobactam | Ceftolozane/Tazobactam + Aztreonam | 4 | No synergy | [209] |
| 8. | Polymyxin B + Tetracycline | Polymyxin B + Doxcycline | 3 | Moderate synergy (TK) | [210] |
| Trial Number | Phase/Participants | Study Design | Type of Infection | Test Therapy | Result |
|---|---|---|---|---|---|
| NCT03140085 | P2,3/113 | Cohort (Randomized, Double-blinded, Parallel 3 arm) | Urinary tract infections in patients undergoing trans-urethral resection of the prostate. | Intravesical bacteriophage treatment with PYO phage. | Intravesical bacteriophage therapy was non-inferior to standard-of-care antibiotic treatment, but was not superior to placebo bladder irrigation in treating UTIs in patients undergoing TURP. Bacteriophage safety profile seems to be favorable. |
| NCT04803708 | P I/IIa/20 | Cohort (Randomized, Double-blinded) | Non-infected and infected diabetic foot ulcers with P. aeruginosa, Staphylococcus aureus and/or Acinetobacter baumanni. | Topical bacteriophage cocktail (TP-102). | (Not published) |
| NCT01818206 | -/60 | Interventional | Cystic fibrosis with P. aeruginosa infection. | A cocktail of 10 bacteriophages. | (Not published) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kothari, A.; Kherdekar, R.; Mago, V.; Uniyal, M.; Mamgain, G.; Kalia, R.B.; Kumar, S.; Jain, N.; Pandey, A.; Omar, B.J. Age of Antibiotic Resistance in MDR/XDR Clinical Pathogen of Pseudomonas aeruginosa. Pharmaceuticals 2023, 16, 1230. https://doi.org/10.3390/ph16091230
Kothari A, Kherdekar R, Mago V, Uniyal M, Mamgain G, Kalia RB, Kumar S, Jain N, Pandey A, Omar BJ. Age of Antibiotic Resistance in MDR/XDR Clinical Pathogen of Pseudomonas aeruginosa. Pharmaceuticals. 2023; 16(9):1230. https://doi.org/10.3390/ph16091230
Chicago/Turabian StyleKothari, Ashish, Radhika Kherdekar, Vishal Mago, Madhur Uniyal, Garima Mamgain, Roop Bhushan Kalia, Sandeep Kumar, Neeraj Jain, Atul Pandey, and Balram Ji Omar. 2023. "Age of Antibiotic Resistance in MDR/XDR Clinical Pathogen of Pseudomonas aeruginosa" Pharmaceuticals 16, no. 9: 1230. https://doi.org/10.3390/ph16091230