


Galactoside-Based Molecule Enhanced Antimicrobial Activity through Acyl Moiety Incorporation: Synthesis and In Silico Exploration for Therapeutic Target

 , ,
, ,  ,
,  , , , and
, , , and 
Abstract
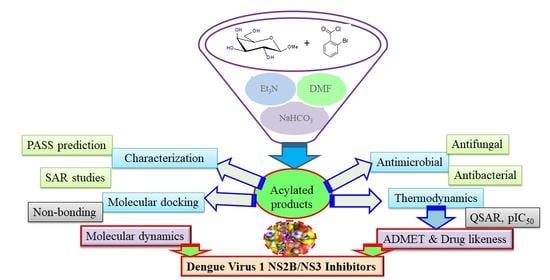
:
1. Introduction
2. Results and Discussion
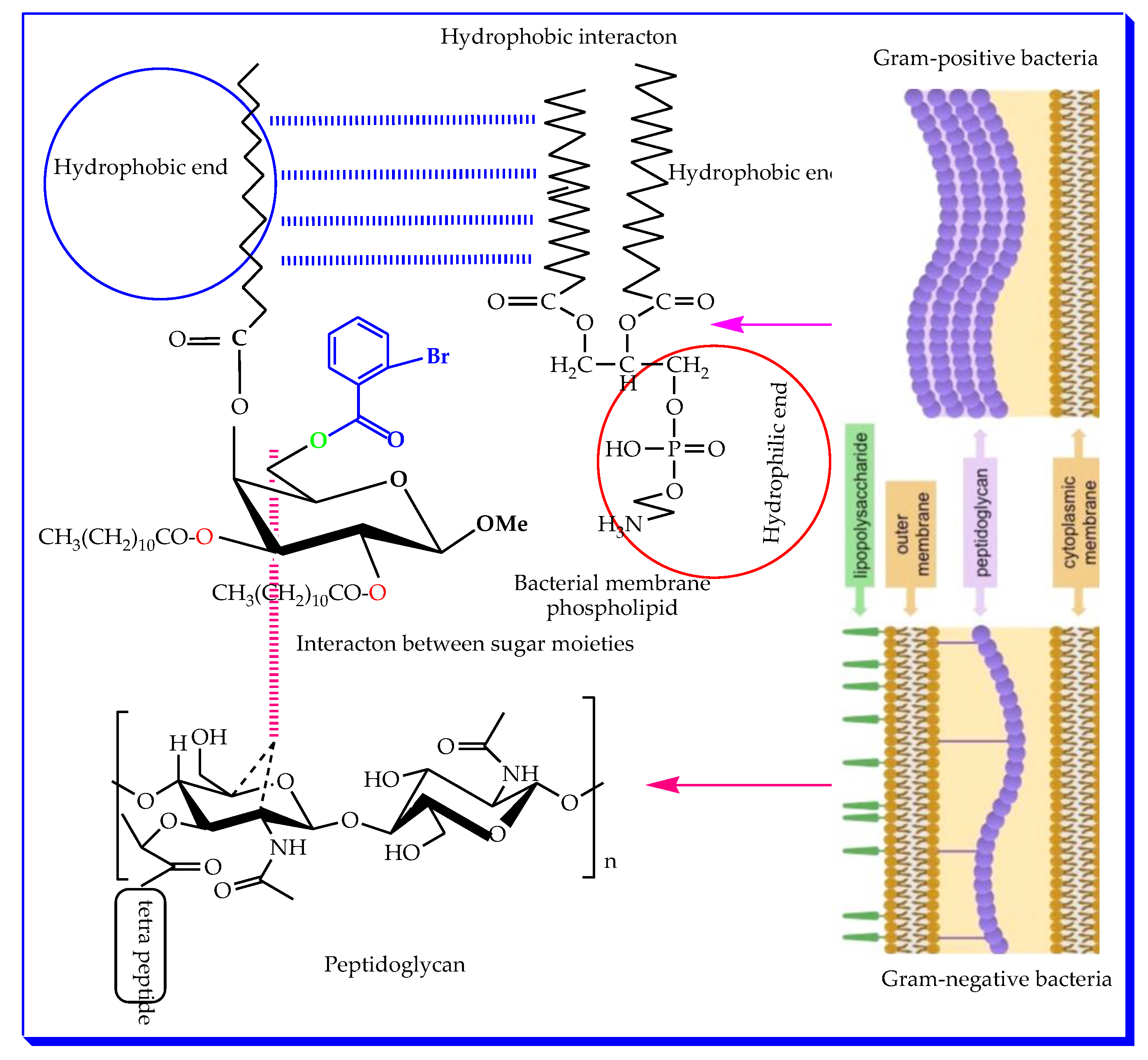
2.1. Chemistry
2.2. Characterization
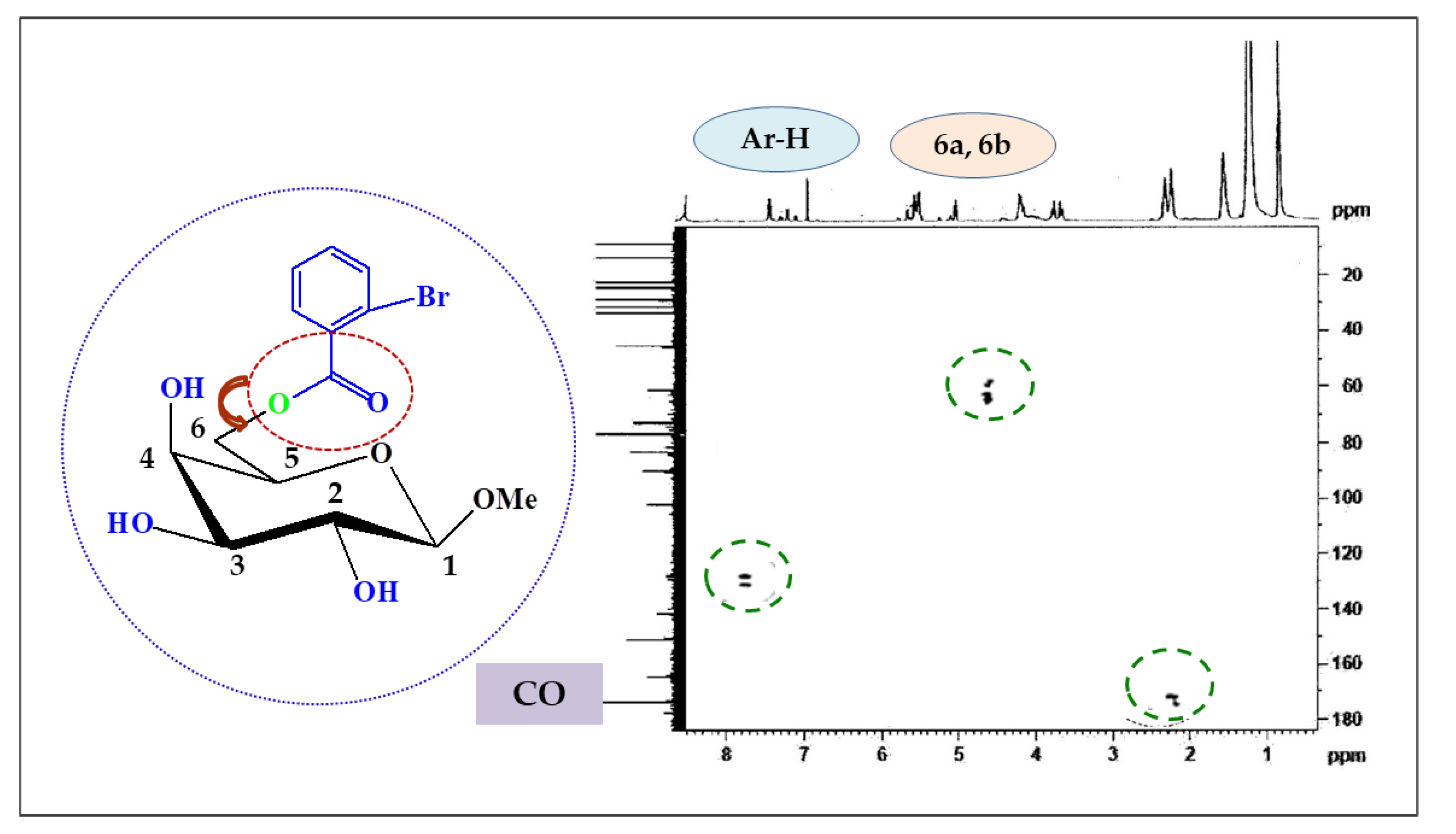
2.3. Two-Dimensional NMR
2.4. Antibacterial Susceptibility
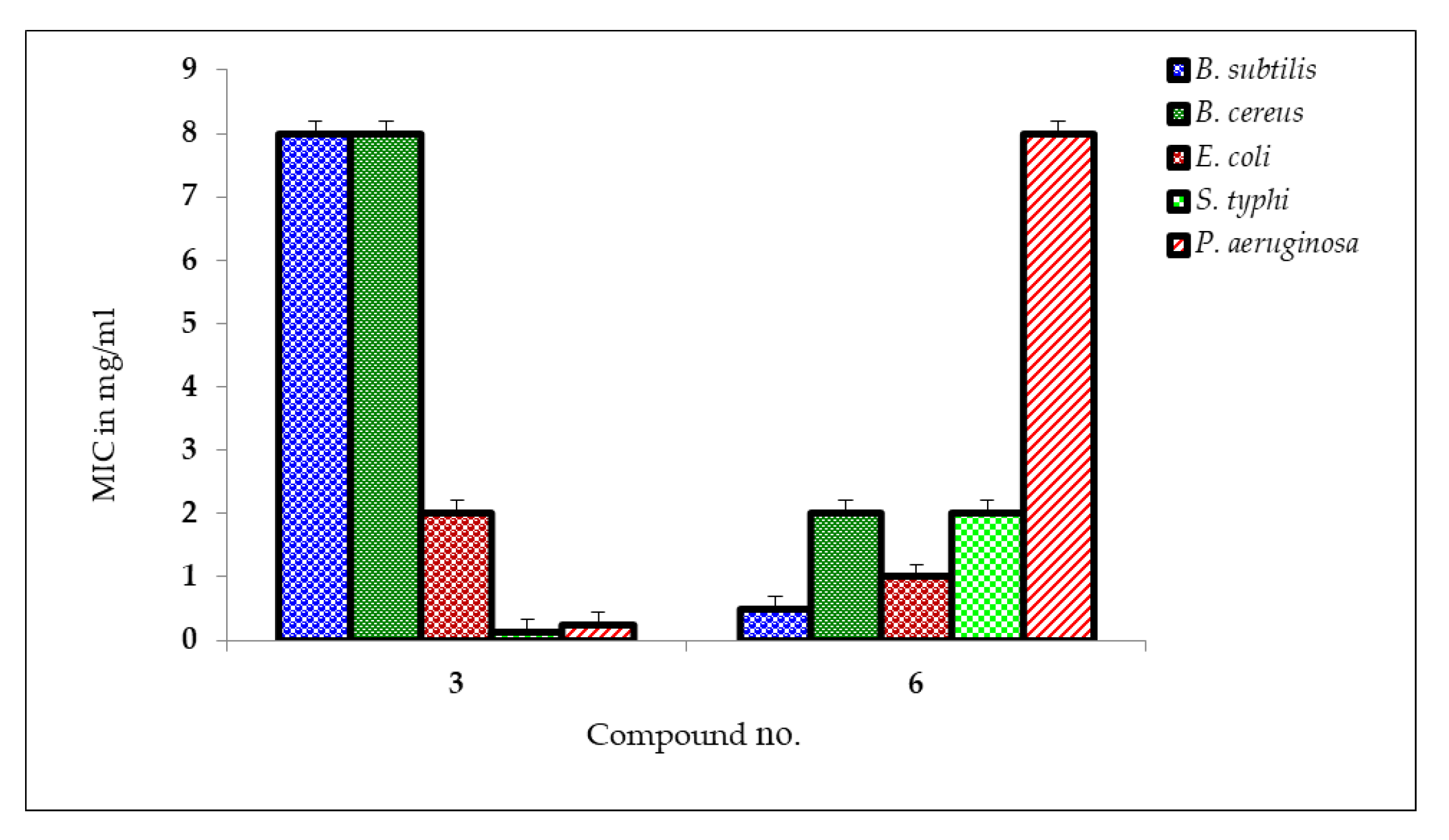
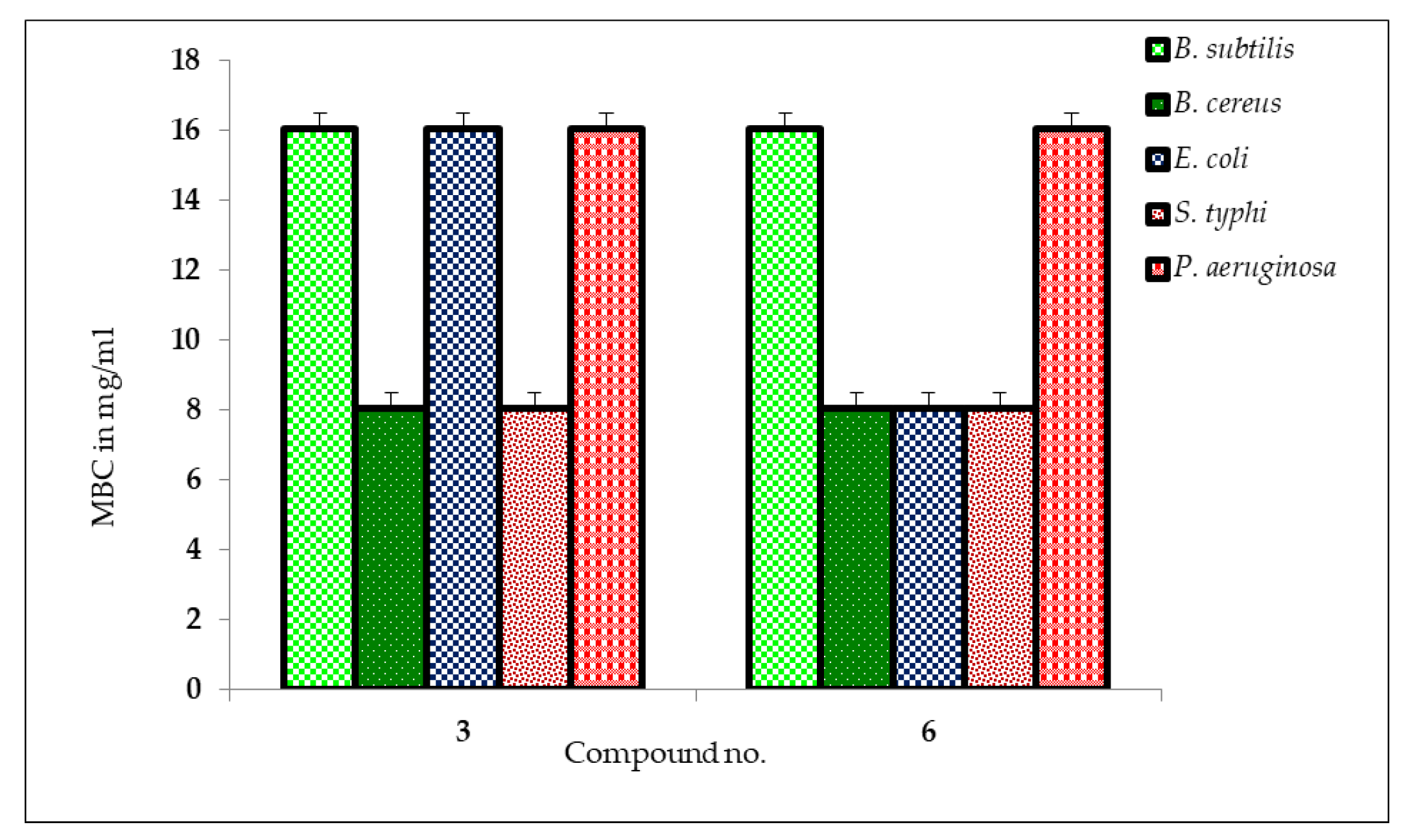
2.5. MIC and MBC Measurement
2.6. Antifungal Potential
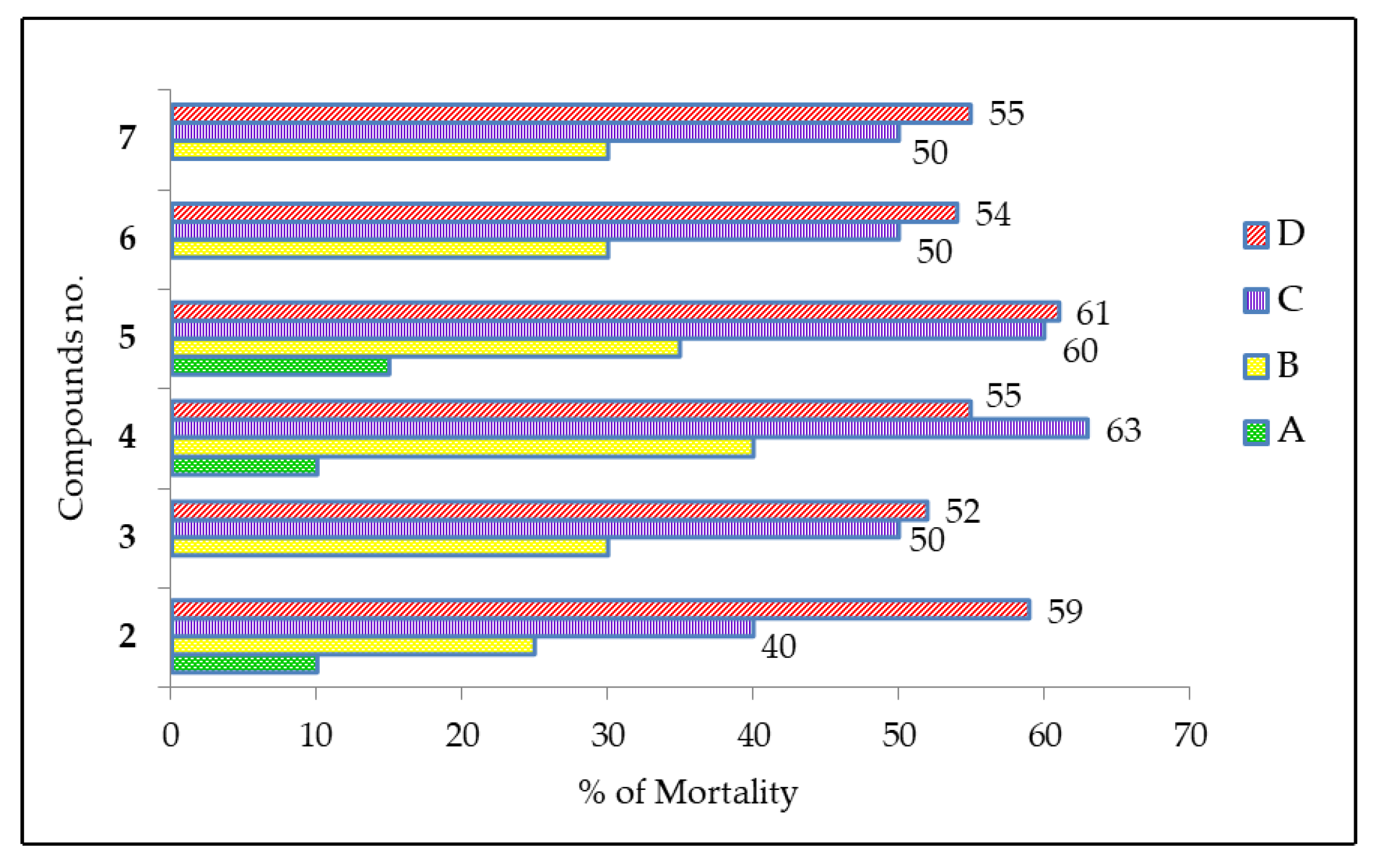
2.7. Cytotoxic Activity of MDGP Compounds
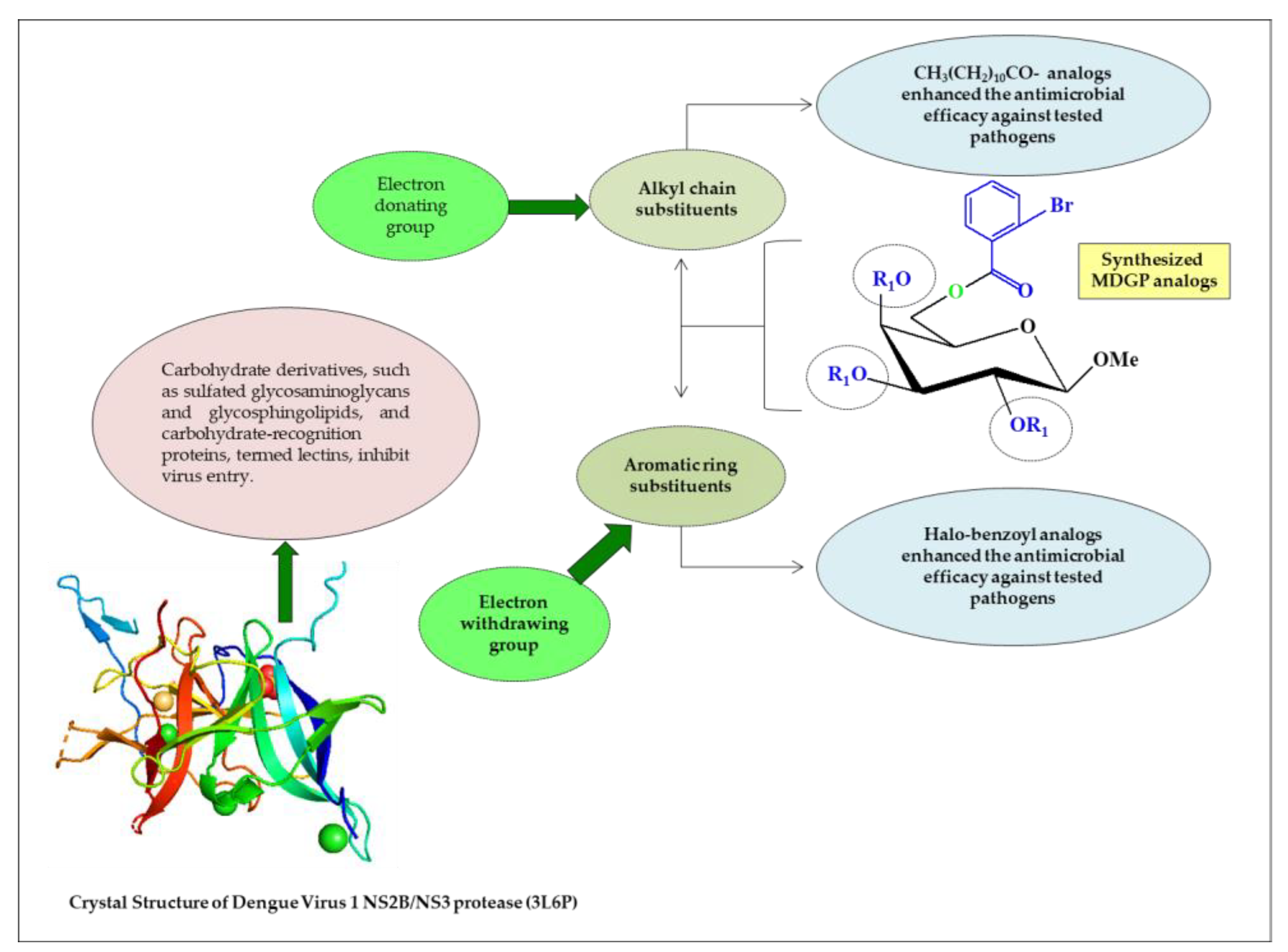
2.8. Structure–Activity Relationship
2.9. In Silico Studies
2.10. Assessment of Antimicrobial Activity by PASS and Bioactivity
2.11. Thermodynamic Analysis
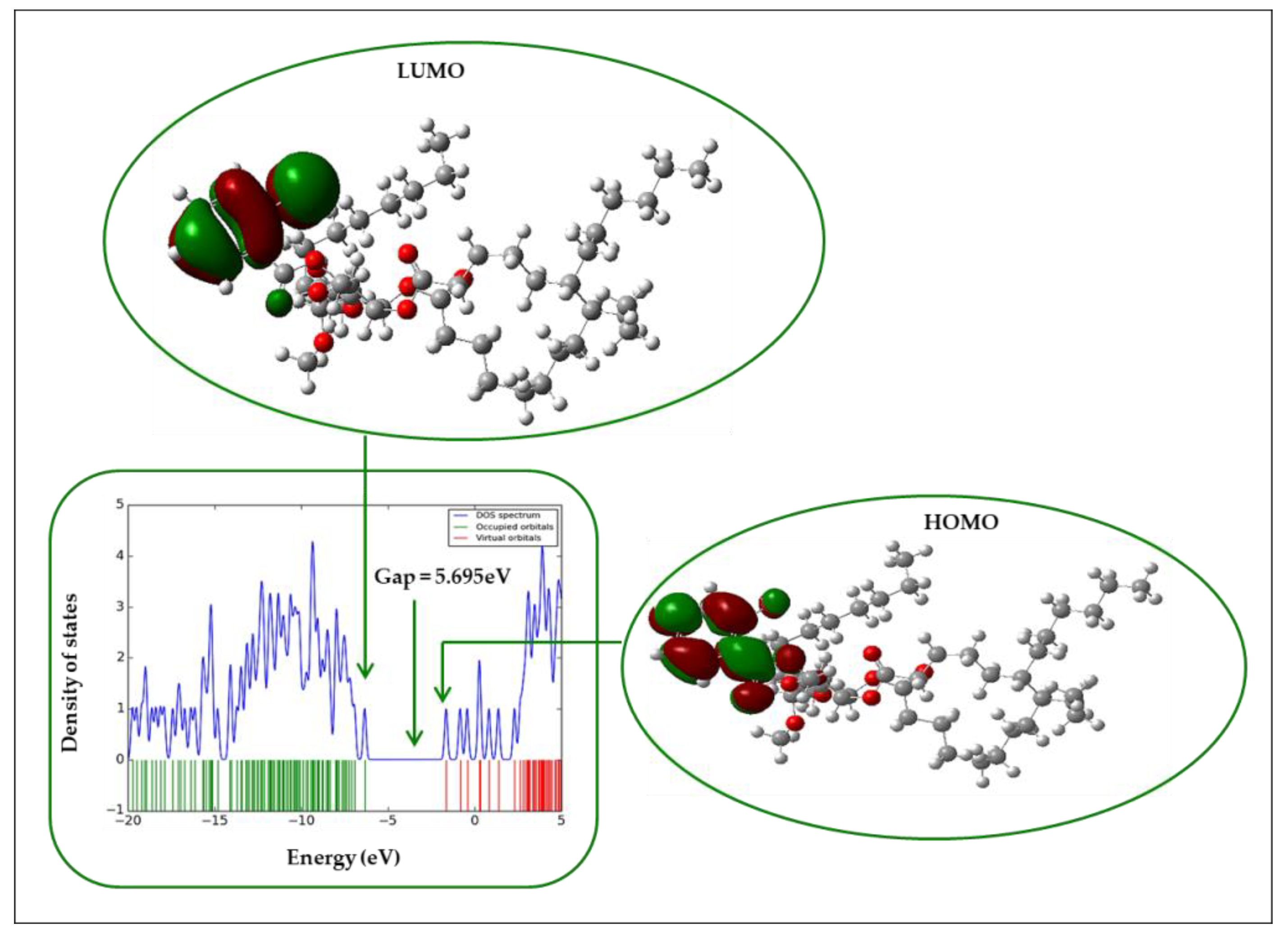
2.12. Frontier Molecular Orbital (FMO)
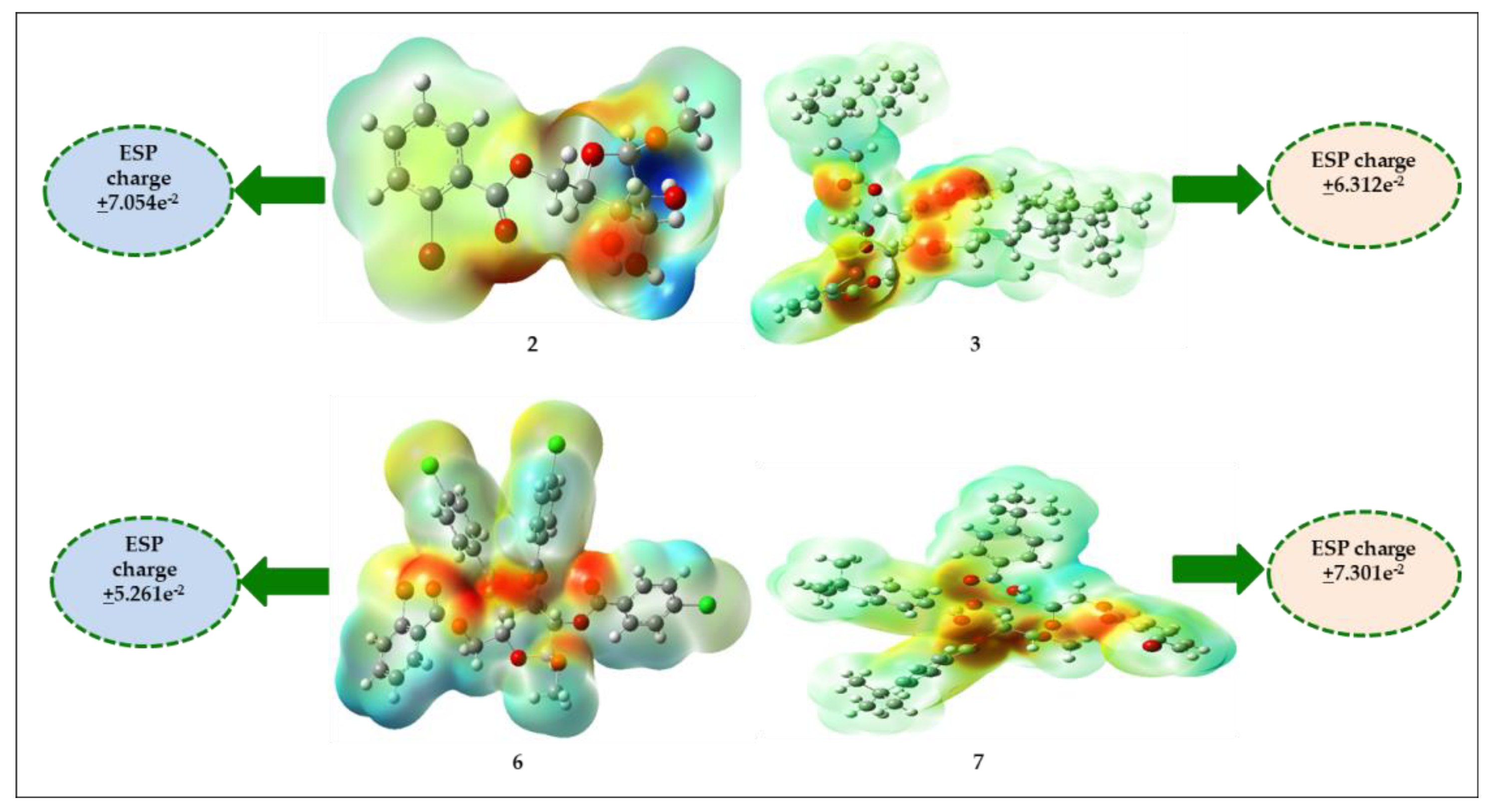
2.13. Molecular Electrostatic Potential (MEP)
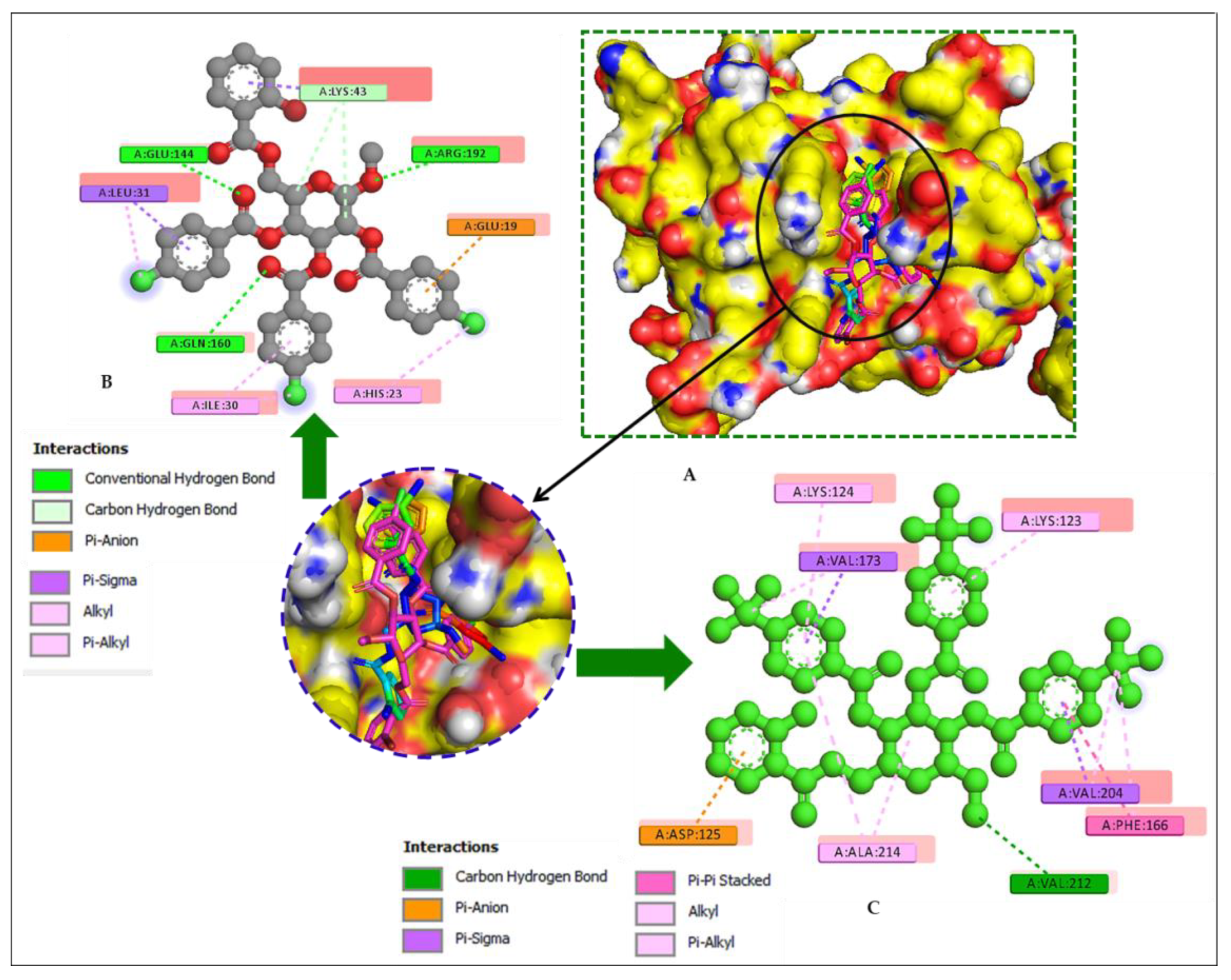
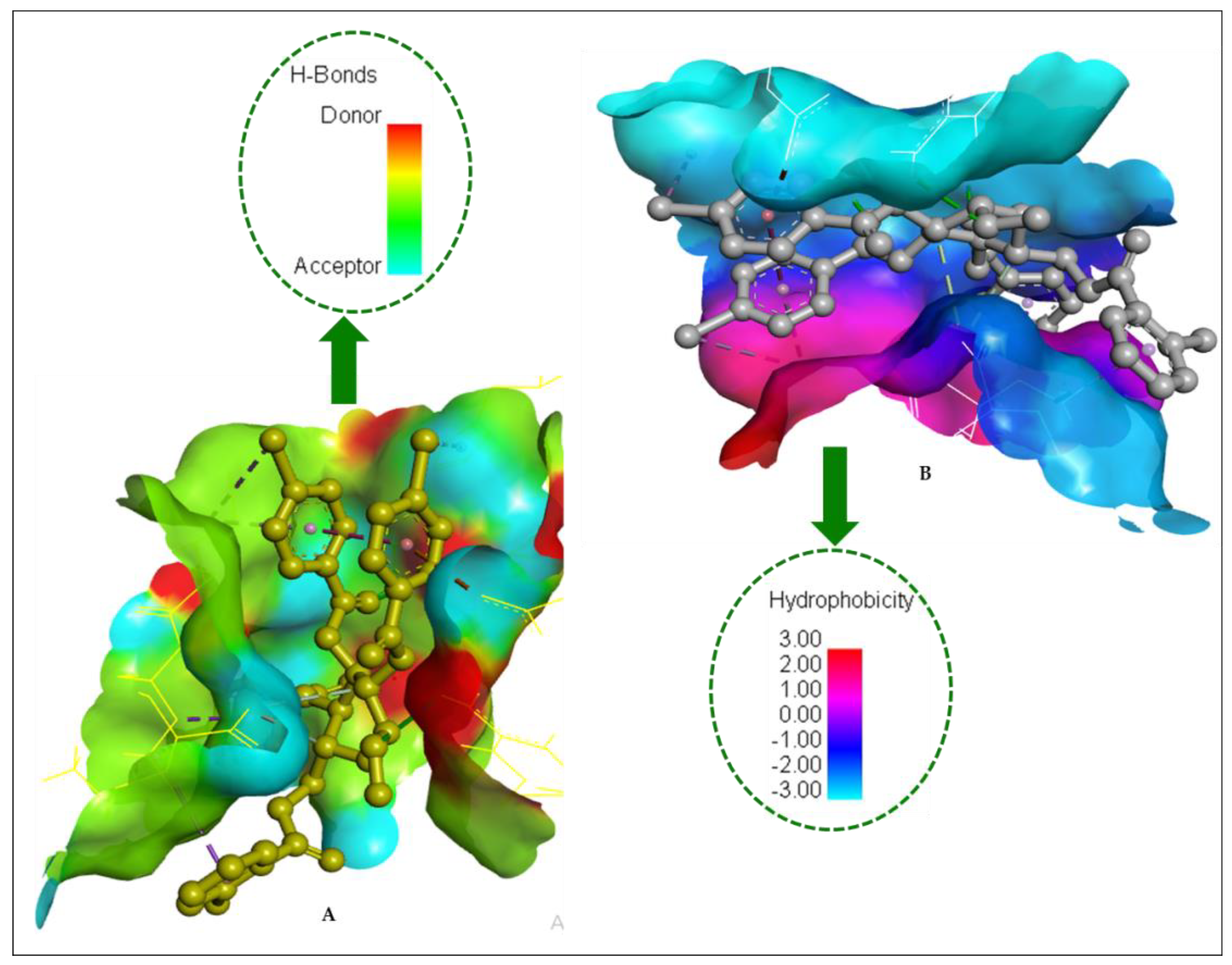
2.14. Molecular Docking
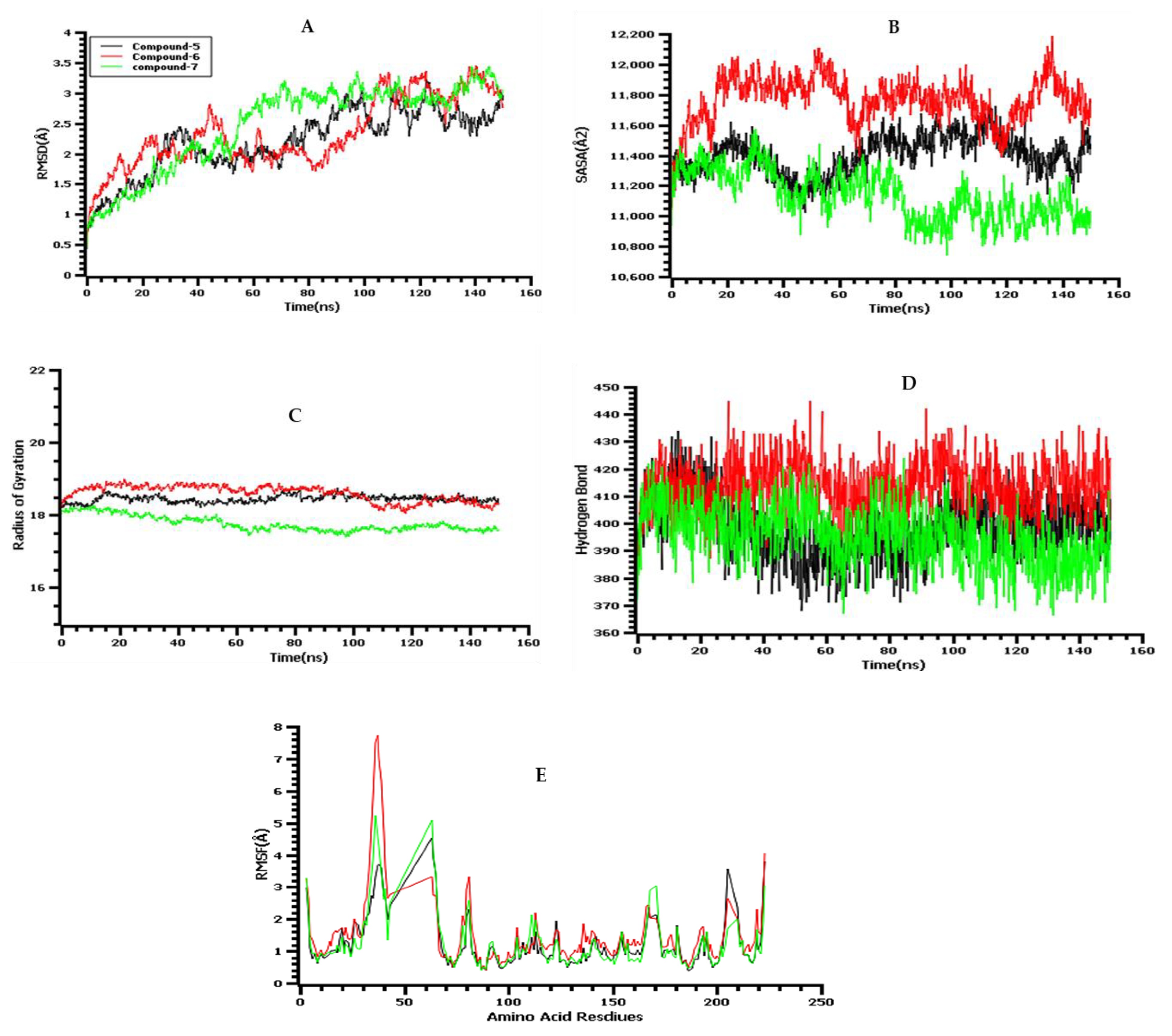
2.15. Molecular-Dynamics (MD) Simulations
2.16. ADMET Profile and Drug Likeness
2.17. Calculation of QSAR and pIC50
3. Materials and Methods
3.1. Reagents and Instrumentation
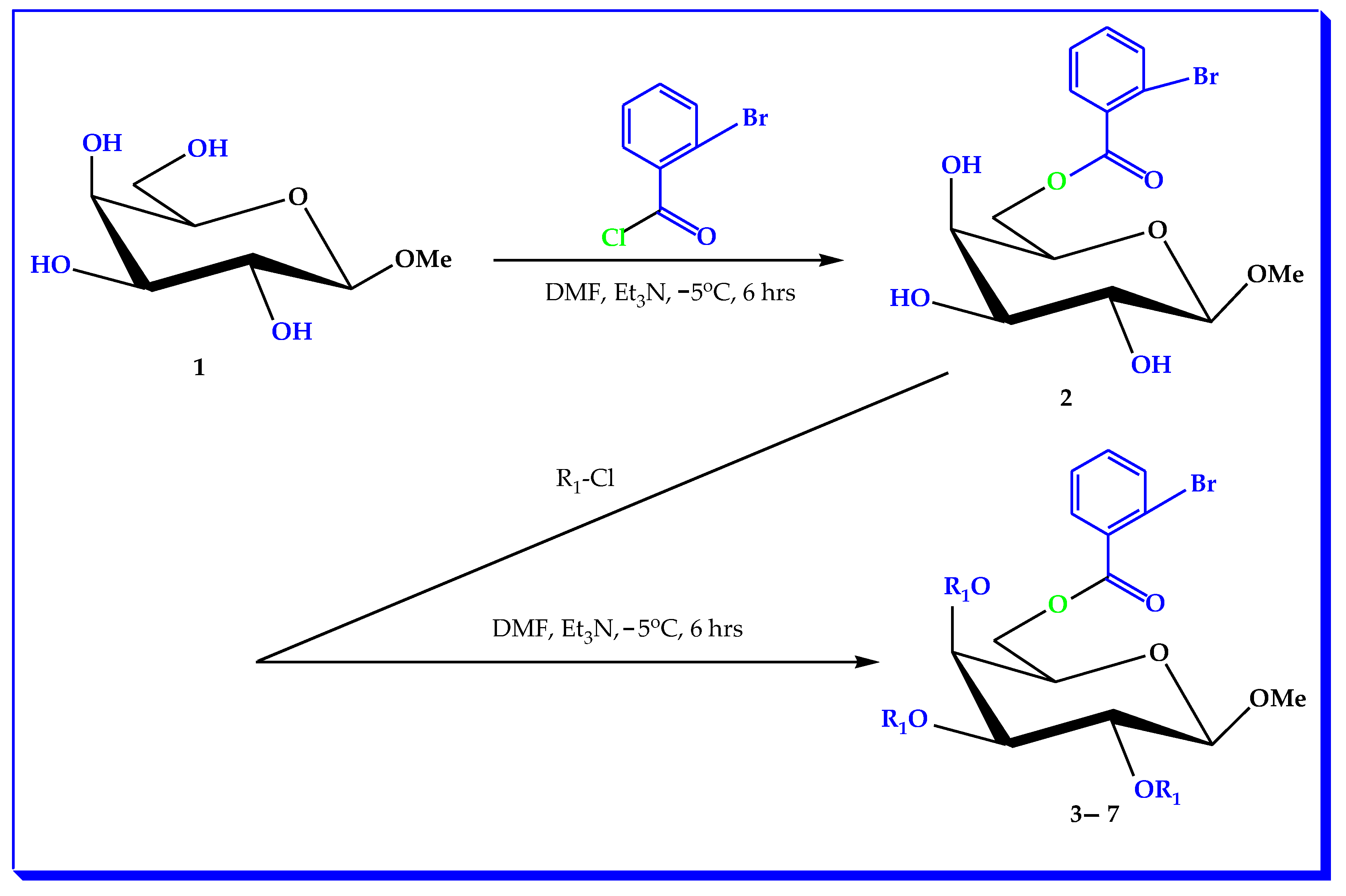
3.2. Synthesis of (MDGP, 1) Analogs
3.3. General Procedure for the Synthesis of (2-Bromobenzoyl) Analogs 3–7
3.4. Microorganisms
3.5. Antibacterial Activity
3.6. Determination of MIC and MBC
3.7. Antifungal Activity
3.8. Cytotoxic Activity Evaluation
3.9. Structure–Activity Relationship (SAR)
3.10. PASS Prediction and Bioactivity
3.11. Geometry DFT Optimization
3.12. Protein Selection and Molecular Docking
3.13. Molecular-Dynamics Simulation
3.14. Pharmacokinetic and Drug-Likeness Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cowan, M.M. Plants Products as Antimicrobial Agents. Clin. Microbiol Rev. 1999, 12, 564–582. [Google Scholar] [CrossRef] [Green Version]
- Coates, A.; Hu, Y.; Bax, R.; Page, C. The Future Challenges Facing the Development of New Antimicrobial Drugs. Nat. Rev. Drug Discov. 2002, 1, 895–910. [Google Scholar] [CrossRef]
- Wright, P.M.; Seiple, I.B.; Myers, A.G. The Evolving Role of Chemical Synthesis in Antibacterial Drug Discovery. Angew. Chem. Int. Edit. 2014, 53, 8840–8869. [Google Scholar] [CrossRef] [Green Version]
- Arshad, M.; Bhat, A.; Athar, F. Heterocyclic Azoles and their Biological Application as Antimicrobials. J. Nat. Sci. Biol. Med. 2011, 2, 131. [Google Scholar]
- Kato, K.; Ishiwa, A. The Role of Carbohydrates in Infection Strategies of Enteric Pathogens. Trop. Med. Health 2015, 43, 41–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, P.; Wong, C.H. Intervention of Carbohydrate Recognition by Proteins and Nucleic Acids. Proc. Natl. Acad. Sci. USA 1996, 93, 12086–12093. [Google Scholar] [CrossRef]
- Seeberger, P.H.; Werz, D.B. Synthesis and Medical Applications of Oligosaccharides. Nature 2007, 446, 1046–1051. [Google Scholar] [CrossRef]
- Chen, S.; Fukuda, M. Cell Type-specific roles of carbohydrates in tumor metastasis. Meth. Enzymol. 2006, 416, 371–380. [Google Scholar]
- Kabir, A.K.M.S.; Kawsar, S.M.A.; Bhuiyan, M.M.R.; Rahman, M.S.; Banu, B. Biological evaluation of some octanoyl derivatives of methyl 4,6-O-cyclohexylidene-α-D-glucopyranoside. Chittagong Univ. J. Biol. Sci. 2008, 3, 53–64. [Google Scholar] [CrossRef]
- Gubler, D.J. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 1998, 11, 480–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monath, T.P. Dengue: The risk to developed and developing countries. Proc. Natl. Acad. Sci. USA 1994, 91, 2395–2400. [Google Scholar] [CrossRef]
- Shagir, A.C.; Bhuiyan, M.M.R.; Ozeki, Y.; Kawsar, S.M.A. Simple and rapid synthesis of some nucleoside derivatives: Structural and spectral characterization. Curr. Chem. Lett. 2016, 5, 83–92. [Google Scholar]
- Meadows, D.C.; Sanchez, T.; Neamati, N.; North, T.W.; Gervay-Hague, J. Ring Substituents Effects on Biological Activity of Vinyl Sulfones as Inhibitors of HIV-1. Bioorg. Med. Chem. 2007, 15, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Bulbul, M.Z.H.; Chowdhury, T.S.; Misbah, M.M.H.; Ferdous, J.; Dey, S.; Hasan, I.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Synthesis of new series of pyrimidine nucleoside derivatives bearing the acyl moieties as potential antimicrobial agents. Pharmacia 2021, 68, 23–34. [Google Scholar] [CrossRef]
- Klekota, J.; Roth, F.P. Chemical Substructures that Enrich for Biological Activity. Bioinformatics 2008, 24, 2518–2525. [Google Scholar] [CrossRef] [Green Version]
- Maowa, J.; Alam, A.; Rana, K.M.; Hosen, A.; Dey, S.; Hasan, I.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Synthesis, characterization, synergistic antimicrobial properties and molecular docking of sugar modified uridine derivatives. Ovidius. Univ. Ann. Chem. 2021, 32, 6–21. [Google Scholar] [CrossRef]
- Kabir, A.K.M.S.; Kawsar, S.M.A.; Bhuiyan, M.M.R.; Islam, M.R.; Rahman, M.S. Biological Evaluation of Some Mannopyranoside Derivatives. Bull. Pure Appl. Sci. 2004, 23, 83–91. [Google Scholar]
- Alam, A.; Hosen, M.A.; Hosen, A.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Synthesis, characterization, and molecular docking against a receptor protein FimH of Escherichia coli (4XO8) of thymidine derivatives. J. Mex. Chem. Soc. 2021, 65, 256–276. [Google Scholar] [CrossRef]
- Islam, M.; Zzaman, A.; Rahman, M.; Rahman, M.A.; Kawsar, S.M.A. Novel methyl 4,6-O-benzylidene-α-D-glucopyranoside derivatives: Synthesis, structural characterization and evaluation of antibacterial activities. Hacettepe J. Biol. Chem. 2019, 47, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Insuasty, D.; Castillo, J.; Becerra, D.; Rojas, H.; Abonia, R. Synthesis of Biologically Active Molecules through Multicomponent Reactions. Molecules 2020, 25, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odds, F.; Brown, A.J.P.; Gow, N.A.R. Antifungal Agents: Mechanism of Action. Trends Microbiol. 2003, 6, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Kawsar, S.M.A.; Hosen, M.A.; Fujii, Y.; Ozeki, Y. Thermochemical, DFT, molecular docking and pharmacokinetic studies of methyl β-D-galactopyranoside esters. J. Comput. Chem. Mol. Model 2020, 4, 452–462. [Google Scholar] [CrossRef]
- Maowa, J.; Hosen, M.A.; Alam, A.; Rana, K.M.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Pharmacokinetics and molecular docking studies of uridine derivatives as SARS- COV-2 Mpro inhibitors. Phys. Chem. Res. 2021, 9, 385–412. [Google Scholar]
- Jumina, J.; Mutmainah, M.; Purwono, B.; Kuniawan, Y.S.; Syah, Y.M. Antibacterial and Antifungal Activity of Three Monosaccharide Monomyristate Derivatives. Molecules 2019, 24, 3692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucarini, S.; Fagioli, L.; Campana, R.; Cole, H.; Duranti, A.; Baffone, W.; Vllasaliu, D.; Casettari, L. Unsaturated fatty acids lactose esters: Cytotoxicity, permeability enhancement and antimicrobial activity. Eur. J. Pharm. Biopharm. 2016, 107, 88–96. [Google Scholar] [CrossRef]
- Kawsar, S.M.A.; Hosen, M.A. An optimization and pharmacokinetic studies of some thymidine derivatives. Turkish Comp. Theo. Chem. 2020, 4, 59–66. [Google Scholar] [CrossRef]
- Spriha, S.E.; Rahman, S.M.A. A Review on Biological Activities of Sugars and Sugar Derivatives. Dhaka Univ. J. Pharm. Sci. 2022, 20, 381–394. [Google Scholar] [CrossRef]
- Wang, G.; Dyatkina, N.; Prhavc, M.; Williams, C.; Serebryany, V.; Hu, Y.; Huang, Y.; Wu, X.; Chen, T.; Huang, W.; et al. Synthesis and anti-HCV activity of sugar-modified guanosine analogues: Discovery of AL-611 as an HCV NS5B polymerase inhibitor for the treatment of chronic hepatitis C. J. Med. Chem. 2020, 63, 10380–10395. [Google Scholar] [CrossRef]
- Frommer, J.; Karg, B.; Weisz, K.; Muller, S. Preparation and Characterization of Pyrene Modified Uridine Derivatives as Potential Electron Donors in RNA. Org. Biomol. Chem. 2018, 16, 7663–7673. [Google Scholar] [CrossRef]
- Staro, J.; Dbrowski, J.M.; Guzik, M. Lactose esters: Synthesis and Biotechnological Applications. Crit. Rev. Biotechnol. 2018, 38, 1–14. [Google Scholar] [CrossRef]
- ALrajhi, M.; Al-Rasheedi, M.; Eltom, S.E.M.; Alhazmi, Y.; Mustafa, M.M.; Ali, A.M. Antibacterial Activity of Date Palm Cake Extracts (Phoenix dactylifera). Cogent Food Agric. 2019, 5, 1625479. [Google Scholar] [CrossRef]
- Alfindee, M.N.; Zhang, Q.; Subedi, Y.P.; Shrestha, J.P.; Kawasaki, Y.; Grilley, M.; Takemoto, J.Y.; Chang, C.T. One Step Synthesis of Carbohydrate Esters as Antibacterial and Antifungal Agent. Bioorg. Med. Chem. 2018, 26, 765–774. [Google Scholar] [CrossRef]
- Mutmainah, J.; Purwono, B. Chemical synthesis of monosaccharide lauric acid esters as antibacterial and antifungal agents. Mater. Sci. Forum. 2019, 948, 63–68. [Google Scholar] [CrossRef]
- LeTourneau, N.; Vimal, P.; Klepacki, D.; Mankin, A.; Melman, A. Synthesis and Antibacterial Activity of Desosamine-Modified Macrolide Derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 4575–4578. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef]
- Mulcahy, L.R.; Isabella, V.M.; Lewis, K. Pseudomonas aeruginosa biofilms in disease. Micro. Ecol. 2014, 68, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Martins, P.M.; Merfa, M.V.; Takita, M.A.; De Souza, A.A. Persistence in phytopathogenic bacteria: Do we know enough? Front. Microbiol. 2018, 9, 1099. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Isono, K. Nucleoside antibiotics: Structure, biological activity, and biosynthesis. J. Antibiot. 1988, 41, 1711–1739. [Google Scholar] [CrossRef]
- Aljeldah, M.M.; Yassin, M.T.; Mostafa, A.A.F.; Aboul-Soud, M.A.M. Synergistic Antibacterial Potential of Greenly Synthesized Silver Nanoparticles with Fosfomycin against some Nosocomial Bacterial Pathogens. Infect. Drug Resist. 2023, 2023, 125–142. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Zheng, X.; Yang, X.; Ma, P.; Cai, Y.; Chen, Y. Design of novel analogues of short antimicrobial peptide anoplin with improved antimicrobial activity. J. Peptide Sci. 2014, 20, 945–951. [Google Scholar] [CrossRef]
- Salehi, P.; Babanezhad-Harikandei, K.; Bararjanian, M.; Al-Harrasi, A.; Esmaeili, M.-A.; Aliahmadi, A. Synthesis of novel 1, 2, 3-triazole tethered 1, 3-disubstituted β-carboline derivatives and their cytotoxic and antibacterial activities. Med. Chem. Res. 2016, 25, 1895–1907. [Google Scholar] [CrossRef]
- Yoon, B.K.; Jackman, J.A.; Valle-Gonzalez, E.R.; Cho, N.J. Antibacterial free fatty acids and monoglycerides: Biological activities, experimental testing, and therapeutic applications. Int. J. Mol. Sci. 2018, 19, 1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawsar, S.M.A.; Matsumoto, R.; Fujii, Y.; Matsuoka, H.; Masuda, N.; Iwahara, C.; Yasumitsu, H.; Kanaly, R.A.; Sugawara, S.; Hosono, M.; et al. Cytotoxicity and Glycan-Binding Profile of α-D-Galactose-Binding Lectin from the Eggs of a Japanese Sea Hare (Aplysia kurodai). Protein J. 2011, 30, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.D.; Han, N.; Perez, Y.; Kirkpatrick, J.; Wang, S.; Sheridan, Y.M.C. Design, synthesis, and nanostructure-dependent antibacterial activity of cationic peptide amphiphiles. ACS Appl. Mat. Interfaces 2018, 11, 2790–2801. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Nobmann, P.; Henehan, G.; Bourke, P.; Dunne, J. Synthesis and Antimicrobial Evaluation of Carbohydrate and Polyhydroxylated Non-Carbohydrate Fatty Acid Ester and Ether Derivatives. Carbohydr. Res. 2008, 343, 2557–2566. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Culebras, P.V.; Gandía, M.; Boronat, A.; Marcos, J.F.; Manzanares, P. Differential susceptibility of mycotoxin-producing fungi to distinct antifungal proteins (AFPs). Food Microbiol. 2021, 97, 103760. [Google Scholar] [CrossRef]
- Perfect, J.R. The Antifungal Pipeline: A Reality Check. Nat. Rev. Drug Discov. 2017, 16, 603–616. [Google Scholar] [CrossRef] [Green Version]
- de Souza, L.S.; Tosta, C.L.; Borlot, J.R.P.O.; Varricchio, M.C.B.N.; Kitagawa, R.R.; Filgueiras, P.R.; Kuster, R.M. Chemical Profile and Cytotoxic Evaluation of Aerial Parts of Euphorbia tirucalli L. on Gastric Adenocarcinoma (AGS Cells). Nat. Prod. Res. 2023, 37, 1–7. [Google Scholar] [CrossRef]
- Saleh, S.S.; Salihi, S.S.A.; Mohammed, I.A. Biological activity Study for some heterocyclic compounds and their impact on the gram positive and negative bacteria. Energy Procedia 2019, 157, 296–306. [Google Scholar] [CrossRef]
- Li, W.R.; Xie, X.B.; Shi, Q.S.; Zeng, H.Y.; Ou-Yang, Y.S.; Chen, Y.B. Antibacterial activity and mechanism of silver nanoparticles on Escherichia coli. Appl. Microbiol. Biotechnol. 2010, 85, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Judge, V.; Narasimhan, B.; Ahuja, M.; Sriram, D.; Yogeeswari, P.; Clercq, E.D.; Pannecouque, C.; Balzarini, J. Synthesis, antimycobacterial, antiviral, antimicrobial activities, and QSAR studies of isonicotinic acid-1-(substituted phenyl)-ethylidene/cycloheptylidene hydrazides. Med. Chem. Res. 2012, 21, 1935–1952. [Google Scholar] [CrossRef]
- Cohen, N.; Benson, S.W. Estimation of heats of formation of organic compounds by additivity methods. Chem. Rev. 1993, 93, 2419–2438. [Google Scholar] [CrossRef]
- Lien, E.J.; Guo, Z.R.; Li, R.L.; Su, C.T. Use of dipole moment as a parameter in drug-receptor interaction and quantitative structure-activity relationship studies. J. Pharm. Sci. 1982, 71, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, S.; Balachandran, V. Quantum chemical studies, natural bond orbital analysis and thermodynamic function of 2,5-di-chlorophenylisocyanate. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 120, 351–364. [Google Scholar] [CrossRef]
- Amin, M.L. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insight 2013, 7, 27–34. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Molecular electrostatic potentials and chemical reactivity. Rev. Comput. Chem. 1991, 2, 273–312. [Google Scholar]
- Liu, X.; Wang, X.J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genom. 2020, 7, 119–121. [Google Scholar] [CrossRef]
- Kawsar, S.M.A.; Mamun, S.M.A.; Rahman, M.S.; Yasumitsu, H.; Ozeki, Y. In Vitro Antibacterial and Antifungal Effects of a 30 kDa D-Galactoside-Specific Lectin from the Demosponge, Halichondria okadai. Int. J. Biol. Life Sci. 2011, 6, 31–37. [Google Scholar]
- Hirata, K.; Uchida, T.; Nakajima, Y.; Maekawa, T.; Mizuki, T. Chemical Synthesis and Cytotoxicity of Neo-Glycolipids; Rare Sugar-Glycerol-Lipid Compounds. Heliyon 2018, 4, e00861. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, B.D. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.B.D.; Gaudio, A.C. BuildQSAR: A new computer program for QSAR analysis. Mol. Inform. 2001, 19, 599–601. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, J.; Zhao, Y. High Intensity Ultrasound Assisted Heating to Improve Solubility, Antioxidant and Antibacterial Properties of Chitosan-Fructose Maillard Reaction Products. LWT Food Sci. Technol. 2015, 60, 253–262. [Google Scholar] [CrossRef]
- Jumina, N.A.; Fitria, A.; Pranowo, D.; Sholikhah, E.N.; Kurniawan, Y.S.; Kuswandi, B. Monomyristin and Monopalmitin Derivatives: Synthesis and Evaluation as Potential Antibacterial and Antifungal Agents. Molecules 2018, 23, 3141. [Google Scholar] [CrossRef] [Green Version]
- Grover, R.K.; Moore, J.D. In-vitro efficacy of certain essential oils and plant extracts against three major pathogens of Jatropha curcas L. Phytopathology 1962, 52, 876–879. [Google Scholar]
- McLaughlin, J.L. Crown-Gall Tumors in Potato Discs and Brine Shrimp Lethality: Two Simple Bioassays for Higher Plant Screening and Fractionation. In Methods in Plant Biochemistry: Assays for Bioactivity; Hostettmann, K., Ed.; Academic Press: Cambridge, MA, USA, 1991; Volume 6, pp. 1–32. [Google Scholar]
- Hunt, W.A. The effects of aliphatic alcohols on the biophysical and biochemical correlates of membrane function. Adv. Exp. Med. Biol. 1975, 56, 195–210. [Google Scholar]
- Kim, Y.M.; Farrah, S.; Baney, R.H. Structure–antimicrobial activity relationship for silanols, a new class of disinfectants, compared with alcohols and phenols. Int. J. Antimicrob. Agents 2007, 29, 217–222. [Google Scholar] [CrossRef]
- Kumaresan, S.; Senthilkumar, V.; Stephen, A.; Balakumar, B.S. GC-MS analysis and pass-assisted prediction of biological activity spectra of extract of Phomopsis sp. isolated from Andrographis paniculata. World J. Pharm. Res. 2015, 4, 1035–1053. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, M.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, R.G. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Nat. Acad. Sci. USA 1986, 83, 8440–8441. [Google Scholar] [CrossRef] [PubMed]
- Ouassaf, M.; Belaidi, S.; Khamouli, S.; Belaidi, H.; Chtita, S. Combined 3D-QSAR and Molecular Docking Analysis of Thienopyrimidine Derivatives as Staphylococcus aureus Inhibitors. Acta. Chim. Slov. 2021, 68, 289–303. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delano, W.I. The PyMOL Molecular Graphics System; De-Lano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Aouidatea, A.; Ghaleba, A.; Ghamalia, M.; Chtitaa, S.; Choukrada, M.; Sbaia, A.; Bouachrineb, M.; Lakhlifi, M. Combined 3D-QSAR and Molecular Docking Study on 7,8-dialkyl-1,3-diaminopyrrolo-[3,2-f] Quinazoline Series Compounds to understand the binding mechanism of DHFR Inhibitors. J. Mol. Strutc. 2017, 1139, 319–327. [Google Scholar] [CrossRef]
- Belhassana, A.; Chtita, S.; Zaki, H.; Alaqarbehd, M.; Alsakhene, N.; Almohtasebf, F.; Lakhlifia, T.; Bouachrinea, M. In silico detection of potential inhibitors from vitamins and their derivatives compounds against SARS-CoV-2 main protease by using molecular docking, molecular dynamic simulation and ADMET profiling. J. Mol. Strutc. 2022, 1258, 132652. [Google Scholar] [CrossRef]
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. Methods Mol. Biol. 2018, 1685, 43–67. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Ouassaf, M.; Belaidi, S.; Al Mogren, M.M.; Chtita, S.; Khan, S.U.; Htar, T.T. Combined docking methods and molecular dynamics to identify effective antiviral 2, 5-diaminobenzophenonederivatives against SARS-CoV-2. J. King Saud Univ. Sci. 2021, 33, 101352. [Google Scholar] [CrossRef] [PubMed]
- Ouassaf, M.; Belaidi, S.; Chtita, S.; Lanez, T.; Qais, F.A.; Amiruddin, H.M. Combined molecular docking and dynamics simulations studies of natural compounds as potent inhibitors against SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2022, 40, 11264–11273. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Chemical Structure | Mol. Formula |
|---|---|---|
| 2 |  | C14H17O7Br |
| 3 |  | C50H83O10Br |
| 4 |  | C56H95O10Br |
| 5 |  | C35H26O10Br.Cl |
| 6 |  | C35H26O10Br.Cl |
| 7 |  | C47H53O10Br |
| Position | δH (ppm) (J Hz) | (HSQC) δC (ppm) | HMBC |
|---|---|---|---|
| Ar-H | 7.81 (d, J = 7.6) | 136.3 | H : Ar |
| Ar-H | 7.63 (d, J = 7.4) | 132.4 | H : Ar |
| H-1 | 5.10 (d, J = 8.1) | 104.1 | H : 2, OCH3 |
| H-6a | 4.52 (dd, J = 11.0 and 6.4) | 63.1 | H : 5, CO |
| H-6b | 4.50 (dd, J = 11.0 and 6.6) | 63.0 | H : 5, CO |
| H-4 | 4.19 (d, J = 3.6) | 77.0 | H : 3, 5 |
| H-3 | 4.00 (dd, J = 3.1 and 10.2) | 75.2 | H : 2, 4 |
| H-2 | 3.87 (dd, J = 8.1 and 10.3) | 77.2 | H : 1, 3 |
| H-5 | 3.76 (m) | 69.1 | H : 4, 6a, 6b |
| 1-OCH3 | 3.16 (s) | 57.0 | H : 1 |
| 2-Br.C6H4CO- | 179.0 | H : 6a, 6b |
| Entry | Solvent and Rf | FTIR (KBr, νmax) cm−1 | LC–MS [M + 1]+ | mp. (°C) | Yield (%) | Found (Calculated) | |
|---|---|---|---|---|---|---|---|
| %C | %H | ||||||
| 2 | CH3OH-CHCl3 (1:6) (Rf = 0.51) | 1724 (C=O), 3404~3507 cm−1 (br) (-OH) | 378.14 | 101–102 | 92 | 44.56 (44.55) | 4.56 (4.54) |
| 3 | CH3OH-CHCl3 (1:7) (Rf = 0.52) | 1715 (C=O) | 924.93 | 107–108 | 90 | 64.93 (64.94) | 9.08 (9.06) |
| 4 | CH3OH-CHCl3 (1:6) (Rf = 0.53) | 1700 (C=O) | 1009.08 | 154–155 | 70 | 66.67 (66.66) | 9.52 (9.50) |
| 5 | CH3OH-CHCl3 (1:5) (Rf = 0.51) | 1707 (-CO) | 793.65 | 136–137 | 77 | 52.98 (52.99) | 3.32 (3.31) |
| 6 | CH3OH-CHCl3 (1:5) (Rf = 0.54) | 1711 (-CO) | 793.65 | 184–185 | 77 | 52.98 (52.99) | 3.33 (3.31) |
| 7 | CH3OH-CHCl3 (1:6) (Rf = 0.50) | 1718 (-CO) | 858.66 | 122–123 | 53 | 65.77 (65.76) | 6.22 (6.23) |
| Diameter of Inhibition Zones (In mm) | |||||
|---|---|---|---|---|---|
| Entry | B. subtilis (G + ve) (ATCC 6633) | S. aureus (G + ve) (BTCC 19) | E. coli (G − ve) (ATCC 8739) | S. typhi (G − ve) (AE 14612) | P. aeruginosa (G − ve) (ATCC 9027) |
| 1 | NI | NI | NI | NI | NI |
| 2 | NI | 10.50 ± 0.2 | NI | 11.25 ± 0.2 | * 15.00 ± 0.1 |
| 3 | * 13.00 ± 0.1 | * 15.50 ± 0.3 | 11.00 ± 0.2 | * 12.25 ± 0.1 | 11.25 ± 0.3 |
| 4 | NI | NI | NI | NI | NI |
| 5 | NI | NI | NI | NI | * 18.25 ± 0.3 |
| 6 | * 12.75 ± 0.1 | 11.50 ± 0.3 | 11.25 ± 0.2 | * 13.00 ± 0.3 | 11.50 ± 0.1 |
| 7 | 7.75 ± 0.1 | NI | 8.50 ± 0.1 | 9.00 ± 0.3 | 7.50 ± 0.3 |
| Azithromycin | ** 18.25 ± 0.2 | ** 17.50 ± 0.2 | ** 17.25 ± 0.2 | ** 18.0 ± 0.1 | ** 18.5 ± 0.3 |
| Entry | % Inhibition of Fungal Mycelial Growth in mm | |
|---|---|---|
| Aspergillus niger (ATCC 16404) | Aspergillus flavus (ATCC 204304) | |
| 1 | NI | NI |
| 2 | * 72.88 ± 1.0 | * 85.66 ± 0.9 |
| 3 | * 64.83 ± 1.1 | * 84.02 ± 1.3 |
| 4 | * 71.19 ± 1.0 | NI |
| 5 | 48.73 ± 1.1 | * 81.97 ± 1.0 |
| 6 | * 78.81 ± 1.0 | * 81.97 ± 1.2 |
| 7 | 46.61 ± 0.7 | NI |
| Nystatin | ** 66.7 ± 1.1 | ** 65.2 ± 1.2 |
| Diameter of Inhibition Zone In mm | ||||||
|---|---|---|---|---|---|---|
| Entry | Antiviral | Antibacterial | Antifungal | |||
| Pa | Pi | Pa | Pi | Pa | Pi | |
| 1 | 0.511 | 0.021 | 0.414 | 0.036 | 0.374 | 0.091 |
| 2 | 0.607 | 0.011 | 0.502 | 0.024 | 0.440 | 0.032 |
| 3 | 0.411 | 0.107 | 0.487 | 0.022 | 0.473 | 0.024 |
| 4 | 0.393 | 0.131 | 0.487 | 0.022 | 0.473 | 0.024 |
| 5 | 0.571 | 0.045 | 0.533 | 0.024 | 0.504 | 0.048 |
| 6 | 0.554 | 0.129 | 0.539 | 0.062 | 0.567 | 0.048 |
| 7 | 0.548 | 0.066 | 0.551 | 0.039 | 0.536 | 0.21 |
| Entry | GPCR Ligand | Ion Channel Modulator | Kinase Inhibitor | Nuclear Receptor Ligand | Protease Inhibitor | Enzyme Inhibitor |
|---|---|---|---|---|---|---|
| 1 | −0.13 | −0.24 | −0.17 | −1.19 | −0.71 | 0.88 |
| 2 | 0.04 | 0.23 | 0.09 | 0.47 | 0.21 | 0.55 |
| 3 | 0.19 | −0.46 | −0.23 | −0.58 | −0.23 | 0.11 |
| 4 | −0.21 | −0.91 | −0.49 | −0.77 | −0.19 | −0.06 |
| 5 | −0.05 | −1.09 | 0.63 | −1.11 | 0.27 | 0.24 |
| 6 | 0.27 | 0.42 | 0.14 | 0.51 | 0.27 | 0.24 |
| 7 | 0.36 | 0.40 | −0.11 | 0.34 | 0.26 | 0.23 |
| Entry | HOMO | LUMO | Gap (∆Ԑ) | η | S | µ | χ | ω |
|---|---|---|---|---|---|---|---|---|
| 1 | −6.021 | −0.391 | 5.630 | 2.815 | 0.355 | 3.206 | −3.206 | 2.317 |
| 2 | −6.100 | −0.863 | 5.237 | 2.618 | 0.381 | 3.481 | −3.481 | 2.314 |
| 3 | −6.233 | −0.925 | 5.308 | 2.654 | 0.376 | 3.579 | −3.579 | 2.413 |
| 4 | −6.589 | −0.897 | 5.692 | 2.846 | 0.351 | 3.743 | −3.743 | 2.461 |
| 5 | −5.148 | −0.203 | 4.945 | 2.472 | 0.404 | 2.675 | −2.675 | 1.447 |
| 6 | −6.236 | −0.758 | 5.478 | 2.739 | 0.365 | 3.497 | −3.497 | 2.232 |
| 7 | −5.850 | −0.726 | 5.124 | 2.562 | 0.390 | 3.288 | −3.288 | 2.019 |
| Entry | 3L6P (kcal/mol) |
|---|---|
| 1 | −5.5 |
| 2 | −6.6 |
| 3 | −6.1 |
| 4 | −5.1 |
| 5 | −8.1 |
| 6 | −8.0 |
| 7 | −8.3 |
| Entry | Bond Category | Residues in Contact | Interaction Type | Distance (Å) |
|---|---|---|---|---|
| 1 | H | Gly201 | CH | 2.4942 |
| H | Phe180 | CH | 2.4973 | |
| H | Gly183 | CH | 2.0541 | |
| H | Thr184 | CH | 2.5129 | |
| H | Tyr211 | CH | 1.9715 | |
| H | Lys181 | C | 3.6387 | |
| 2 | H | Gln217 | CH | 2.0536 |
| H | Gln217 | CH | 2.1626 | |
| H | Gln217 | CH | 2.3156 | |
| H | Lys124 | C | 3.5204 | |
| H | Thr168 | C | 3.5964 | |
| Hydrophobic | Ile215 | PA | 5.2676 | |
| 3 | H | Lys123 | CH | 2.1638 |
| H | Gly171 | C | 3.4559 | |
| Hydrophobic | Val204 | A | 5.0692 | |
| Hydrophobic | Ala141 | A | 3.8337 | |
| Hydrophobic | Lys123 | PA | 4.0691 | |
| Hydrophobic | Phe166 | PA | 4.5004 | |
| 4 | H | Gly171 | C | 3.73352 |
| Hydrophobic | Lys219 | A | 3.7839 | |
| Hydrophobic | Val173 | A | 5.4094 | |
| Hydrophobic | Lys124 | A | 4.8909 | |
| 5 | H | Glu20 | CH | 2.6152 |
| H | Leu31 | CH | 2.1156 | |
| H | Gln160 | CH | 2.0824 | |
| H | Arg192 | CH | 1.9956 | |
| H | Arg192 | CH | 2.5099 | |
| H | Arg192 | CH | 2.0967 | |
| Electrostatic | Glu19 | PAn | 4.1395 | |
| Hydrophobic | Trp17 | PPT | 5.2745 | |
| Hydrophobic | Arg192 | A | 3.9082 | |
| Hydrophobic | Leu31 | A | 4.4797 | |
| Hydrophobic | Arg192 | PA | 4.0916 | |
| Hydrophobic | Leu31 | PA | 4.5535 | |
| 6 | H | Glu144 | CH | 2.1561 |
| H | Gln160 | CH | 2.9897 | |
| H | Gln160 | CH | 2.9917 | |
| H | Arg192 | CH | 2.7495 | |
| H | Lys43 | C | 3.7800 | |
| H | Lys43 | C | 3.6630 | |
| Electrostatic | Glu19 | PAn | 3.6453 | |
| Hydrophobic | Leu31 | PS | 3.4595 | |
| Hydrophobic | Lys43 | PS | 3.7162 | |
| Hydrophobic | Leu31 | A | 4.2217 | |
| Hydrophobic | Ile30 | A | 4.7386 | |
| Hydrophobic | His23 | PA | 4.2129 | |
| Hydrophobic | Ile30 | PA | 3.8409 | |
| 7 | H | Val212 | C | 3.2112 |
| Electrostatic | Asp125 | PAn | 4.3311 | |
| Hydrophobic | Val173 | PS | 3.9352 | |
| Hydrophobic | Val204 | PS | 3.8277 | |
| Hydrophobic | Phe166 | PPS | 4.8680 | |
| Hydrophobic | Ala214 | A | 4.3635 | |
| Hydrophobic | Val173 | A | 5.0337 | |
| Hydrophobic | Val204 | A | 5.0532 | |
| Hydrophobic | Phe166 | PA | 5.0407 | |
| Hydrophobic | Lys124 | PA | 4.7050 | |
| Hydrophobic | Ala214 | PA | 5.1764 | |
| Hydrophobic | Lys123 | PA | 5.3628 |
| Entry | Water Solubility (log mol/L) | Caco-2 Permeability | Intestinal Absorption | Skin Permeability |
|---|---|---|---|---|
| 1 | −3.01 | −0.658 | 59.069 | −3.118 |
| 2 | −4.450 | 0.360 | 77.541 | −2.032 |
| 3 | −4.759 | 0.407 | 81.201 | −2.237 |
| 4 | −5.151 | 0.593 | 86.001 | −2.370 |
| 5 | −5.257 | 0.421 | 91.379 | −2.561 |
| 6 | −5.369 | 0.664 | 93.907 | −2.791 |
| 7 | −5.857 | 0.487 | 96.325 | −2.811 |
| Entry | Distribution | Execration | |||
|---|---|---|---|---|---|
| Vdss | BBB Permeability | CNS Permeability | Total Clearance | Renal OCT2 Substrate | |
| 1 | −0.204 | −0.877 | −4.087 | 0.321 | No |
| 2 | −0.315 | −1.114 | −3.324 | 1.874 | No |
| 3 | −0.473 | −1.137 | −3.344 | 1.850 | No |
| 4 | 0.223 | −1.265 | −3.231 | 1.379 | No |
| 5 | −0.417 | −1.301 | −3.122 | 1.198 | No |
| 6 | −0.079 | −1.210 | −3.036 | 0.057 | No |
| 7 | −0.055 | −1.354 | −3.047 | 0.089 | No |
| Entry | Chiv5 | (bcutm1) | (MRVSA9) | (MRVSA6) | (PEOEVSA5) | GATSv4 | PIC50 |
|---|---|---|---|---|---|---|---|
| 1 | 0.494 | 2.343 | 0.000 | 0.00 | 0.00 | 0.92 | 4.78 |
| 2 | 1.112 | 2.884 | 7.299 | 0.00 | 58.270 | 0.99 | 4.25 |
| 3 | 1.710 | 4.917 | 15.109 | 73.32 | 68.807 | 1.07 | 4.24 |
| 4 | 2.873 | 4.816 | 28.237 | 81.41 | 77.896 | 1.15 | 3.91 |
| 5 | 4.027 | 3.201 | 35.342 | 93.71 | 80.100 | 1.21 | 5.66 |
| 6 | 5.630 | 4.663 | 55.317 | 98.22 | 107.636 | 1.23 | 6.25 |
| 7 | 6.449 | 4.371 | 39.441 | 107.11 | 144.675 | 1.37 | 6.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmmed, F.; Al-Mijalli, S.H.; Abdallah, E.M.; Eissa, I.H.; Ali, F.; Bhat, A.R.; Jamalis, J.; Ben Hadda, T.; Kawsar, S.M.A. Galactoside-Based Molecule Enhanced Antimicrobial Activity through Acyl Moiety Incorporation: Synthesis and In Silico Exploration for Therapeutic Target. Pharmaceuticals 2023, 16, 998. https://doi.org/10.3390/ph16070998
Ahmmed F, Al-Mijalli SH, Abdallah EM, Eissa IH, Ali F, Bhat AR, Jamalis J, Ben Hadda T, Kawsar SMA. Galactoside-Based Molecule Enhanced Antimicrobial Activity through Acyl Moiety Incorporation: Synthesis and In Silico Exploration for Therapeutic Target. Pharmaceuticals. 2023; 16(7):998. https://doi.org/10.3390/ph16070998
Chicago/Turabian StyleAhmmed, Faez, Samiah Hamad Al-Mijalli, Emad M. Abdallah, Ibrahim H. Eissa, Ferdausi Ali, Ajmal R. Bhat, Joazaizulfazli Jamalis, Taibi Ben Hadda, and Sarkar M. A. Kawsar. 2023. "Galactoside-Based Molecule Enhanced Antimicrobial Activity through Acyl Moiety Incorporation: Synthesis and In Silico Exploration for Therapeutic Target" Pharmaceuticals 16, no. 7: 998. https://doi.org/10.3390/ph16070998