

Bioanalytical LC-QTOF/MS Method for a N-phenylpiperazine Derivate (LQFM05): An Anxiolytic- and Antidepressant-like Prototype Drug Applied to Pharmacokinetic and Biodistribution Studies
 , , , and
, , , and
Abstract
:
1. Introduction
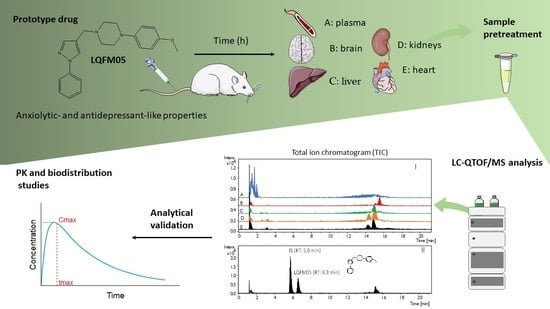
2. Results and Discussion
2.1. Method Development and Sample Preparation Strategies
2.2. Analytical Validation
2.3. Tissue Distribution Study
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Animals
3.3. LQFM05 Dosage Form Preparation
3.3.1. Drug Administration to Pharmacokinetic Studies
3.3.2. Drug Administration to Tissue Distribution Study
3.4. Preparation of Samples for LC-QTOF/MS Analysis
3.4.1. Stock Solutions, Calibration Standards and Quality Control Samples
3.4.2. Unkown Rat Plasma and Tissue Samples
3.5. Instrumentation
LC-QTOF/MS Conditions
3.6. Analytical Validation
3.6.1. Selectivity
3.6.2. Linearity
3.6.3. Precision and Accuracy
3.6.4. Recovery of the Extraction Procedure
3.6.5. Carry-Over Effects
3.6.6. Matrix Effect
3.6.7. Stability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

References
- Chen, X.-W.; Sun, Y.-Y.; Fu, L.; Li, J.-Q. Synthesis and Pharmacological Characterization of Novel N -(Trans -4-(2-(4-(Benzo[d]Isothiazol-3-Yl)Piperazin-1-Yl)Ethyl)Cyclohexyl) Amides as Potential Multireceptor Atypical Antipsychotics. Eur. J. Med. Chem. 2016, 123, 332–353. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.Y.; Teoh, S.L.; Wu, D.B.-C.; Kotirum, S.; Chiou, C.-F.; Chaiyakunapruk, N. Global Economic Burden of Schizophrenia: A Systematic Review. Neuropsychiatr. Dis. Treat. 2016, 12, 357–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mena, C.; Gonzalez-Valderrama, A.; Iruretagoyena, B.; Undurraga, J.; Crossley, N.A. Early Treatment Resistance in a Latin-American Cohort of Patients with Schizophrenia. Schizophr. Res. 2018, 199, 380–385. [Google Scholar] [CrossRef] [PubMed]
- da Silva Moreira, L.K.; Silva, R.R.; da Silva, D.M.; Mendes, M.A.S.; de Brito, A.F.; de Carvalho, F.S.; Sanz, G.; Rodrigues, M.F.; da Silva, A.C.G.; Thomaz, D.V.; et al. Anxiolytic- and Antidepressant-like Effects of New Phenylpiperazine Derivative LQFM005 and Its Hydroxylated Metabolite in Mice. Behav. Brain Res. 2022, 417, 113582. [Google Scholar] [CrossRef]
- Magotra, A.; Sharma, A.; Singh, S.; Ojha, P.K.; Kumar, S.; Bokolia, N.; Wazir, P.; Sharma, S.; Khan, I.A.; Singh, P.P.; et al. Physicochemical, Pharmacokinetic, Efficacy and Toxicity Profiling of a Potential Nitrofuranyl Methyl Piperazine Derivative IIIM-MCD-211 for Oral Tuberculosis Therapy via in-Silico—In-Vitro—In-Vivo Approach. Pulm. Pharmacol. Ther. 2018, 48, 151–160. [Google Scholar] [CrossRef]
- Pires, C.C.; Kaiser, M.; Grünspan, L.D.; Barreto, F.; Innocente, A.; Gnoatto, S.; Laureano, J.V.; Araujo, B.V.; Dalla Costa, T.; Tasso, L. Development and Validation of a Sensitive and Selective LC—MS/MS Method for the Determination of an Antimalarial Drug Candidate in Rat Plasma, and Its Application to a Preclinical Pharmacokinetic Study. Acta Chromatogr. 2016, 28, 439–453. [Google Scholar] [CrossRef] [Green Version]
- Verbanac, D. Predictive Methods as a Powerful Tool in Drug Discovery. Biochem. Med. 2010, 20, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Conrado, D.J.; Palma, E.C.; Fraga, C.A.M.; Barreiro, E.J.; Rates, S.M.K.; Dalla Costa, T. Development and Validation of a LC-MS/MS Method with Electrospray Ionization for Determination of LASSBio-579 in Rat Plasma. J. Pharm. Biomed. Anal. 2007, 43, 677–682. [Google Scholar] [CrossRef]
- de Freitas, D.F.; Porto, C.E.D.; Vieira, E.P.; de Siqueira, M.E.P.B. Three-Phase, Liquid-Phase Microextraction Combined with High Performance Liquid Chromatography-Fluorescence Detection for the Simultaneous Determination of Fluoxetine and Norfluoxetine in Human Plasma. J. Pharm. Biomed. Anal. 2010, 51, 170–177. [Google Scholar] [CrossRef]
- Siva Selva Kumar, M.; Ramanathan, M. Concurrent Determination of Olanzapine, Risperidone and 9-Hydroxyrisperidone in Human Plasma by Ultra Performance Liquid Chromatography with Diode Array Detection Method: Application to Pharmacokinetic Study. Biomed. Chromatogr. 2016, 30, 263–268. [Google Scholar] [CrossRef]
- Tasso, L.; Neves, G.; Menegatti, R.; Fraga, C.A.M.; Barreiro, E.; Eifler-Lima, V.; Rates, S.M.K.; Costa, T.D. Pharmacokinetics and Tissue Distribution of a New Heterocyclic N-Phenylpiperazine Derivative (LASSBio-581) in Rats. Eur. J. Pharm. Sci. 2005, 26, 194–202. [Google Scholar] [CrossRef]
- Haas, S.E.; Brum, L.; de Andrade, C.; Azeredo, F.J.; Pigatto, M.; Silva Torres, B.G.; Guterres, S.S.; Costa, T.D. Highly Sensitive LC-MS/MS Method for the Determination of Clozapine in Rat Plasma: Application to a Preclinical Pharmacokinetic Study. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 2873–2883. [Google Scholar] [CrossRef]
- Marghade, S.; Musmade, P.B.; Moorkoth, S. High-Performance Liquid Chromatographic Assay for Ziprasidone in Plasma Samples: Application to Pharmacokinetic Studies in Rats. J. Chromatogr. Sci. 2012, 50, 902–908. [Google Scholar] [CrossRef] [Green Version]
- Petruczynik, A.; Wróblewski, K.; Szultka-Młyńska, M.; Buszewski, B.; Karakuła-Juchnowicz, H.; Gajewski, J.; Morylowska-Topolska, J.; Waksmundzka-Hajnos, M. Determination of Some Psychotropic Drugs in Serum and Saliva Samples by HPLC-DAD and HPLC MS. J. Pharm. Biomed. Anal. 2016, 127, 68–80. [Google Scholar] [CrossRef]
- Silva, B.J.G.; Lanças, F.M.; Queiroz, M.E.C. Determination of Fluoxetine and Norfluoxetine Enantiomers in Human Plasma by Polypyrrole-Coated Capillary in-Tube Solid-Phase Microextraction Coupled with Liquid Chromatography-Fluorescence Detection. J. Chromatogr. A 2009, 1216, 8590–8597. [Google Scholar] [CrossRef]
- Brazil Agência Nacional de Vigilância Sanitária (ANVISA). Farmacopeia Brasileira, Monografias: Insumos e Especialidades, 6th ed.; Agência Nacional de Vigilância Sanitária SIA Trecho 5: Brasília, Brazil, 2019; Volume 2. [Google Scholar]
- Chemicalize. Calculate Properties Instantly, Search Chemical Data, and Draw Molecules Online. Available online: www.chemicalize.com (accessed on 30 November 2017).
- Araujo, P. Key Aspects of Analytical Method Validation and Linearity Evaluation. J. Chromatogr. B 2009, 877, 2224–2234. [Google Scholar] [CrossRef]
- Miller, J.N.; Miller, J.C. Statistics and Chemometrics for Analytical Chemistry, 5th ed.; Pearson Education Limited: Edinburgh Gate, London, UK, 2005; ISBN 0 131 29192 0. [Google Scholar]
- Raposo, F. Evaluation of Analytical Calibration Based on Least-Squares Linear Regression for Instrumental Techniques: A Tutorial Review. TrAC Trends Anal. Chem. 2016, 77, 167–185. [Google Scholar] [CrossRef]
- Jurado, J.M.; Alcázar, A.; Muñiz-Valencia, R.; Ceballos-Magaña, S.G.; Raposo, F. Some Practical Considerations for Linearity Assessment of Calibration Curves as Function of Concentration Levels According to the Fitness-for-Purpose Approach. Talanta 2017, 172, 221–229. [Google Scholar] [CrossRef]
- Kadian, N.; Raju, K.S.R.; Rashid, M.; Malik, M.Y.; Taneja, I.; Wahajuddin, M. Comparative Assessment of Bioanalytical Method Validation Guidelines for Pharmaceutical Industry. J. Pharm. Biomed. Anal. 2016, 126, 83–97. [Google Scholar] [CrossRef]
- Chambers, E.; Wagrowski-Diehl, D.M.; Lu, Z.; Mazzeo, J.R. Systematic and Comprehensive Strategy for Reducing Matrix Effects in LC/MS/MS Analyses. J. Chromatogr. B 2007, 852, 22–34. [Google Scholar] [CrossRef]
- Moein, M.M.; El Beqqali, A.; Abdel-Rehim, M. Bioanalytical Method Development and Validation: Critical Concepts and Strategies. J. Chromatogr. B 2017, 1043, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.; Tozer, T.N. Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications, 4th ed.; Lippincott William & Wilkins: Philadelphia, PA, USA, 2011; ISBN 9780781750097. [Google Scholar]
- Conrado, D.J.; Verli, H.; Neves, G.; Fraga, C.A.M.; Barreiro, E.J.; Rates, S.M.K.; Costa, T.D. Pharmacokinetic Evaluation of LASSBio-579: An N -Phenylpiperazine Antipsychotic Prototype. J. Pharm. Pharmacol. 2008, 60, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Carreño, F.; Paese, K.; Silva, C.M.; Guterres, S.S.; Dalla Costa, T. Pre-Clinical Investigation of the Modulation of Quetiapine Plasma Pharmacokinetics and Tissues Biodistribution by Lipid-Core Nanocapsules. J. Pharm. Biomed. Anal. 2016, 119, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Laxman, T.S.; Puttrevu, S.K.; Pradhan, R.; Mishra, A.; Verma, S.; Chhonker, Y.S.; Srivastava, S.; Singh, S.P.; Sashidhara, K.V.; Bhatta, R.S. Pharmacokinetics, Metabolism, Bioavailability, Tissue Distribution and Excretion Studies of 16α-Hydroxycleroda-3, 13(14) Z -Dien-15, 16-Olide—A Novel HMG-CoA Reductase Inhibitor. Naunyn. Schmiedebergs. Arch. Pharmacol. 2018, 391, 965–973. [Google Scholar] [CrossRef]
- Brazil Ministério da Saúde; ANVISA. RESOLUÇÃO RDC No 166, DE 24 DE JULHO DE 2017. Available online: https://www.in.gov.br/materia/-/asset_publisher/Kujrw0TZC2Mb/content/id/19194581/do1-2017-07-25-resolucao-rdc-n-166-de-24-de-julho-de-2017-19194412 (accessed on 27 September 2022).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Plasma | Brain | Heart | Liver | Kidneys |
|---|---|---|---|---|---|
| Slope a (±SD, n = 4) | 1.761 × 10−3± 1.587 × 10−5 | 9.882 × 10−4 ± 8.168 × 10−6 | 1.239 × 10−3 ± 1.985 × 10−5 | 1.692 × 10−3 ± 7.928 × 10−6 | 1.127 × 10−3 ± 1.655 × 10−5 |
| Intercept a (±SD, n = 4) | 1.081 × 10−3 ± 7.043 × 10−3 | 0.905 × 10−2 ± 0.362 × 10−2 | −0.7043 × 10−3 ± 0.8809 × 10−2 | 7.30 × 10−3 ± 3.518 × 10−3 | 8.292 × 10−3 ± 7.345 × 10−3 |
| r2 b | 0.9969 | 0.9974 | 0.9903 | 0.9992 | 0.9919 |
| RSDslope (%) c | 0.901 | 0.827 | 1.602 | 0.469 | 1.469 |
| Fcalculated d | 1.351 | 1.257 | 0.285 | 1.062 | 0.641 |
| Ccalculated e | 0.201 | 0.226 | 0.204 | 0.073 | 0.191 |
| Matrix | Cspiked (ng/mL) | Intra (n = 6) | Inter (n = 12) | ||||
|---|---|---|---|---|---|---|---|
| CM Mean ± SD (ng/mL) | RSD% | RE% | CM Mean ± SD (ng/mL) | RSD% | RE% | ||
| Plasma | 10 | 9.66 ± 0.51 | 5.3 | −3.4 | 9.98 ± 0.72 | 7.2 | −0.2 |
| 30 | 31.00 ± 1.37 | 4.4 | 3.3 | 30.71 ± 1.57 | 5.1 | 2.4 | |
| 450 | 455.58 ± 9.50 | 2.1 | 1.2 | 453.78 ± 8.97 | 2.0 | 0.8 | |
| 750 | 754.80 ± 10.30 | 1.4 | 0.6 | 754.24 ± 10.06 | 1.3 | 0.6 | |
| Brain | 10 | 9.44 ± 0.56 | 6.0 | −0.2 | 9.76 ± 0.79 | 8.1 | −2.4 |
| 30 | 29.31 ± 1.47 | 5.0 | −0.1 | 29.36 ± 1.39 | 4.7 | −2.1 | |
| 450 | 432.76 ± 3.20 | 0.7 | −0.2 | 441.38 ± 9.34 | 2.1 | −1.9 | |
| 750 | 727.68 ± 7.53 | 1.0 | −4.2 | 743.67 ± 18.95 | 2.5 | −0.8 | |
| Heart | 10 | 9.85 ± 0.30 | 3.4 | −1.5 | 9.92 ± 0.25 | 2.6 | −0.8 |
| 30 | 30.06 ± 0.12 | 0.4 | 0.2 | 30.22 ± 0.42 | 1.4 | 0.7 | |
| 450 | 451.04 ± 8.07 | 1.8 | 0.2 | 449.65 ± 6.04 | 1.3 | −0.1 | |
| 750 | 750.11 ± 11.61 | 1.5 | 0.0 | 751.43 ± 9.65 | 1.3 | 0.2 | |
| Liver | 10 | 10.22 ± 0.37 | 3.6 | 2.2 | 10.30 ± 0.35 | 3.4 | 3.0 |
| 30 | 30.16 ± 0.86 | 2.9 | 0.5 | 30.36 ± 0.76 | 2.5 | 1.2 | |
| 450 | 449.78 ± 7.87 | 1.8 | 0.1 | 449.19 ± 6.16 | 1.4 | −0.2 | |
| 750 | 751.24 ± 8.68 | 1.2 | 0.2 | 749.57 ± 8.92 | 1.2 | −0.1 | |
| Kidneys | 10 | 10.59 ± 0.71 | 6.7 | 5.9 | 10.49 ± 0.71 | 6.7 | 4.9 |
| 30 | 29.84 ± 0.77 | 2.6 | −0.5 | 29.94 ± 0.75 | 2.5 | −0.2 | |
| 450 | 452.49 ± 10.95 | 2.4 | 0.6 | 453.60 ± 8.57 | 1.9 | 0.8 | |
| 750 | 756.41 ± 10.11 | 1.3 | 0.9 | 752.52 ± 8.93 | 1.2 | 0.3 | |
| Matrix | Cspiked (ng/mL) | Recovery | Matrix Effect (NMF) | |||
|---|---|---|---|---|---|---|
| Mean ± SD (%) | RSD (%) | Mean ± SD | RSD (%) | |||
| Plasma | LQFM05 | 30 | 55.53 ± 4.63 | 8.3 | 1.02 ± 0.09 | 8.8 |
| 450 | 56.82 ± 5.0 | 8.7 | 1.04 ± 0.06 | 5.8 | ||
| 750 | 56.22 ± 3.70 | 6.6 | 1.03 ± 0.03 | 2.9 | ||
| IS | 250 | 79.50 ± 4.93 | 6.2 | 1.09 ± 0.04 | 4.1 | |
| Brain | LQFM05 | 30 | 45.84 ± 3.82 | 8.3 | 1.09 ± 0.04 | 4.1 |
| 450 | 47.54 ± 2.03 | 4.3 | 1.05 ± 0.04 | 3.6 | ||
| 750 | 43.26 ± 1.62 | 3.7 | 1.06 ± 0.03 | 2.7 | ||
| IS | 250 | 94.38 ± 4.50 | 4.8 | 0.99 ± 0.07 | 7.3 | |
| Heart | LQFM05 | 30 | 68.94 ± 3.87 | 5.6 | 1.04 ± 0.04 | 3.5 |
| 450 | 63.47 ± 1.84 | 2.9 | 1.00 ± 0.05 | 4.5 | ||
| 750 | 65.28 ± 3.05 | 4.7 | 0.98 ± 0.04 | 4.4 | ||
| IS | 250 | 99.61 ± 5.89 | 5.9 | 1.02 ± 0.05 | 5.1 | |
| Liver | LQFM05 | 30 | 71.82 ± 4.79 | 6.7 | 0.99 ± 0.04 | 3.7 |
| 450 | 59.13 ± 2.92 | 4.9 | 0.98 ± 0.06 | 6.3 | ||
| 750 | 70.75 ± 5.03 | 7.1 | 1.00 ± 0.04 | 3.7 | ||
| IS | 250 | 82.38 ± 7.04 | 8.5 | 1.00 ± 0.03 | 3.0 | |
| Kidneys | LQFM05 | 30 | 63.77 ± 6.97 | 10.9 | 1.05 ± 0.04 | 3.6 |
| 450 | 55.40 ± 2.53 | 4.6 | 1.06 ± 0.03 | 2.7 | ||
| 750 | 66.50 ± 2.38 | 3.6 | 0.99 ± 0.07 | 7.3 | ||
| IS | 250 | 98.31 ± 3.70 | 3.8 | 1.04 ± 0.04 | 3.5 | |
| Matrix | AUC(0–∞) (h*ng/g) | tmax (h) | Cmax (ng/g) | Kβ (h−1) | t1/2β (h) | MRT (h) | Kp b |
|---|---|---|---|---|---|---|---|
| Plasma | 5366.7 a | - | - | 0.31 | 2.3 | 1.7 | - |
| Brain | 10,460.6 | 0.08 | 12,357.0 | 0.28 | 2.5 | 2.0 | 1.9 |
| Heart | 10,919.6 | 0.08 | 7430.9 | 0.33 | 2.1 | 2.1 | 2.0 |
| Liver | 8235.3 | 0.17 | 4922.9 | 0.19 | 3.6 | 3.1 | 1.5 |
| Kidneys | 14,595.2 | 0.08 | 8768.0 | 0.30 | 2.3 | 2.4 | 2.7 |
| No. | Compounds | MRM Transition (m/z) | CE *(eV) | RT ** (min) | |
|---|---|---|---|---|---|
| Quantifier | Qualifier | ||||
| 1 | Diazepam (IS) | 285.08 → 257.09 | 20 | 5.6 | |
| 2 | LQFM05 | 349.21 → 157.08 | 349.21 → 191 | 15 | 6.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos, A.C.M.; Rezende, K.R.; Teixeira, C.M.; Fernandes, A.R.; Santos, H.; Machado, R.D.; Menegatti, R.; Vaz, B.G.; Chaves, A.R. Bioanalytical LC-QTOF/MS Method for a N-phenylpiperazine Derivate (LQFM05): An Anxiolytic- and Antidepressant-like Prototype Drug Applied to Pharmacokinetic and Biodistribution Studies. Pharmaceuticals 2023, 16, 930. https://doi.org/10.3390/ph16070930
Ramos ACM, Rezende KR, Teixeira CM, Fernandes AR, Santos H, Machado RD, Menegatti R, Vaz BG, Chaves AR. Bioanalytical LC-QTOF/MS Method for a N-phenylpiperazine Derivate (LQFM05): An Anxiolytic- and Antidepressant-like Prototype Drug Applied to Pharmacokinetic and Biodistribution Studies. Pharmaceuticals. 2023; 16(7):930. https://doi.org/10.3390/ph16070930
Chicago/Turabian StyleRamos, Ana Cláudia M., Kênnia R. Rezende, Carolina M. Teixeira, Aline R. Fernandes, Heloa Santos, Rúbia Darc Machado, Ricardo Menegatti, Boniek G. Vaz, and Andréa R. Chaves. 2023. "Bioanalytical LC-QTOF/MS Method for a N-phenylpiperazine Derivate (LQFM05): An Anxiolytic- and Antidepressant-like Prototype Drug Applied to Pharmacokinetic and Biodistribution Studies" Pharmaceuticals 16, no. 7: 930. https://doi.org/10.3390/ph16070930